
ABSTRACT
Introduction: Biologics are highly complex drugs and many of their characteristics are defined by the manufacturing process. In recent years, several monoclonal antibody (mAb) biologics, which were hitherto only available for intravenous (IV) administration, have been reformulated for subcutaneous (SC) administration. Reformulation involves alterations to the established manufacturing process and it has been argued that a SC formulation should therefore be considered a biosimilar version of its previously approved IV formulation.
Areas covered: Developing an SC version of an approved IV mAb product requires the adaptation of its formulation and route of administration. This process is illustrated via case examples of trastuzumab and rituximab and summarize what data requirements support the regulatory approval of these new formulations. Furthermore, we discuss similarities and differences between the development of an SC product vs. the development of a biosimilar.
Expert opinion: The production process of a biosimilar is independently established from the very beginning, while the development of an SC formulation affects later stages of an already established production process and builds on extensive experience with the IV formulation. Considering these fundamentally different situations, a biologic product reformulated for SC administration should not be considered a ‘biosimilar’ version of itself.
1. Introduction
Therapeutic proteins such as monoclonal antibodies (mAbs) are highly complex drugs in terms of both molecular structure and manufacturing processes. Unlike small molecule drugs which are chemically defined entities, therapeutic proteins are large in size and have great structural complexity that defines their biologic activity. Therefore, all therapeutic proteins can only be produced in living cells and are highly sensitive to their manufacturing process.
Setting up the manufacturing process for a therapeutic protein requires the development of a cell line which is genetically modified to express the protein of interest. The production of the protein of interest is based on cells from a unique and proprietary master cell bank, from which the protein is harvested, and subsequently purified in a complex multi-step process [Citation1,Citation2]. The master cell bank and the manufacturing conditions used in this production process define many physical, chemical, biological and microbiological product attributes of the therapeutic protein, so-called quality attributes (QAs) [Citation3,Citation4]. Importantly, a subset of these QAs can have a direct impact on the efficacy and safety of a product. Any changes to the manufacturing process, even when very minor, can therefore potentially affect the structural characteristics of the finished product and, most importantly, the way it functions in the body [Citation5]. It is therefore crucial that manufacturers maintain the quality of their product over time by tightly controlling the manufacturing process.
Technical advances or changes in regulatory requirements may prompt a manufacturer to improve or adapt a product’s manufacturing process after its approval. Such incremental adaptations are common and often intended to benefit patients. However, they may potentially have a clinically relevant impact on product attributes and therefore, manufacturers and regulators will carefully assess the risk of such a manufacturing process change prior to its implementation [Citation6–8]. A so-called comparability exercise must be conducted to demonstrate that pre-and post-change product are comparable. This assessment is based on stringent regulatory standards as defined by the International Council of Harmonization (ICH) and adopted by leading regulatory authorities such as the European Medicines Agency (EMA) and the United States Food and Drug Administration (FDA) [Citation6,Citation9,Citation10].
An example for adapting an authorized product’s production process is modifying the formulation and route of administration of an intravenously (IV) administered product for subcutaneous (SC) administration. The SC route of administration can provide significant benefits for patients, e.g. in terms of treatment duration and convenience [Citation11,Citation12]. Examples of therapeutic proteins which have originally been formulated for IV administration and were subsequently developed for SC administration include abatacept (Orencia®, Bristol-Myers Squibb), alemtuzumab (Lemtrada®, Sanofi Genzyme), rituximab (MabThera®, F. Hoffmann-La Roche), tocilizumab (Actemra®, F. Hoffmann-La Roche) and trastuzumab (Herceptin®, F. Hoffmann-La Roche) [Citation13–15].
In a different setting, once the exclusivity of an established IV administered therapeutic protein approaches its end, third party manufacturers can develop follow-on versions (biosimilars) of an originator (or ‘reference product’) using manufacturing processes developed independently of the originator manufacturer’s proprietary knowledge. Biosimilars result from a ‘reverse engineering’ approach aimed at matching the originator’s QA profile as close as possible by means of de novo developed master cell banks, manufacturing conditions and process controls. The EMA and FDA only approve a product as biosimilar once its similarity to the reference product has been demonstrated by means of thorough biosimilarity assessment including comparative analytical-, non-clinical and clinical studies.
Demonstrating comparability of an authorized IV product and its corresponding SC formulation is a contextually different exercise from demonstrating biosimilarity between a biosimilar and its reference product. However, because the development of an approved IV product for SC administration requires some adjustments of its established production process, opinions have been voiced that SC products could be considered a biosimilar of the respective IV product [Citation16]. To critically assess this notion, we present case examples of Herceptin and MabThera based on which we discuss similarities and differences between the development of an SC product vs. the development of a biosimilar.
2. Development of therapeutic proteins for SC administration
Many currently approved therapeutic proteins are administered by IV infusion, which is time-consuming and requires (costly) medical supervision and infrastructure. Therefore, SC administration has been developed as an alternative delivery method for some of these therapeutic proteins [Citation17–19]; as oral administration is usually not feasible for proteins due to low intestinal absorption and degradation. SC administration can significantly reduce the treatment burden for patients and several clinical studies have demonstrated that patient preference for receiving the SC instead of the IV formulation is high [Citation12,Citation20–22]. As acknowledged in the US prescribing information for an SC mAb product, patients prefer the SC formulation because of less time spent in the clinic, greater comfort during administration, less emotional distress while receiving treatment, and lower levels of injection site pain [Citation12,Citation20,Citation21,Citation23]. On top of these benefits, the convenience and speed of SC administration can lead to time- and cost-savings in the health care system, relieve strain on infusion centers and allow greater patient access [Citation11].
SC formulations need to deliver a dose of the therapeutic protein that produces a treatment effect comparable to the IV formulation, i.e. the minimum therapeutic protein concentration at the site of action after administration needs to be comparable between the two formulations. A challenge for SC formulations is that this dose must be administered in a much lower volume compared to IV administration. Furthermore, they must ensure appropriate absorption of the protein from the site of administration to obtain clinical activity comparable to that of the IV formulation. The extracellular matrix in the hypodermis represents a significant barrier to SC drug delivery and usually limits administration volumes to 1–2 mL. While this is for example sufficient for SC administration of insulin, larger volumes are typically needed for SC administration of mAbs. One option to overcome this limitation is a technology involving recombinant human hyaluronidase (rHuPH20) [Citation24]. rHuPH20 is an enzyme which temporarily degrades the main component of the extracellular matrix, hyaluronic acid. The component has been approved in multiple markets globally including EU and US, and its safety and effectiveness have been established [Citation24]. Co-formulation of drugs with rHuPH20 makes the relatively pain-free administration of larger fluid volumes and thus the SC administration of mAbs possible. Examples of mAbs which have been developed for SC administration using rHuPH20 include Herceptin® and MabThera® [Citation17–19,Citation25].
2.1. Case examples: Herceptin® and MabThera®
2.1.1. How does an SC formulation differ from the original IV formulation
The main difference between an SC and IV version of a therapeutic protein product is the route of administration. The manufacturing process of the therapeutic protein for an SC product is usually identical to the one for the IV product; with the exception of final steps where adjustments are needed to achieve desired formulation, strength and pharmaceutical form. This has been illustrated below with case examples of Herceptin and MabThera.
Herceptin is indicated for the treatment of adult patients with HER2+metastatic and early breast cancer, and reconstituted Herceptin solution for IV infusion contains 21 mg/mL of trastuzumab. MabThera is indicated for the treatment of certain blood cancers and inflammatory conditions, and its IV formulation is available as 10 mg/mL concentrate for solution for infusion. Administered doses of Herceptin and MabThera need to be individually adapted based on body weight and body surface area, respectively, and administration is associated with considerable infusion times (Herceptin: initial loading dose as 90-minute infusion; subsequent doses as 30-minute infusion, 2-hour follow-up; MabThera in non-Hodgkin’s lymphoma [NHL] and chronic lymphocytic leukemia [CLL]: 4–6 hours for first dose, 2–4 hours for subsequent infusions) [Citation26,Citation27].
In recent years, both of these products have been reformulated for SC administration. Compared to the IV presentation, Herceptin SC has a different strength (600 mg in 5 mL solution) and pharmaceutical form (solution for injection), and can be administered within 2–5 minutes as a fixed dose irrespective of the patient’s body weight [Citation17]. In order to develop this high concentrated, ready to use, liquid SC formulation of Herceptin, excipient concentrations and pH value were adapted and a stabilizer in liquid formulation was added. Further, rhuPH20 was added and the final formulation step was adapted to achieve a trastuzumab concentration of 120 mg/ml. Beside this, the main part of the manufacturing process, including the master cell bank with the trastuzumab-encoding genetic sequence and its growth conditions remained unchanged. The same harvest and purification methods are applied for the active component in the SC and IV formulation and it can be asserted that the same quality is obtained.
Similarly, the MabThera SC formulation has a different strength (NHL: 1400 mg in 11.7 mL solution; CLL: 1600 mg in 13.4 mL solution) compared to its IV presentation and can be administered over approximately 5 min as a fixed dose irrespective of the patient’s body surface area [Citation18,Citation19]. Analogous to Herceptin, the SC formulation of MabThera was developed by minor adaptations of the composition to stabilize the antibody, and by adding rHuPH20.
2.1.2. What are the data requirements for approval of SC formulations
Pre- and post-manufacturing change products (e.g. IV and SC versions of a mAb product) are assessed in comparability exercises to evaluate the potential impact of any adjustments of the manufacturing process and/or formulation on clinical safety and efficacy of the product and characterize potential new components [Citation6,Citation9,Citation10] (). This assessment is based on a hierarchical risk-based approach to determine the need for supportive pre-clinical or clinical data. It may comprise analytical analyses, biological characterization and, if required non-clinical or clinical studies assessing PK, PD, safety and/or efficacy of the post-change product [Citation28].
Table 1. Comparison of biosimilarity assessment vs. evaluation of a licensed biologic developed for SC administration.
Incremental manufacturing process changes that occur at later steps of production are usually of low or medium risk, i.e. the cell line and active substance typically remain unaffected by such an adaptation [Citation8]. By these criteria, above described adaptations of the formulations of MabThera and Herceptin could have been assessed by means of quality and analytical analyses. However, a change in the route of administration of a mAb product generally bears higher risks of producing clinically meaningful differences between the pre- and post-change product (compared to the formulation change alone). For some drugs, the SC route of administration can be associated with a higher risk of immunogenicity than the IV route [Citation29]. Therefore, the development program for an SC formulation of an approved IV therapeutic protein includes both pre-clinical and clinical assessments.
Development of an SC product typically aims at achieving comparable pharmacokinetic (PK) characteristics between the SC and IV formulation. This development approach is also referred to as ‘PK bridging’ and is commonly used to achieve the well-established safety and efficacy profile of the approved IV product in another formulation. The main objective of this approach is to select and confirm an SC dose that yields drug exposure at least as high as the IV dose and is therefore expected to produce an analogous treatment effect. The clinical development and regulatory pathways for SC formulations of Herceptin and MabThera followed such a PK bridging approach. The obtained pre-clinical and clinical data which formed the basis for approval of MabThera SC are summarized below as an illustrative example [Citation18,Citation19].
Technical and pre-clinical development of MabThera SC included e.g. analytical studies to demonstrate comparability of MabThera SC and IV active component after the above described adaptations of the manufacturing process, evaluation of quality and safety of the novel component rHuPH20 as well as animal studies that assessed toxicity, local tolerance, PK and anti-tumor efficacy of the SC vs. IV presentation [Citation18,Citation19].
The PK bridging approach for the clinical development of MabThera SC included two Phase 1b clinical studies (SparkThera in FL and SAWYER in CLL) and one Phase 3 study (SABRINA in FL) aimed to confirm comparability of MabThera SC vs. IV in terms of PK. Clinical pharmacology data from these studies demonstrated a statistically non-inferior exposure (Ctrough) with fixed doses of 1400 mg and 1600 mg MabThera SC compared with the established MabThera IV body surface area-adjusted doses in NHL and CLL, respectively [Citation30–32]. Based on extensive scientific knowledge on rituximab’s mechanism of action and decades of clinical knowledge, achieving comparable rituximab exposure is expected to produce comparable target site saturation and B-cell depletion, resulting in analogous efficacy.
The MabThera SC development approach expanded the framework of PK bridging to include supportive clinical evaluations of efficacy and safety. In the SABRINA and SAWYER studies, MabThera SC and IV formulations showed comparable efficacy in patients with FL and CLL in terms of complete and overall response rates [Citation20,Citation33].
In addition, safety data were available from all three studies and no new adverse drug reactions were observed with MabThera SC vs. IV. The incidence of injection site/infusion-related reactions was higher in the MabThera SC cohorts than in the MabThera IV cohorts, driven by a higher incidence of local cutaneous reactions (none of them serious or severe). The potential of MabThera SC to induce an unwanted immune response was low and consistent with that observed in patients receiving MabThera IV. In the few patients who developed anti-rituximab antibodies, these produced no apparent clinical consequences.
In Europe, MabThera SC was initially approved for NHL based on the SparkThera and SABRINA studies in 2014, and subsequently for CLL based on the SAWYER study in 2016 [Citation34,Citation35]. The more convenient route of administration offers an added benefit of MabThera SC vs. MabThera IV [Citation12,Citation36]. Both formulations contain the same active component originating from the same master cell bank and potential impacts of any changes to the manufacturing process were thoroughly assessed. Overall, the benefit/risk assessment of MabThera SC was considered positive and comparable to that of the IV formulation and MabThera SC was approved for all hematological indications of MabThera IV based on the PK bridging approach.
3. Biosimilar development
3.1. How do biosimilars differ from the originator biologics
Therapeutic proteins are inherently variable because their QAs are susceptible to biological processes inside the production cell line. Even though the primary amino acid sequence of a therapeutic protein remains the same, so-called post-translational modifications are imprinted on proteins via intracellular enzymatic processes that may be specific to a certain cell line or impacted by manufacturing conditions [Citation4]. This variability can be controlled by means of robust quality systems that ensure well-controlled manufacturing processes to maintain QAs within narrow, pre-defined ranges.
A biosimilar’s manufacturer has to use a ‘reverse engineering’ approach to (1) analyze the reference product’s QAs and how these are affected by the manufacturing process, and (2) match the reference product’s QA profile as closely as possible while developing a new manufacturing process from the very beginning () [Citation37]. By default, this approach necessitates the establishment of a new cell line. Therefore, based on the inherent variability of biologics, biosimilars and reference products derived from independently developed cell lines and manufacturing processes cannot be identical.
Figure 1. Schematic depiction of manufacturing changes that are required to develop an established IV product into an SC product (upper part) vs. the de novo set up of a manufacturing process for a biosimilar of an established IV product (lower part).
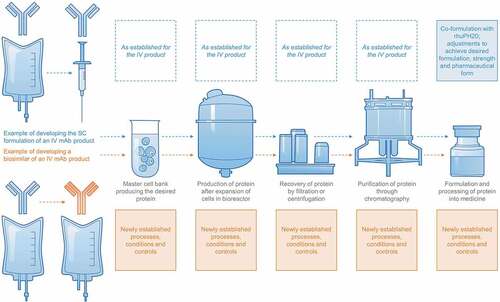
In contrast, the development of an SC formulation of an IV product is based on deep knowledge of the IV product’s QA profile, manufacturing process and process controls, and affects only later stages of production (). The active component in an SC and IV version of one therapeutic protein product therefore generally remains identical.
Grounded in these fundamental differences, the development of biosimilars follows a regulatory pathway that is distinct from the evaluation and approval of SC formulations of IV products [Citation9,Citation10,Citation38,Citation39]. The EMA and FDA define biosimilars as being ‘highly similar’ to their reference products. Accordingly, some differences between a biosimilar and its reference product with regards to QAs are expected and may be acceptable if verified as non-critical and therefore not causing clinically relevant differences in the safety and efficacy of the product [Citation39,Citation40].
3.2. What are the data requirements for approval of a biosimilar
A biosimilarity assessment is required to demonstrate comparability between the biosimilar candidate and its originator product. Because biosimilars are developed independently from the originator manufacturer’s proprietary knowledge of the reference product, the potential for clinically relevant differences between a biosimilar and the reference product may be greater than between the IV and SC formulation of one established product [Citation37]. Generally, the FDA anticipate data requirements for establishing biosimilarity to be higher than for establishing comparability between a pre- and post-manufacturing change product [Citation39].
Therefore, a biosimilarity assessment typically includes all of the following: head-to-head comparative analytical, pre-clinical, and clinical evaluation () [Citation39,Citation40]. Pre-clinical development of a biosimilar comprises extensive analytical and functional studies as well as limited assessments in animals [Citation18,Citation19]. Clinical trials are specifically designed as a final comparative evaluation step and aim to resolve residual uncertainties that remain following pre-clinical development, regarding the similarity of the proposed biosimilar with the reference product. The study population and selected endpoint(s) need to be adequately sensitive to detect potential differences in efficacy, safety, or immunogenicity between the biosimilar and originator. Development programs for the recently approved biosimilars of MabThera and Herceptin included large Phase 3 studies randomizing up to 827 patients [Citation41,Citation42].
Regulators will take into consideration the ‘totality of evidence’ available for a proposed biosimilar when making their decision on its approval, including analytical, pre-clinical, and clinical data packages [Citation37,Citation39,Citation40]. Any observed differences have to be duly justified with regard to their potential impact on safety and efficacy. Once approval is granted, the benefits and risks of a biosimilar are inferred from the similarity to the originator in terms of quality, efficacy and safety.
Importantly, once a biosimilar has been approved, there is no regulatory requirement to demonstrate its similarity to the reference product again in the post-approval period [Citation43]. The biosimilar is considered similar to the reference product throughout its life-cycle. Accordingly, the manufacturer of an IV biosimilar may adapt it for SC administration independently of the reference product. While the comparability between the IV and SC biosimilar will have to be demonstrated, an analytical or clinical comparison of the SC biosimilar with an SC version of the reference product will not be required. As an example, the IV administered infliximab biosimilar CT-P13 (Remsima®, Celltrion Healthcare) was initially approved as biosimilar of the IV administered originator (Remicade®, Janssen Biotech, Inc.) and is now in development for the SC route of administration independently of its reference product [Citation44].
Independent life-cycles of biosimilars and reference products may initiate a process referred to as ‘divergence’ which suggests that clinically relevant differences in QA profiles could accumulate over time [Citation45,Citation46]. This is of relevance in clinical practice as physicians may consider switching patients who are stable on one treatment with a therapeutic protein (IV or SC) to a biosimilar (IV or SC), or from one biosimilar to another. In this real-world setting, full transparency and traceability of administered products is highly important to know which product a patient received and thereby differentiate safety signals that may arise between different products.
4. Conclusion
There are important differences between the development of an approved therapeutic protein product for SC administration and the development of a biosimilar which impact the type and amount of data needed to evaluate quality, safety and efficacy of the product. The active component in the therapeutic protein product is essentially identical in the SC and IV formulation provided that it is derived from the same master cell bank using the same growth conditions and purification methods; adjustments of the IV product’s formulation and manufacturing generally affect later stages of the production process. In contrast, a biosimilar is based on a newly established master cell bank as well as independently developed manufacturing conditions and process controls. No product-specific process history resides with the developer of a biosimilar and a link to clinical experience must be established.
Clinical development programs to support approval of an SC formulation in the context of a change in the route of administration for an existing therapeutic protein product are based on a PK bridging approach with the primary aim to show comparability of the PK profile, thereby applying the well-established safety and efficacy profile of an approved product to the adjusted formulation.
Clinical studies to support approval of a biosimilar need to directly compare the efficacy and safety of two different products, i.e. the biosimilar candidate and its reference product. By default, a biosimilarity assessment requires comparative analytical/functional, pre-clinical and clinical studies with the reference product, and hence in general more extensive data needs to be generated to demonstrate biosimilarity.
In conclusion, different factors need to be considered in the assessment of an approved IV product that has been developed for SC administration vs. the assessment of a biosimilar. While the active component in an SC formulation is essentially identical to that of the IV formulation, a biosimilar can only be ‘similar’ to the reference product. Accordingly, the biosimilar development pathway is fundamentally different from the development of an SC formulation of an approved IV product and the SC formulation of a therapeutic protein is not a ‘biosimilar’ version of its IV formulation.
5. Expert opinion
Since the advent of biosimilars, knowledge gaps remain in the broader stakeholder community regarding their development, including the differentiation between biosimilarity assessments (i.e. demonstrating similarity between a proposed biosimilar and its reference product) vs. comparability exercises (i.e. the assessment of a licensed biologic that has undergone a manufacturing process change) [Citation16,Citation47,Citation48]. Educational materials released by the European Medicines Agency (EMA) and the United States Food and Drug Administration (FDA), as well as initiatives from medical societies emphasize the need to increase understanding of biosimilars among patients and healthcare professionals [Citation49–51].
mAbs such as Herceptin and MabThera are produced by living organisms and manufactured in a complex multi-step process. Each stage of this process confers unique properties to the resulting product. Even small adaptations of the manufacturing process can therefore affect product quality and potentially impact its quality, safety, and/or efficacy. Because during a mAb product’s life-cycle, manufacturing process changes can potentially result in a product quality attribute shifting outside its originally established margins, the notion emerged that originator products that have undergone manufacturing changes become biosimilars of themselves [Citation16,Citation48,Citation52]. Relating to the presented case examples of Herceptin and MabThera, this notion also implies that SC products could be considered biosimilars of the previously approved IV products.
However, there are key regulatory and contextual differences between a biosimilar vs. an established product that underwent a manufacturing change. The development of an SC formulation of an approved biologic has been considered a manufacturing process change by regulatory authorities and was thoroughly assessed in a comparability exercise prior to its implementation. Requirements of such an exercise are outlined in the internationally agreed ICH Q5E guideline which is applied by the EMA and FDA [Citation6,Citation9,Citation10]. Because a change in the route of administration can potentially produce clinically meaningful differences between the pre- and post-change product, non-clinical studies and Phase 1b and 3 clinical studies were conducted in case of MabThera to link the efficacy and safety profile of the IV formulation to the SC formulation. Importantly, the active substance of both formulations remained essentially identical considering that only later steps of the production process were affected by the manufacturing change (leaving early components such as the cell line unchanged). Subsequently, significant patient benefits were shown for the approved SC formulation, e.g. in terms of treatment duration and convenience [Citation11,Citation12].
Biosimilars can present less expensive alternatives to their respective originator biologics. The active substance of a biosimilar is considered a version of the active substance of the originator biologic, taking into account that a biosimilar’s manufacturing process, including the cell line, is developed independently from the originator manufacturer’s proprietary knowledge [Citation38]. A biosimilar’s assessment and approval follow regulatory pathways that are distinct from establishing comparability after a manufacturing change, as stipulated by leading regulatory authorities such as the EMA and FDA. The FDA acknowledge that, although some scientific principles in ICH Q5E may apply in the demonstration of biosimilarity, they anticipate the data requirements to be higher for establishing biosimilarity compared to showing comparability between a pre- and post-manufacturing change product [Citation39].
We therefore consider the distinction between a biosimilar vs. an approved biologic that has undergone a manufacturing change imperative for healthcare professionals and the medical community to recognize contextual differences between the two situations. Accordingly, a biologic reformulated for SC administration should not be considered a ‘biosimilar’ version of itself.
Article highlights
Biologics are highly complex drugs that can be affected by biological processes inside the cells in which they are produced and are therefore particularly sensitive to their manufacturing process.
For the purpose of significant patient benefit, a manufacturer may choose to develop an SC formulation of an authorized IV product.
The SC formulation of a therapeutic protein is not a ‘biosimilar’ version of its IV formulation; biosimilars are developed by third party manufacturers once the exclusivity of the IV originator product approaches its end.
While the development of an SC formulation affects only later stages of an already established production process leaving the active component typically unaffected, the production process of a biosimilar is independently established from the very beginning, including a new cell line.
Based on fundamental differences with regards to context and data requirements, the development of biosimilars follows a regulatory pathway that is distinct from the evaluation and approval of SC formulations of IV products.
Declaration of interest
AZ, DH, GB and TS are F. Hoffmann-La Roche Ltd employees. BB is a contractor at F. Hoffmann-La Roche Ltd. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have received an honorarium from Expert Review of Precision Medicine and Drug Development for their review work. A reviewer on this manuscript has disclosed that they have served as a consultant for AbbVie, Roche, MSD, Pfizer, Sandoz, Fresenius, Biogen, and Sanofi-Genzyme. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Additional information
Funding
References
- Sekhon BP, Saluja V. Biosimilars: an overview. Biosimilars. 2011;1:1–11.
- Liu HF, Ma J, Winter C, et al. Recovery and purification process development for monoclonal antibody production. mAbs. 2010;2(5):480–499.
- Kuriakose A, Chirmule N, Nair P. Immunogenicity of biotherapeutics: causes and association with post-translational modifications. J Immunol Res. 2016;2016:1298473.
- Vulto AG, Jaquez OA. The process defines the product: what really matters in biosimilar design and production? Rheumatology (Oxford). 2017;56(suppl_4):iv14–iv29.
- Reusch D, Tejada ML. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology. 2015;25(12):1325–1334.
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH harmonised tripartite guideline: comparability of biotechnological/biological products subject to changes in their manufacturing process - Q5E: 2004. [updated 2004 Nov 18]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5E/Step4/Q5E_Guideline.pdf.
- Schneider CK. Biosimilars in rheumatology: the wind of change. Ann Rheum Dis. 2013;72(3):315–318.
- Vezer B, Buzas Z, Sebeszta M, et al. Authorized manufacturing changes for therapeutic monoclonal antibodies (mAbs) in European Public Assessment Report (EPAR) documents. Curr Med Res Opin. 2016;32(5):829–834.
- European Medicines Agency. Guideline on comparability of biotechnology-derived medicinal products after a change in the manufacturing process: non-clinical and clinical issues: 2007 [ updated 2007 Jul 19]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003935.pdf.
- Food and Drug Administration. Q5E comparability of biotechnological/biological products subject to changes in their manufacturing process: 2005. [updated 2005 Jun]. Available from: http://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm073476.pdf.
- Dychter SS, Gold DA, Haller MF. Subcutaneous drug delivery: a route to increased safety, patient satisfaction, and reduced costs. J Infus Nurs. 2012;35(3):154–160.
- Rummel M, Kim TM, Aversa F, et al. Preference for subcutaneous or intravenous administration of rituximab among patients with untreated CD20+ diffuse large B-cell lymphoma or follicular lymphoma: results from a prospective, randomized, open-label, crossover study (PrefMab). Ann Oncol. 2017;28(4):836–842.
- Bittner B, Richter W, Schmidt J. Subcutaneous administration of biotherapeutics: an overview of current challenges and opportunities. BioDrugs. 2018;32(5):425–440.
- Jackisch C, Muller V, Maintz C, et al. Subcutaneous administration of monoclonal antibodies in oncology. Geburtshilfe Frauenheilkd. 2014;74(4):343–349.
- Kivitz A, Olech E, Borofsky MA, et al. Two-year efficacy and safety of subcutaneous tocilizumab in combination with disease-modifying antirheumatic drugs including escalation to weekly dosing in rheumatoid arthritis. J Rheumatol. 2018;45(4):456–464.
- Mehr SR, Zimmerman MP. Is a biologic produced 15 years ago a biosimilar of itself today? Am Health Drug Benefits. 2016;9(9):515–518.
- Committee for Medicinal Products for Human Use (CHMP). Assessment report - Herceptin (EMA/CHMP/751770/2012/corr1): 2013. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/000278/WC500153233.pdf.
- Committee for Medicinal Products for Human Use (CHMP). Assessment report MabThera SC (EMA/CHMP/71722/2014): 2014. [EPAR Basis for approval in NHL]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/000165/WC500168097.pdf.
- Committee for Medicinal Products for Human Use (CHMP). Assessment report MabThera SC (EMA/276108/2016): 2016. [EPAR, basis for approval in CLL]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/000165/WC500208542.pdf.
- Davies A, Berge C, Boehnke A, et al. Subcutaneous rituximab for the treatment of B-cell hematologic malignancies: a review of the scientific rationale and clinical development. Adv Ther. 2017;34(10):2210–2231.
- Pivot X, Gligorov J, Muller V, et al. Preference for subcutaneous or intravenous administration of trastuzumab in patients with HER2-positive early breast cancer (PrefHer): an open-label randomised study. Lancet Oncol. 2013;14(10):962–970.
- Pivot X, Gligorov J, Muller V, et al. Patients’ preferences for subcutaneous trastuzumab versus conventional intravenous infusion for the adjuvant treatment of HER2-positive early breast cancer: final analysis of 488 patients in the international, randomized, two-cohort PrefHer study. Ann Oncol. 2014;25(10):1979–1987.
- Food and Drug Administration. US prescribing information RITUXAN HYCELA: 2017. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761064s000lbl.pdf.
- Wasserman RL. Recombinant human hyaluronidase-facilitated subcutaneous immunoglobulin infusion in primary immunodeficiency diseases. Immunotherapy. 2017;9(12):1035–1050.
- Leveque D. Subcutaneous administration of anticancer agents. Anticancer Res. 2014;34(4):1579–1586.
- Roche Registration Ltd. Herceptin® (trastuzumab). Summary of Product Characteristics [cited 2016 Sept]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000278/WC500074922.pdf
- Roche Registration Ltd. MabThera - Summary of product characteristics: 2017. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000165/WC500025821.pdf.
- Putnam WS, Prabhu S, Zheng Y, et al. Pharmacokinetic, pharmacodynamic and immunogenicity comparability assessment strategies for monoclonal antibodies. Trends Biotechnol. 2010;28(10):509–516.
- Pimentel FF, Morgan G, Tiezzi DG, et al. Development of new formulations of biologics: expectations, immunogenicity, and safety for subcutaneous trastuzumab. Pharmaceut Med. 2018;32(5):319–325.
- Davies A, Merli F, Mihaljevic B, et al. Pharmacokinetics and safety of subcutaneous rituximab in follicular lymphoma (SABRINA): stage 1 analysis of a randomised phase 3 study. Lancet Oncol. 2014;15(3):343–352.
- Assouline S, Buccheri V, Delmer A, et al. Pharmacokinetics, safety, and efficacy of subcutaneous versus intravenous rituximab plus chemotherapy as treatment for chronic lymphocytic leukaemia (SAWYER): a phase 1b, open-label, randomised controlled non-inferiority trial. Lancet Haematol. 2016;3(3):e128–e138.
- Salar A, Avivi I, Bittner B, et al. Comparison of subcutaneous versus intravenous administration of rituximab as maintenance treatment for follicular lymphoma: results from a two-stage, phase IB study. J Clin Oncol. 2014;32(17):1782–1791.
- Davies A, Merli F, Mihaljevic B, et al. Efficacy and safety of subcutaneous rituximab versus intravenous rituximab for first-line treatment of follicular lymphoma (SABRINA): a randomised, open-label, phase 3 trial. Lancet Haematol. 2017;4(6):e272–e82.
- European Medicines Agency. EPAR MabThera EMA/276108/2016: procedural steps taken and scientific information after the authorisation: 2016 [updated 2016 Aug 2]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Procedural_steps_taken_and_scientific_information_after_authorisation/human/000165/WC500025820.pdf.
- European Medicines Agency. EPAR MabThera EMA/CHMP/71722/2014: procedural steps taken and scientific information after the authorisation. 2014. Available from: https://www.ema.europa.eu/documents/procedural-steps-after/mabthera-epar-procedural-steps-taken-scientific-information-after-authorisation_en.pdf.
- De Cock E, Kritikou P, Sandoval M, et al. Time savings with rituximab subcutaneous injection versus rituximab intravenous infusion: a time and motion study in eight countries. PLoS One. 2016;11(6):e0157957.
- Lee JF, Litten JB, Grampp G. Comparability and biosimilarity: considerations for the healthcare provider. Curr Med Res Opin. 2012;28(6):1053–1058.
- European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues (EMEA/CHMP/BMWP/42832/2005 Rev1): 2014. [ updated 2014 Dec 18]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf.
- Food and Drug Administration. Guidance for industry: scientific considerations in demonstrating biosimilarity to a reference product: 2015 [ updated 2015 Apr]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf%202.%20Clinical%20Immunogenicity%20Assessment3.
- European Medicines Agency. Guideline on similar biological products (CHMP/437/04 Rev 1): 2014 [ updated 2014 Oct 23]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf.
- Stebbing J, Baranau Y, Baryash V, et al. CT-P6 compared with reference trastuzumab for HER2-positive breast cancer: a randomised, double-blind, active-controlled, phase 3 equivalence trial. Lancet Oncol. 2017. 18(7):917–928.
- von Minckwitz G, Colleoni M, Kolberg HC, et al. Efficacy and safety of ABP 980 compared with reference trastuzumab in women with HER2-positive early breast cancer (LILAC study): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2018. 19(7):987–998.
- Kang HN, Knezevic I. Regulatory evaluation of biosimilars throughout their product life-cycle. Bull World Health Organ. 2018;96(4):281–285.
- Westhovens R, Yoo D, Jaworski J. THU0191 Novel formulation of ct-p13 for subcutaneous administration in patients with rheumatoid arthritis: initial results from a phase i/iii randomised controlled trial. Ann Rheum Dis. 2018;77:315.
- Eleryan MG, Akhiyat S, Rengifo-Pardo M, et al. Biosimilars: potential implications for clinicians. Clin Cosmet Investig Dermatol. 2016;9:135–142.
- Ramanan S, Grampp G. Drift, evolution, and divergence in biologics and biosimilars manufacturing. BioDrugs. 2014;28(4):363–372.
- European Biopharmaceutical Enterprises. Position paper - biosimilarity and comparability after manufacturing changes: can a biologic become a biosimilar of itself? 2016 [ updated 2016 Feb 22]. Available from: http://www.ebe-biopharma.eu/uploads/Modules/Documents/ebe-briefing-biosimilarity-versus-manufacturing-changes-public-2016-02-22-2.pdf.
- Rugo HS, Rifkin RM, Declerck P, et al. Demystifying biosimilars: development, regulation and clinical use. Future Oncol. 2018.
- European Medicines Agency Biosimilars in the EU - Information guide for healthcare professionals: 2017 [cited 2017 Jul 10]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Leaflet/2017/05/WC500226648.pdf.
- Food and Drug Administration. Biosimilars: new educational materials: 2017. Available from: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/default.htm.
- Lyman GH, Balaban E, Diaz M, et al. American society of clinical oncology statement: biosimilars in oncology. J Clin Oncol. 2018;36(12):1260–1265.
- McCamish M, Woollett G. Worldwide experience with biosimilar development. mAbs. 2011;3(2):209–217.