
Abstract
The present study firstly reported a complete mitochondrial genome of Minois paupera (Alphéraky, 1888), a Satyrinae species endemic to China. This mitogenome is circular, 15,213 bp in length, and consists of 37 typical mitochondrial genes, including 13 protein-coding genes (PCGs), 22 tRNAs, and two rRNAs. The phylogenetic position was inferred using 31 previously published complete mitogenomes, and the results reveal that M. paupera is the most closely related to Minois dryas. The complete mitogenome of M. paupera provides useful genetic information for further research on the phylogeography and phylogeny of the genus Minois.
1. Introduction
The genus Minois Hübner, which belongs to ‘Satyrinae,’ known as the mimicry eye spots, is mostly distributed in Palearctic, including one widespread species and several regional endemic species. The genetic relationships of species under this genus are still unclear (Sbordoni et al. Citation2018). In addition, the unique geographical distribution pattern of this population may be related to historical geological events, which can be used as an excellent research material for biogeography and species differentiation. However, up to now, there are only one complete mitochondrial genome of the dispersed species Minois had been reported, hindering further studies of phylogenetic and phylogeography.
Minois paupera (Alphéraky, 1888), belongs to Minois and is endemic to China (including in Gansu, Qinghai, Sichuan, N.W. Yunnan, S.E. Tibet) (Lang Citation2022). The previous studies mainly focused on taxonomy and mostly based on morphological features, lacking the support of molecular evidence. In this study, we sequenced the complete mitochondrial DNA genome of M. paupera to provide baseline data for better understanding its relationship within the genus Minois.
2. Materials and methods
2.1. Sample collection and preservation
The specimen was collected from Yongjing County, Gansu Province (103°23'32.65"E, 36°4'5.50"N, 2,160 m). Three legs on the same side were extracted and preserved in ethanol. In terms of morphological characteristics, the dorsal surface yellowish or orange rings around the postdiscal ocelli present on both wings. In addition, male dorsal forewing brand well developed in spaces 1b and 2, and often extending into space 3. The dorsal hindwing postdiscal ocellus in space 2 well developed (Lang Citation2022). The spread male specimens () were deposited in the Natural History Museum of Sichuan University, Chengdu, China (specimen numbers: SZM742205, contact person: Liang Dou, [email protected]).
Figure 1. The specimen of Minois paupera (Alphéraky, 1888) used in this study, upperside on the left, underside on the right, scale bar = 10 mm. Photographed and processed by wen-qian Hu.

2.2. DNA extraction sequencing and genomic assembling
Genomic DNA was extracted from the three legs of a single individual butterfly using the Sangon Rapid Animal Genome DNA Isolation Kit (Shanghai, China). The library preparation and next generation sequencing was finished by Sangon Biotech (Shanghai) Co., Ltd. The libraries were pooled and loaded on Novaseq 6000 (Illumina, San Diego, USA) sequencer by 2 × 150bp paired end sequence kit according to the manufacture’s instructions.
Rawbases yielded at least 6 GB were used for downstream analysis. All of the raw reads were trimmed by Fastqc 0.11.2 and assembled the raw sequence reads into contigs by SPAdes 3.15 (Bankevich et al. Citation2012). The coverage depth is 1x∼500x, mean: 190x (Figure S1, Table S1). Finally, complete mitochondrial genome was achieved using the contigs hit against the reference mitochondrial genome as seed sequence through the software MITObim 1.9.1 (Hahn et al. Citation2013).
2.3. Annotation and phylogenetic analysis
The CDS gene boundary was obtained by reverse comparison with the reference genomes of closely related species through NCBI Blast+ 2.28 and GeneWise (Birney et al. 2004). MiTFi (Jühling et al. Citation2012) was used to obtain tRNA sequence annotation, cmsearch (rfam.cm) identifies non-coding RNA by comparison. Finally, summarizes and collates the complete annotation results. The phylogenetic tree was constructed using PhyloSuite 1.2.3 (Zhang et al. Citation2020) based on concatenated nucleotide sequences of 13 PCGs and two rRNAs of M. paupera, other 29 representatives from 3 subfamilies of the Nymphalidae, and two outgroup species from Lycaenidae. The nucleotide sequences datasets were initially aligned in batches with MAFFT v7.505 (Katoh and Standley Citation2013). Ambiguously aligned fragments within the alignments of the 13 PCGs and gap sites within the rRNA sequences were subsequently removed using Gblocks 0.91b (Talavera and Castresana Citation2007) and trimAl v1.2rev57 (Capella-Gutiérrez et al. Citation2009), respectively. Maximum likelihood (ML) phylogenies were inferred using IQ-TREE v2.2.0 (Nguyen et al. Citation2015) under the optimal GTR + F + R5 model identified by ModelFinder (Kalyaanamoorthy et al. Citation2017) for 5000 ultrafast (Minh et al. Citation2013) bootstraps. In addition, CodonW (http://codonw.sourceforge.net//culong.html) was used to analyze The Relative Synonymous Codon Usage (RSCU).
3. Results and discussion
3.1. Characteristics of M. paupera genome
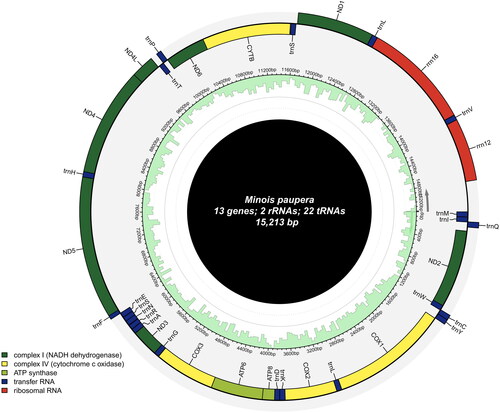
The mitochondrial genome of M. paupera is 15,213 bp in length (GenBank accession number: OR944656), with AT content (80.2%) significantly higher than GC Ratio (19.81%). The genome contains 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), and two ribosomal RNA genes (rRNAs), The plus (+) strand encodes nad2, cox1, cox2, atp8, atp6, cox3, nad3, nad6, and cytb, while the minus (−) strand encodes nad1, nad4l, nad4 and nad5 (Figure S2). The gene content and arrangement are highly conserved, exhibiting typical characteristics of other Minois species (Yang et al. Citation2020) (Figure S2, Table S2). In addition, codon bias analysis reveals that among the amino acids, arginine, leucine, and serine exhibit the highest codon bias scores, indicating the most pronounced codon preference. These amino acids show the highest level of genetic variation within the genome (Figure S3) ().
Figure 2. The circular-mapping mitochondrial genome of Minois paupera: the total length was 15,213 bp, which was divided into 37 genes, including 13 PCGs, 22 tRNAs, two rRNAs.
3.2. Phylogenetic position
The ML phylogenetic tree reveals that M. paupera is most closely related to M. dryas, while the genus Minois forms a cluster with Oeneis and Davidina, both supported by 100% values. Furthermore, the monophyly of the genus Minois is well supported in phylogenetic analyses, which aligns with previous phylogenetic studies (Yang et al. Citation2020; Zhou et al. Citation2020) and morphological classification research (medium or large size butterflies; antennal club slender, not spatulate as in genus Hipparchia; forewing subcostal vein dilated at base, vein 1b not thickened; hindwing outer margin scalloped, slightly in males, more deeply in females; male upf androconial patch present or absent; forewing two large single-pupilled postdiscal ocelli always present in space 2 andspace 5) (Lang Citation2022). Notably, Minois species have traditionally been classified within the genus Satyrus, however, there is a dearth of comprehensive molecular data for further validation. Moreover, except for M. dryas, all species in this genus are exclusively found in China and exhibit predominantly regional distributions, but the genetic relationship between these species is still unclear. Therefore, it is imperative to conduct further molecular investigations at the mitochondrial genome level or beyond to elucidate the phylogenetic relationships within this genus ().
Figure 3. The Maximum likelihood (ML) phylogenetic tree contains Minois paupera (Alphéraky, 1888) (marked with bold font), 29 nymphalidae and 2 lycaenidae, The numbers on each branch are bootstrap support values out of 5000 replicates. The GenBank accession number followed by each species name, and the vertical lines on the side indicate families and subfamilies. The following sequences were used: MN242789 (Yang et al. Citation2020), KF590543 (Wu et al. Citation2014), NC026838 (Fan et al. Citation2016), KJ547676 (Teixeira da Costa Citation2016), MN242790 (Yang et al. Citation2020), NC025762 (Tang et al. Citation2014), MN756798 (Zhou et al. Citation2020), KF906487 (Zhang et al. Citation2016), MN917147 (Zhou et al. Citation2020), MN242787 (Yang et al. 2020), GQ868707 (Kim et al. 2010), NC024550 (Huang et al. Citation2016), KF590553 (Wu et al. Citation2014), MN242788 (Yang et al. Citation2020), NC021370 (Shi et al. Citation2013), KF590538 (Wu et al. Citation2014), NC016196 (Qin et al. Citation2012).

4. Conclusions
In the present study, the complete mitogenome of M. paupera was assembled and analyzed. It will provide useful information for improving the taxonomic system of Minois. We found the phylogenetic position of M. paupera within the subfamily of Satyrinae was determined, and the results showed that M. paupera was closely related to M. dryas, and the gene content and arrangement of the newly sequenced mitogenome are similar to those of other determined mitogenomes of Satyrinae. It also provides baseline data for further molecular verification of the biogeographical evolution and phylogenetic relationships within the Satyrinae.
Authors’ contributions
DL and MKS conceptualized and designed this study, HWQ analyzed the data and drafted the manuscript, DL and MKS modified the final manuscript. All authors agree to be accountable for all aspects of the work.
Ethical approval
This work does not require Ethical approval or specific permissions according to the recommendations of the Research Ethic Committee of Sichuan University.
Supplemental Material
Download ()Supplemental Material
Download ()Supplemental Material
Download ()Supplemental Material
Download (){kind=link}
Supplemental Material
Download (){kind=link}
Supplemental Material
Download (){kind=link}
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at (https://www.ncbi.nlm.nih.gov/nuccore/OR944656) under the accession no. OR944656. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA1053844, SRR27238771, and SAMN38873769 respectively.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477. doi:10.1089/cmb.2012.0021.
- Birney E, Clamp M, Durbin R. 2004. GeneWise and genomewise. Genome Res. 14(5):988–995. doi:10.1101/gr.1865504.
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 25(15):1972–1973. doi:10.1093/bioinformatics/btp348.
- Fan C, Xu C, Li J, Lei Y, Gao Y, Xu C, Wang R. 2016. Complete mitochondrial genome of a satyrid butterfly, Ninguta schrenkii (Lepidoptera: nymphalidae). Mitochondrial DNA A DNA Mapp Seq Anal. 27(1):80–81. doi:10.3109/19401736.2013.873909.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads–a baiting and iterative mapping approach. Nucleic Acids Res. 41(13):e129–e129. doi:10.1093/nar/gkt371.
- Huang D, Hao J, Zhang W, Su T, Wang Y, Xu X. 2016. The complete mitochondrial genome of Melanargia asiatica (Lepidoptera: nymphalidae: satyrinae). Mitochondrial DNA A DNA Mapp Seq Anal. 27(2):806–808. doi:10.3109/19401736.2014.919452.
- Jühling F, Pütz J, Bernt M, Donath A, Middendorf M, Florentz C, Stadler PF. 2012. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res. 40(7):2833–2845. doi:10.1093/nar/gkr1131.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589. doi:10.1038/nmeth.4285.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. doi:10.1093/molbev/mst010.
- Lang S. 2022. The Nymphalidae of China (Lepidoptera, Rhopalocera) Part III. Pardubice, Czechia, Tshikolovets Pubulications.
- Kim, Min Jee, et al. 2010. Complete nucleotide sequence and organization of the mitogenome of the endangered Eumenis autonoe (Lepidoptera: nymphalidae). Afr J Biotechnol. 9(5):735–754. doi:10.5897/AJB09.1486.
- Minh BQ, Nguyen MA, von Haeseler A. 2013. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. 30(5):1188–1195. doi:10.1093/molbev/mst024.
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274. doi:10.1093/molbev/msu300.
- Qin XM, Guan QX, Zeng DL, Qin F, Li HM. 2012. Complete mitochondrial genome of Kallima inachus (Lepidoptera: nymphalidae: nymphalinae): comparison of K. inachus and Argynnis hyperbius. Mitochondrial DNA. 23(4):318–320. doi:10.3109/19401736.2012.684093.
- Sbordoni V, Cesaroni D, Coutsis JG, et al. 2018. Guide to the Butterflies of the Palearctic Region Satyrinae part V. Milano: Omnes Artes.
- Shi Q-H, Zhao F, Hao J-S, Yang Q. 2013. Complete mitochondrial genome of the Common Evening Brown, Melanitis leda Linnaeus (Lepidoptera: nymphalidae: satyrinae). Mitochondrial DNA. 24(5):492–494. doi:10.3109/19401736.2013.770501.
- Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 56(4):564–577. doi:10.1080/10635150701472164.
- Tang M, Tan M, Meng G, Yang S, Su X, Liu S, Song W, Li Y, Wu Q, Zhang A, et al. 2014. Multiplex sequencing of pooled mitochondrial genomes-a crucial step toward biodiversity analysis using mito-metagenomics. Nucleic Acids Res. 42(22):e166–e166. doi:10.1093/nar/gku917.
- Teixeira da Costa LF. 2016. The complete mitochondrial genome of Parage aegeria (Insecta: lepidoptera: papilionidae). Mitochondrial DNA A DNA Mapp Seq Anal. 27(1):551–552. doi:10.3109/19401736.2014.905853.
- Wu L-W, Lin L-H, Lees DC, Hsu Y-F. 2014. Mitogenomic sequences effectively recover relationships within brush-footed butterflies (Lepidoptera: nymphalidae). BMC Genomics. 15(1):468. doi:10.1186/1471-2164-15-468.
- Xiang C, Gao F, et al. 2023. Using PhyloSuite for molecular phylogeny and tree‐based analyses. iMeta. 2:e87. doi:10.1002/imt2.87.
- Yang M, Song L, Zhou L, Shi Y, Song N, Zhang Y. 2020. Mitochondrial genomes of four satyrine butterflies and phylogenetic relationships of the family Nymphalidae (Lepidoptera: papilionoidea). Int J Biol Macromol. 145:272–281. doi:10.1016/j.ijbiomac.2019.12.008.
- Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, Wang GT. 2020. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 20(1):348–355. doi:10.1111/1755-0998.13096.
- Zhang W, Gan S, Zuo N, Chen C, Wang Y, Hao J. 2016. The complete mitochondrial genome of Triphysa phryne (Lepidoptera: nymphalidae: satyrinae). Mitochondrial DNA A DNA Mapp Seq Anal. 27(1):474–475. doi:10.3109/19401736.2014.900673.
- Zhou L, Yang C, Zhai Q, Zhang Y. 2020. The complete mitochondrial genome sequence of Coenonympha amaryllis and monophyly of Satyrinae (Lepidoptera: nymphalidae). Mitochondrial DNA B Resour. 5(2):1223–1224. doi:10.1080/23802359.2020.1730729.
- Zhou Y, Liang Z, Wang S, Zhong H, Wang N, Liang B. 2020. A mitogenomic phylogeny of satyrid butterflies and complete mitochondrial genome of Oeneis urda (Lepidoptera: nymphalidae: satyrinae). Mitochondrial DNA Part B. 5(2):1344–1345. doi:10.1080/23802359.2020.1735272.