
ABSTRACT
A healthy 15-year-old male spilled elemental mercury contaminating his garage and bedroom. The patient developed new onset hypertension, significant weight loss, pain (muscular, testicular, and abdominal), insomnia, delusions, hallucinations, tachycardia, palmar desquamation, diaphoresis, tremor, and ataxia leading to two consecutive hospitalizations. Blood and urine mercury were 23 and 330 µg/L, respectively. He received 21 days of chelation with 2,3-Dimercaptosuccinic acid during his second hospital stay. He continued to deteriorate. Three weeks post-chelation, he was transferred to our facility and his exam was unchanged. He could not stand or feed himself unassisted. He was started on selenium 500 mcg/day and N-acetylcysteine (NAC) 50 mg/kg/day. By day 3 of Se and NAC, he showed noticeable improvement, and by day 11, delusions, delirium, tachycardia, and abdominal pain resolved. Muscle strength, weight gain, speech, unassisted ambulation, and emotional liability improved. After five months with Se and NAC (1) he had regained 45 pounds, (2) restored to premorbid emotional, academic, and athletic performance, and (3) tachycardia, hypertension, rash, palmar skin changes, tremor, and insomnia had resolved. Features of this case include (1) improvement after selenium and NAC supplementation (2) contrasted with continued deterioration after DMSA chelation.
Introduction
In recent years, there has been an important shift in the understanding of the mechanisms of toxicity of mercury (Hg) both at the cellular and organism level. The shift in a large part has occurred from a long-held focus on the covalent binding of mercury to sulfur in the body's ubiquitous sulfhydryl groups. There is convincing evidence that the pathophysiological target of mercury is not the in vivo binding of sulfur, but rather selenium (Se). Recent evidence suggests that the mechanism of toxicity of mercury is the selenium-based proteins thioredoxin reductase and glutathione peroxidase [Citation1,Citation2]. We report a case of severe mercury poisoning unchanged by chelation who later achieved significant improvement related to selenium and N-acetylcysteine (NAC) supplementation.
Case report
Without informing his family, a healthy 15-year-old 70-kg (154 lb) athletic male spilled elemental mercury contaminating his garage, bedroom, and personal items. Over the following three weeks, the patient developed muscle pain, testicular pain, and new onset hypertension. After six weeks of continued intermittent mercury vapor exposure, worsening muscle pain, hypertension, mood changes, abdominal pain, anorexia, and a 14 pound (6.4 kg) weight loss, he was admitted to the hospital to rule out pheochromocytoma/paraganglioma. Results were negative on metaiodobenzylguanidine scan, normal 24 hour urine metanephrines, and a normal echocardiogram. He continued to have tachycardia, emotional lability, profound insomnia, delirium, and delusions. A CT of the head/neck/chest/abdomen/pelvis revealed an enlarged colon with fecal matter. He was started on lisinopril and clonidine, evaluated by psychiatry, and discharged home with no clear diagnosis. One week later, he was admitted to a second hospital for worsening abdominal pain, continued weight loss, auditory hallucinations, persistent tachycardia, hypertension, palmar desquamation, diaphoresis, tremor of his hands, ataxia, constipation, and dyschezia. His weight had declined to 52.45 kg (115.4 lb).
Initial management focused on the hypertension and maintaining food intake to stop the weight loss. There was concern of a psychiatric component to the sudden change. Multiple imaging and laboratory studies including chemistries, repeated renal function, thyroid studies, and cerebral spinal fluid (CSF) studies were evaluated and were unremarkable. Investigation of metal exposure included copper, arsenic, selenium, lead, and mercury. Nine days after admission to the second hospital, blood and urine mercury were 23 (normal < 10 µg/L) and 330 µg/L (normal < 20 µg/L), respectively. Chelation with 2,3-Dimercaptosuccinic acid (DMSA) for 21 days was initiated on day 9 of the second hospitalization (10 mg/kg every 8 hours for 7 days, and 10 mg/kg every 12 hours for 14 days). The patient continued to deteriorate and on day 18 of hospitalization (day 9 of chelation) he was transferred to the intensive care unit due to continued autonomic instability and concern for aspiration due to difficulty swallowing.
Prior to and post chelation, extensive laboratory values remained unremarkable including a blood urea nitrogen (BUN) of 10, creatinine of 0.6, blood lead concentration of 3 µg/dL, serum zinc concentration of 103 µg/dL, and arsenic of <3 µg/L. During chelation urine total porphyrins were 179.6 µg/L. Approximately three weeks after cessation of chelation with DMSA urine mercury rose from 44 to 71.3 µg/L, while blood Hg fell from 19 to 11 µg/L, without evidence of clinical improvement. Transaminases rose during this admission from aspartate aminotransferase (AST) 53 U/L and alanine aminotransferase (ALT) 71 U/L to AST 61 U/L and ALT of 102 U/L. An electromyogram showed a demyelinating motor neuropathy suggesting Guillain–Barre Syndrome. He received two doses of IV immunoglobulin with no improvement in his clinical condition. During 54 days in hospital, he failed to improve despite speech therapy, occupational therapy, physical therapy, feeding tube placement, and three weeks of chelation with DMSA.
On arrival to our facility, his exam was unchanged with muscle pain, muscle weakness, testicular pain, persistent sinus tachycardia, diaphoresis, palmar desquamation, rash on trunk and arms, tremor of his hands, ataxia, mood changes, and insomnia. He could not stand or feed himself unassisted. His weight was 49 kg. Sinus tachycardia persisted with a range of 114 to 124 bpm, but hypertension appeared to be controlled with lisinopril 40 mg/day, clonidine 0.1 mg/day, and amlodipine 10 mg/day (BP range 100–120/60–63 mmHg). His gabapentin 200 mg/day from the previous institution was discontinued and he was started on selenium (Se) 500 mcg/day and NAC 50 mg/kg/day PO. He began to make noticeable improvements within three days. His tremor improved and on day 5 he was able to feed himself. His appetite increased, muscle strength improved, and he began to gain weight and ambulate independently. By hospital day 7, delusions, delirium, and abdominal pain had resolved and his insomnia improved. By hospital day 10, he was able to ambulate without assistance 1/3 mile (530 m) across the hospital campus for his favorite meal, which he ate with enthusiasm. His rash and tachycardia (100 bpm) continued for one more week. He was discharged on hospital day 11. Blood Hg, serum Se, and urine Hg initially rose after Se supplementation: blood Hg 11–28.1 µg/L, serum Se 108–135 µg/L, and urine Hg 23.8–48.4 µg/L, respectively. illustrate time courses of blood mercury concentrations, serum selenium concentrations, and urine mercury concentrations.
Figure 1. Blood mercury concentrations.
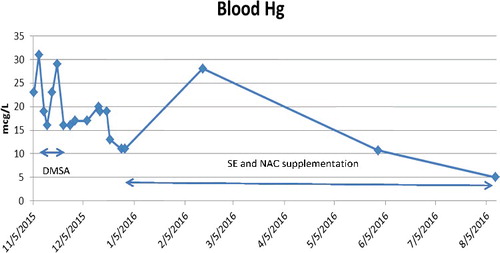
Figure 2. 24 hour urine mercury concentrations.
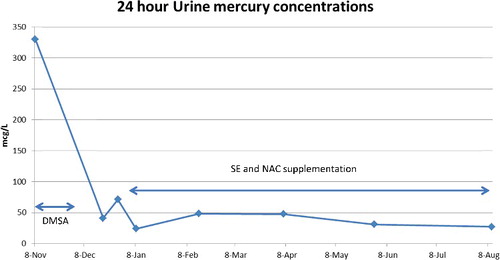
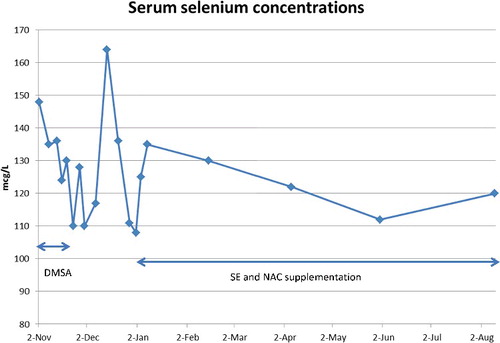
Figure 3. Serum selenium concentrations.
After three months with continued Se and NAC supplementation: (1) he had regained 35 pounds, (2) restored to premorbid emotional and academic performance with direct athletic participation, and (3) tachycardia, rash, palmar skin changes, tremor, and insomnia had resolved. After five months with continued Se and NAC, (1) he had regained 45 pounds, (2) returned to active football, regaining his position on the team, and (3) hypertension had resolved. All cardiovascular and behavioral medications had been discontinued.
Discussion
The primary cellular target of mercury appears to be the selenoproteins of the thioredoxin system. These include thioredoxin reductase1 (TrxR1) in the cytosol and thioredoxin reductase2 (TrxR2) in the mitochondria and the glutathione–glutaredoxin system (glutathione peroxidase, GPx) [Citation1,Citation3–5]. Impairment of the thioredoxin and glutaredoxin systems allows for proliferation of cytosol and mitochondrial reactive oxygen species which in turn leads to a cascade of impaired glutamate uptake, N-methyl-D-aspartate (NMDA) receptor over-stimulation, mitochondrial injury/loss, lipid peroxidation, impairment of protein repair, and apoptosis [Citation6]. The ultimate outcome of mercury toxicity is to produce a severe selenium deficiency state causing oxidative stress from a loss of intracellular redox control [Citation6,Citation7].
The role of the thiol-based chelators is to bind blood mercury and increase urinary elimination [Citation8]. Neither of these roles addresses the primary toxicologic mechanism of mercury: selenoprotein impairment and reduction of selenium availability. Numerous in vitro and animal models have shown selenium supplementation, with limitations, can ameliorate injury, restore mitochondrial content, regenerate intracellular selenoprotein function and restore intracellular redox environment [Citation1,Citation9–15]. In a rat model, Se supplementation showed greater restoration of TxrR and GPx functions and reduction of oxidative stress than a thiol-based chelator [Citation16]. The addition of NAC to Se supplementation was additive to these effects. The combination of Se and NAC was more effective than either agent alone, with Se > NAC [Citation16].
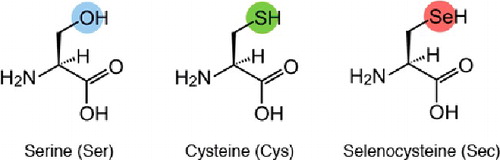
Two likely roles for NAC include (1) as a readily available cysteine donor and (2) increased generation of glutathione. Selenium is incorporated into selenoproteins as the amino acid selenocysteine (Sec). However, in contrast to other amino acids, Sec is not recycled for reincorporation into new proteins but is, instead, degraded to release inorganic Se [Citation7]. All selenium must be degraded and reduced to selenide prior to incorporation in newly formed Sec. Selenide replaces the sulfhydryl group of cysteine or the hydroxyl group of serine to form selenocysteine (). Readily available cysteine from NAC might serve as a donor pool of cysteine to allow more rapid insertion of available selenium into newly formed Sec. The additional glutathione production from NAC would have several potential roles. Excretion of mercury utilizes glutathione-binding during the final excretion transport process [Citation17–19]. NAC supplementation after mercury poisoning appears to increase mercury excretion [Citation20–22]. Additionally, intracellular glutathione is critical to maintaining glutathione peroxidase in the reduced state to allow for the redox/antioxidant activity [Citation7]. The production of additional glutathione in an environment of severe oxidant stress may be partially protective.
Figure 4. Structures of the amino acids serine, cysteine, and selenocysteine.
There were several important changes in mercury and selenium concentrations in our patient following oral selenium and NAC treatment that deserve brief comments. After initiation of oral selenium and NAC supplementation, blood mercury concentration initially rose (from 11 to 28.1 mcg/L), along with a temporal improvement in clinical neurological and cardiovascular status. We believe this may represent a redistribution of mercury to the central compartment from the tissues. [Citation23,Citation24]. Selenium and the selenium–mercury complex (Se:Hg) are primarily bound and transported in blood by selenoprotein-P (Se-P) [Citation23]. Plasma Se-P is a Se-rich glycoprotein (with 10 selenocysteines) produced in the liver. The addition of a robust supply of selenium to the liver and NAC for the multiple cysteines on Se-P may have supported increased Se-P production, with subsequent redistribution/transport of mercury away from the target organs of the brain and kidney.
Despite receiving selenium doses of 500 mcg/day (over 9 times the U.S. recommended daily allowance (RDA) and over 12 times the WHO recommended minimum daily intake), his Se concentrations remained normal. Selenium content of skeletal muscle is approximately 0.4 mcg/g. Our patient in the short period of five months regained 45 pounds (20.45 kg) of body weight, most of which was skeletal muscle regrowth, which would require more than 8 mg of selenium supply. The lack of rise in serum selenium concentration suggests the redistribution of a portion of the oral selenium to the regrowth demands.
The urine mercury concentrations initially fell and then doubled (23.8–48.4 mcg/L) after initiation of oral Se and NAC and remained elevated for four months (). Li et al. [Citation25,Citation26] showed an increase in urinary Hg in humans after supplementation with organoselenium as selenomethionine. The increase in urinary Hg did not occur for 15–30 days post-selenium supplementation, suggesting a redistribution of total body mercury rather than any direct renal effect [Citation25,Citation26]. The urine mercury at five and eight months has fallen. Due to issues of cost and patient compliance, urine mercury was only measured every 6–8 weeks, so we do not have detailed weekly changes. Urinary mercury appears to come from mercury previously deposited in the kidney [Citation27]. We suggest that clinical improvement may be a better measure than urine or blood concentrations.
Conclusion
This is a case of severe elemental mercury poisoning unchanged by chelation who later achieved significant improvement temporally related to selenium and NAC supplementation. Daily large doses of selenium for eight months did not result in elevated serum selenium, suggesting a significant selenium deficit or potential sequestration of Se as an inert Hg:Se complex.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Branco V, Godinho-Santos A, Gonçalves J, et al. Mitochondrial thioredoxin system as a primary target for mercury compounds. Toxicol Lett. 2014;Suppl:S57–S58, 229.
- Branco V, Canario J, Lu J, et al. Mercury and selenium interaction in vivo: effects on thioredoxin reductase and glutathione peroxidase. Free Radical Biol Med. 2012;52:781–793.
- Branco V, Lu J, Holmgren A, et al. Effect of mercury compounds over the redox state of thioredoxin 1 and 2 and its relation with glutathione levels and glutaredoxin activity in human neuroblastoma cells. Toxicol Lett. 2015;Suppl:s314.
- Carvalho CML, Chew EH, Hashemy SI, et al. Inhibition of the human thioredoxin system: a molecular mechanism of mercury toxicity. J Biol Chem. 2008;283:11913–11923.
- Sugiura Y, Tamai Y, Tanaka H. Selenium protection against mercury toxicity: high binding affinity of methylmercury by selenium-containing ligands in comparison with sulfur-containing ligands. Bioinorg Chem. 1978;9:167–180.
- Spiller HA. Rethinking mercury: rethinking mercury: the role of selenium in the pathophysiology of mercury toxicity. Clin Toxicol. 2018:56.
- Ralston NVC, Raymond LJ. Dietray selenium's protective effects against methylmercury toxicity. Toxicology. 2010;278:112–123.
- Kosnett MJ. The role of chelation in the treatment of arsenic and mercury poisoning. J Med Toxicol. 2013;9:347–354.
- Carvalho CML, Lu J, Zhang X, et al. Effects of selenite and chelating agents on mammalian thioredoxin reductase inhibited by mercury: implications for treatment of mercury poisoning. FASEB J. 2011;25:370–381.
- Glaser V, Martins RP, Viera AJH, et al. Diphenyl diselenide administration enhances cortical mitochondrial number and activity by increasing hemeoxygenase type 1 content in a methylmercury-induced neurotoxicity mouse model. Mol Cell Biochem. 2014;390:1–9.
- Mendelev N, Mehta SL, Idris H, et al. Selenite stimulates mitochondrial biogenesis signaling and enhances mitochondrial functional performance in Murine Hippocampal Neuronal Cells. PloS One. 2012;7:e47910.
- Meinerz DF, de Paula MT, Comparsi B, et al. Protective effect of organoselenium compounds against methylmercury-induced oxidative stress in mouse brain mitochondrial-enriched fractions. Braz J Med Biol Res. 2011;44:1156–1163.
- Dalla Corte CL, Soares FAA, Aschner M, et al. Diphenyl diselenide prevents methylmercury-induced mitochondrial dysfunction in rat liver slices. Tetrahedron. 2012;68:10437–10443.
- Penglase S, Hamre K, Ellingsen S. Selenium prevents downregulation of antioxidant selenoprotein genes by methylmercury. Free Radical Biol Med. 2014;75:95–104.
- Guerra I, Branco V, Rodrigues J, et al. Protective effects of selenium compounds in the toxicity of mercury over the thioredoxin system in neuroblastoma cells. Toxicol Lett. 2014;229:S101–S102.
- Joshi D, Mittal DK, Shukla S, et al. Methylmercury toxicity: amelioration by selenium and water-soluble chelators as N-acetyl cysteine and dithiothreitol. Cell Biochem Funct. 2014;32:351–360.
- Dutczak WJ, Ballatori N. Transport of glutathione-methylmercury across liver canalicular membranes on reduced glutathione carriers. J Biol Chem. 1994;269:9746–9751.
- Tanaka-Kagawa T, Naganuma A, Imura N. Tubular excretion and reabsorption of mercury compounds in mouse kidney. J Pharmacol Exp Ther. 1993;264:776–782.
- Wang H, Chen B, He M, et al. Selenocysteine against methylmercury cytotoxicity in HepG2 cells. Sci Rep. 2017;7:147. DOI:10.1038/s41598-017-00231-7
- Koh AS, Simmon-Willis TA, Pritchard JB, et al. Identification of a mechanism by which the methylmercury antidotes N-acetylcyteine and dimercaptopropanesulfonate enhance urinary metal excretion: transport by the renal organic anion transporter-1. Mol Pharmacol. 2002;62:921–926.
- Ballatori N, Lieberman MW, Wang W. N-acetylcysteine as an antidote in methylmercury poisoning. Environ Health Perspect. 1998;106:267–271.
- Madejczyk MS, Aremu DA, Simmon-Willis TA, et al. Accelerated urinary excretion of methylmercury following administration of its antidote N-acetylcysteine requires Mrp2/Abcc2, the atypical multidrug resistant-associated protein. J Pharmacol Exp Ther. 2007;322:378–384.
- Chen C, Yu H, Zhao J, et al. The roles of serum selenium and selenoproteins on mercury toxicity in environmental and occupational exposure. Environ Health Prespect. 2006;114:297–301.
- García-Sevillano MA, Rodriguez-Moro G, Garcia-Barrera T, et al. Biological interactions between mercury and selenium in distribution and detoxification process in mice under controlled exposure. Effects of selenoprotein. Chem Biol Interact. 2015;229:82–90.
- Li YF, Dong Z, Chen C, et al. Organic selenium supplementation increases mercury excretion and decreases oxidative damage in long-term mercury-exposed residents from Wanshan China. Environ Sci Technol. 2012;46:11313–11318.
- Li X, Yin D, Chen Q, et al. Dietary selenium protects against redox-mediated immune suppression induced by methylmercury exposure. Food Chem Toxicol. 2014;72:169–177.
- Hursh JB, Clarkson TW, Nowak TV, et al. Prediction of kidney mercury content by isotype techniques. Kidney Int. 1985;27:898–907.