
Abstract
In the current study, we sequenced the complete plastome of Monstera deliciosa Liebm. (1849), an attractive foliage plant. The total length of the plastome of M. deliciosa is 163,499 bp and it consists of three distinct regions: a large single-copy (90,092 bp), a small single-copy (21,737 bp), and a pair of inverted repeats (IRs, 25,835 bp). The overall GC content is 36.2%, and the genome contains 110 functional genes, excluding pseudogenes. These functional genes encompass 77 protein-coding genes, 29 transfer RNA genes, and four ribosomal RNA genes. Notably, both the infA and rpl23 genes have been identified as pseudogenes. Phylogenetic analysis based on 14 representative plastomes from seven subfamilies indicates that Monsteroideae is monophyletic and sister to Pothoideae. Furthermore, M. deliciosa and M. adansonii were shown to share a recent common ancestor, the finding for which is supported by a strong bootstrap value. The sequenced plastome of M. deliciosa can serve as a valuable resource for establishing phylogenetic relationships and enhancing species identification within the genus Monstera. In addition, it can facilitate investigations into the genetic characteristics of this plant.
Introduction
The genus Monstera Adans. (Araceae) is classified under the subfamily Monsteroideae and tribe Monstereae (Cabrera et al. Citation2008). Comprising over 60 species, the Monstera species is native to tropical America and grows in the regions between Mexico and Southern Brazil (Mayo et al. Citation1998; World Plants Citation2023, www.worldplants.de). New species of Monstera are continuously being reported, as evidenced by studies from Zuluaga and Cameron (Citation2018), Cedeño-Fonseca et al. (Citation2020), and Cedeño-Fonseca et al. (Citation2021). Despite ongoing research, the taxonomy and phylogeny of this genus remain relatively understudied. This is primarily due to the wide infraspecific variation observed within the genus, which presents challenges in species identification using traditionally diagnostic morphological features (Madison Citation1977). Consequently, further investigation and the development of alternative approaches to address these challenges are needed. One promising approach is the use of plastome sequences, which provides genetic insights for species delimitation and the determination of phylogenetic relationships within Monstera.
Monstera deliciosa Liebm. (1849), which originated in Central America between Mexico and Guatemala, has gained popularity as an ornamental foliage plant and has been introduced to many countries, including South Korea, Japan, and China for the same purpose (Lee and Kim Citation1980; Long et al. Citation2017; Sakamoto et al. Citation2022). Recently, there has been growing interest in the chimeric forms of M. deliciosa, characterized by the presence of both green and albino tissues (Li et al. Citation2021). This intriguing phenomenon has sparked increased attention and research in horticultural genetic research (Park et al. Citation2023). Despite the significance of M. deliciosa in horticulture, there has been a lack of research utilizing the whole plastome of M. deliciosa as a molecular genetic resource. Therefore, the objective of this paper is to bridge this gap by providing the complete plastome sequence of M. deliciosa, which allows for further investigations into the genetic traits of this plant.
Materials and methods
Monstera deliciosa was obtained from the Gangwon Provincial Arboretum, South Korea (37°55′22.0″N, 127°43′32.0″E, voucher number PCGE-KHR2023001, ) and grown in a growth room at Kangwon National University until needed for analysis (contact to Hyeran Kim, [email protected]). Genomic DNA was extracted from 100 mg of fresh leaf using the Exgene Plant SV mini kit (GenAll Biotechnology, Seoul, South Korea), following the manufacturer’s instructions. The extracted DNA concentration was determined using a spectrophotometer, while the integrity was confirmed through electrophoresis using a 1% agarose gel.
Figure 1. Monstera deliciosa Liebm. (A) Leaves with natural holes; (B) wrinkled geniculum at the petiole; (C) roots with aerial adventitious roots. All photos were taken by Yun at the Gangwon Provincial Arboretum, South Korea.

Sequencing was performed on the Illumina NovaSeq 6000 platform (Macrogen, Seoul, South Korea). Following this, de novo assembly of the plastome was conducted with NOVOplasty ver. 4.3 (Dierckxsens et al. Citation2017), using a 39 k-mer length as well as a fragment of the rbcL gene from Monstera adansonii as the seed sequence. The coverage depth of completely sequenced M. deliciosa was determined by performing read mapping (Figure S1). The plastome genes to be annotated were identified using CPGAVAS2 (Shi et al. Citation2019) and tRNAscan-SE 2.0 software with the default settings (Schattner et al. Citation2005). The annotation of protein-coding regions was further validated by searching for homologous genes using the NCBI Conserved Domain Database (CDD) (Marchler-Bauer et al. Citation2011). The circular plastome map of M. deliciosa was generated using the OGDRAW ver. 1.3.1 program (Greiner et al. Citation2019).
For phylogenetic analysis, 14 species were included, representing seven subfamilies within the Araceae family. However, the subfamily Gymnostachydoideae was excluded from the analysis due to the unavailability of plastome data. Additionally, Alisma plantago-aquatica from the Alismataceae family was used as an outgroup. The alignment of whole plastome sequences was carried out using MAFFT ver. 7 (Katoh and Standley Citation2013), while gaps and poorly aligned positions were refined using Gblocks v.0.91b (Castresana Citation2000). Lastly, the phylogenetic tree was constructed using the maximum-likelihood method implemented in IQ-TREE v.1.6.7 (Nguyen et al. Citation2015), with 1000 bootstrap replicates. The plastome sequence of M. deliciosa was deposited in GenBank under the accession number OR260879.
Results
The plastome of M. deliciosa is 163,499 bp in size and exhibits a circular quadripartite structure consisting of a large single-copy (LSC, 90,092 bp), a small single-copy (SSC, 21,737 bp), and a pair of inverted repeat regions (IRs, each 25,835 bp) (). The overall GC content is 36.2%, with variation observed among the different regions: 34.6% in the LSC region, 28.9% in the SSC region, and 42.0% in the IR region.
Figure 2. The plastome map of Monstera deliciosa. Genes positioned outside the circle are transcribed in a counterclockwise direction, while genes within the inner circle are transcribed clockwise. The GC content is depicted by the dark grey inner circle.
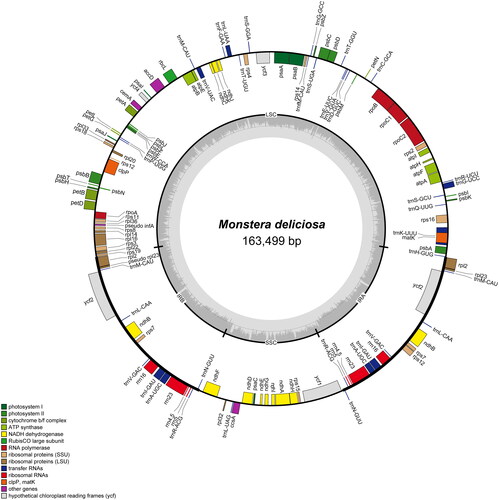
It was found that M. deliciosa has a total of 110 functional genes, excluding pseudogenes. These genes include 77 protein-coding genes, 29 tRNA genes, and four rRNA genes. Among these, 12 protein-coding genes and six tRNA genes possess multiple exons (Figure S2). Notably, the infA gene in the LSC region, as well as the rpl23 gene in both IR regions, each exhibit one internal stop codon, implying that these genes are nonfunctional. The loss of the infA gene has been observed in all subfamilies other than basal group Orontioideae.
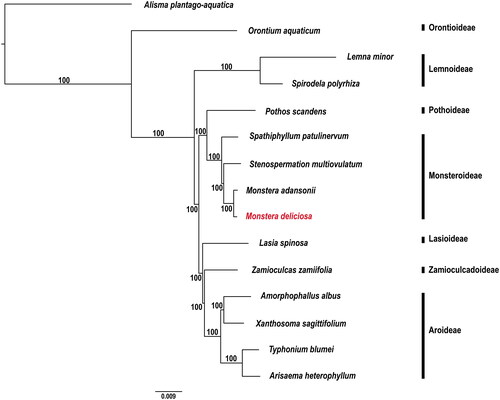
As evidenced by the phylogenetic tree, Monsteroideae forms a monophyletic group and presents as the sister clade to Pothoideae (). Furthermore, within the Monsteroideae clade, it can be noted that M. deliciosa and M. adansonii share a recent common ancestor. All relationships among species in the tree are strongly supported by high bootstrap values (BS = 100).
Figure 3. Phylogenetic tree generated using the maximum-likelihood method implemented in IQ-TREE v.1.6.7, based on complete plastome sequences. The species sequenced in this study is indicated in red and the accession numbers of species used for phylogenetic analysis are as follows: Alisma plantago-aquatica (MK090659), Orontium aquaticum (MT226773), Lemna minor (DQ400350), Spirodela polyrhiza (MN419335), Pothos scandens (MN046891), Spathiphyllum patulinervum (MN477425), Stenospermation multiovulatum (MN046893), Monstera adansonii (MN046888), Monstera deliciosa (OR260879), Lasia spinosa (MT226772), Zamioculcas zamiifolia (MT226775), Amorphophallus albus (OP531918), Xanthosoma sagittifolium (MW628970), Typhonium blumei (MT872311), and Arisaema heterophyllum (ON060885).
Discussion and conclusions
This study reports the first complete plastome sequence of M. deliciosa. It was found that M. deliciosa displays the conserved quadripartite structure that has been observed in previous reports on plastome structure by Palmer (Citation1985). In comparison with other species within the Araceae family, M. deliciosa demonstrated a high level of similarity in terms of gene composition, gene order, and the number of genes (Henriquez et al. Citation2020a). However, in line with previous studies on Monsteroideae (Henriquez et al. Citation2020a, Citation2020b), a nonfunctional infA gene was identified as characterized by the presence of an internal stop codon in the LSC region. The loss of the infA gene, observed in all subfamilies other than Orontioideae, serves as a synapomorphy for the clade consisting of six subfamilies: Lemnoideae to Aroideae. Conversely, the rpl23 gene in M. deliciosa was also shown to exhibit one internal stop codon, which consequently distinguishes it from other Monsteroideae species. Our phylogenetic tree is consistent with a previous study conducted by Henriquez et al. (Citation2014), providing further support for the monophyly of Monsteroideae and its sister relationship with Pothoideae.
Given the advantages of uniparental inheritance and the highly conserved nature of plastomes, which still provide sufficient information to capture evolutionary events (Raubeson and Jansen Citation2005), the complete plastome sequence of M. deliciosa will prove to be of considerable value in various research areas. For example, it can serve as a resource for the establishment of phylogenetic relationships within the Monsteroideae subfamily and facilitate elucidation of its evolutionary history. In addition, the plastome sequence can aid in the development of molecular markers capable of distinguishing between different Monstera species. In turn, these markers can contribute to more accurate species identification and subsequent classification. Ultimately, the plastome of M. deliciosa can facilitate investigations into its own horticultural value and genetic characteristics.
Author contributions
H.K. conceived this study and collected the sample. H.K. and S.Y. performed the experiments. S.Y. analyzed the data and wrote the manuscript. H.K. and S.Y. revised the manuscript.
Supplemental Material
Download MS Word (366.7 KB)Acknowledgements
Both authors thank the PCGE lab members, especially Yeon Hee Hyoung, for their technical assistance. We thank Gangwon Provincial Arboretum for providing the sample of Monstera deliciosa.
Disclosure statement
The authors declare that there is no conflict of interest regarding the publication of this article.
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank on the NCBI database at https://www.ncbi.nlm.nih.gov/ under the accession no. OR260879. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA992674, SRR25232409, and SAMN36379614, respectively.
Additional information
Funding
References
- Cabrera LI, Salazar GA, Chase MW, Mayo SJ, Bogner J, Dávila P. 2008. Phylogenetic relationships of aroids and duckweeds (Araceae) inferred from coding and noncoding plastid DNA. Am J Bot. 95(9):1153–1165. doi: 10.3732/ajb.0800073.
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17(4):540–552. doi: 10.1093/oxfordjournals.molbev.a026334.
- Cedeño-Fonseca M, Croat TB, Zuluaga A, Mittermeier M, Blanco MA. 2020. Two new species of Monstera (Araceae: Monsteroideae) from Costa Rica. Phytotaxa. 461(3):185–194. doi: 10.11646/phytotaxa.461.3.5.
- Cedeño-Fonseca M, Ortiz OO, Zuluaga A, Grayum MH, Croat TB. 2021. Four new species of Monstera (Araceae) from Panama, including one with the largest leaves and another with the largest inflorescences in the genus. Webbia. 76(2):265–279. doi: 10.36253/jopt-10807.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18. doi: 10.1093/nar/gkw955.
- Greiner S, Lehwark P, Bock R. 2019. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47(W1):W59–W64. doi: 10.1093/nar/gkz238.
- Henriquez CL, Arias T, Pires JC, Croat TB, Schaal BA. 2014. Phylogenomics of the plant family Araceae. Mol Phylogenet Evol. 75:91–102. doi: 10.1016/j.ympev.2014.02.017.
- Henriquez CL, Abdullah , Ahmed I, Carlsen MM, Zuluaga A, Croat TB, McKain MR. 2020a. Molecular evolution of chloroplast genomes in Monsteroideae (Araceae). Planta. 251(3):72. doi: 10.1007/s00425-020-03365-7.
- Henriquez CL, Abdullah , Ahmed I, Carlsen MM, Zuluaga A, Croat TB, McKain MR. 2020b. Evolutionary dynamics of chloroplast genomes in subfamily Aroideae (Araceae). Genomics. 112(3):2349–2360. doi: 10.1016/j.ygeno.2020.01.006.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. doi: 10.1093/molbev/mst010.
- Lee JS, Kim MH. 1980. A study on the species of introduced landscape and cultivated plants in Jeju Island, Korea (I). Jeju Natl Univ. 12:97–115.
- Li F, Jin C, Zhang L, Wang J. 2021. Hyper-recombinant plants: an emerging field for plant breeding. Crit Rev Plant Sci. 40(5):446–458. doi: 10.1080/07352689.2021.1985819.
- Long C, Fang Q, Long B, Ji Y, Shu H, Luo B, Liu B. 2017. Ornamental aroids (Araceae) in China. Sbornik naučnyh trudov. 145:125–131.
- Madison M. 1977. A revision of Monstera (Araceae). Contrib Gray Herb Harvard Univ. 206–208(207):3–100. doi: 10.5962/p.336443.
- Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, et al. 2011. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39(Database issue):D225–D229. doi: 10.1093/nar/gkq1189.
- Mayo SJ, Bogner J, Boyce PC. 1998. Araceae. In: Kubitzki K, editor. Flowering plants. Monocotyledons. Berlin, Heidelberg: Springer Berlin Heidelberg; p. 26–74.
- Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274. doi: 10.1093/molbev/msu300.
- Palmer JD. 1985. Comparative organization of chloroplast genomes. Annu Rev Genet. 19(1):325–354. doi: 10.1146/annurev.ge.19.120185.001545.
- Park H-S, Jeon J-H, Cho W, Lee Y, Park JY, Kim J, Park YS, Koo HJ, Kang JH, Lee TJ, et al. 2023. High-throughput discovery of plastid genes causing albino phenotypes in ornamental chimeric plants. Hortic Res. 10(1):uhac246. doi: 10.1093/hr/uhac246.
- Raubeson LA, Jansen RK. 2005. Chloroplast genomes of plants. In: Henry RJ, editor. Plant diversity and evolution: genotypic and phenotypic variation in higher plants. Wallingford (UK): CABI Publishing; p. 45–68.
- Sakamoto A, Uzuhashi S, Hoshi H, Kubota M, Horie H, Kakishima M. 2022. First report of rust caused by Puccinia paullula on Monstera deliciosa and M. adansonii in Japan. Plant Dis. 107(2):570. doi: 10.1094/PDIS-05-22-1035-PDN.
- Schattner P, Brooks AN, Lowe TM. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33(Web Server issue):W686–W689. doi: 10.1093/nar/gki366.
- Shi L, Chen H, Jiang M, Wang L, Wu X, Huang L, Liu C. 2019. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47(W1):W65–W73. doi: 10.1093/nar/gkz345.
- World Plants. 2023. Synonymic checklist and distribution of the world flora. Version 16.1; [updated 2023 Jun 16; accessed 6 Jul 2023]. www.worldplants.de.
- Zuluaga A, Cameron KM. 2018. Two new species of Monstera (Araceae: Monsteroideae) with entire leaves from Panama and Costa Rica. Phytotaxa. 334(1):1–9. doi: 10.11646/phytotaxa.334.1.1.