
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Polymers are becoming more important in all economic sectors, and as environmental concerns grow, biopolymers are replacing metal- or oil-based polymers. Two of these polymers are polyhydroxybutyrate (PHB), which is known for good mechanical characteristics and the manufacturing from renewable resources, and polylactic acid (PLA), which is known for fast degradation rates and great benefits for packaging industry. They exhibit properties that make them competitive alternatives for the less eco-friendly polymers, but processing techniques are not as well-researched. In this study, we performed in vitro degradation and drug release studies with pure PHB and PLA and PHB/PLA blends (1:3). Therefore, polymers were stored at 65°C in a PBS-buffer under rather static conditions to simulate intraossal localization. The mass loss of all samples indicates a degradation of all polymers, and it was confirmed by decreasing molecular weight, decreasing pH, increasing crystallinity, and decreasing water contact angle. Following these measurements, a 60-day drug release study was performed, which revealed a four-phase drug release mechanism, including a diffusion-controlled initial burst release especially elevated for investigated blends due to eased medium interpenetration, and a secondary burst release after 20 days for both blends and the pure PLLA-Biomer with lower molecular weight. The intensity of the secondary burst release corresponded to observed degradation characteristics allowing the conclusion of a degradation controlled drug release here.
Introduction
The polymer market is expanding, and biopolymers are gaining importance as well. In daily life, where climate change is one of the biggest issues for the next decades, biopolymers are an important solution for a great number of challenges. Biopolymers are a subset of polymers, but they are not clearly defined. Biopolymers are polymers that are produced from renewable raw materials, such as wheat or organisms (like bacteria), and/or they can be degraded naturally. This means, that even oil-based polymers can be considered biopolymers if they are also degradable [Citation1]. Polyesters are an example of degradable, oil-based polymers, especially polyethylene terephthalate (PET), which is standardly produced from ethylene glycol and terephthalic acid. Both materials are oil based, but there is a potential to make them more naturally. The first step is to alter the source of ethylene glycol from oil to sugar cane molasses. The second step is to produce terephthalic acid from renewable raw materials. This is a great example for turning a biopolymer into a native biopolymer [Citation2,Citation3]. One of these native biopolymers is polyhydroxybutyrate (PHB). It is biocompatible, biodegradable, and not soluble in water, opening up a wide range of applications. Therefore, PHB is often referred to as the “sleeping giant” [Citation4,Citation5]. There are different kinds of PHBs: e.g. P(3HB) and P(4HB). Structural and atomic differences in the biopolymers result in a variety of different characteristics [Citation6]. Despite the potential usefulness of PHB, manufacturing limitations have prevented its widespread use. Previously, it was only possible to extract PHB from cells as a product of an enzymatic chain reaction [Citation7]. However, methods for its production via renewable raw materials (e.g. wheat or potatoes) have been discovered. To do so, specialized enzymes ferment starches to PHB [Citation4,Citation7–Citation10]. The cost of manufacture remains a challenge, and the process is not well-suited to industrial-scale production of PHB. Most studies and applications of PHB deal with an enzymatic degradation through bacteria or a medium with enzymes. In the absence of enzymes, degradation starts in the amorphous regions of the PHB and will hydrolyze those parts. Additionally, there is the possibility for chain cleavage in the whole polymer. This depends on the medium and other circumstances (e.g. temperature) [Citation7,Citation11,Citation12]. The mechanical characteristics of PHB are quite similar to petrochemical polymers such as polypropylene, increasing the number of possible applications [Citation11–Citation14]. An advantage for biopolymers like PHB is that the required temperatures and pressures are lower for the different manufacturing methods, decreasing the relative costs of production [Citation2,Citation7]. Nevertheless, high production costs of PHB have kept it confined to the medical and aerospace fields [Citation11,Citation15]. However, it can be useful for applications in urban-farming because of its biodegradable character [Citation16,Citation17]. Another, well known, biopolymer is poly(lactic acid) (PLA). Since the 1970s it has been a commonly used polymer and various kinds of PLA are on the market. Either PDLA or PLLA are used, while both base polymers can have different characteristics [Citation18]. In industry, PLA is typically produced via a fermentation process with cereals and corn, which forms lactic acid. With a condensation reaction, it can react to form low molecular weight PLA. Otherwise it is possible to produce higher molecular weight PLA with a depolymerization of the low molecular weight PLA followed by a ring-opening polymerization in which the built lactides will react to form higher molecular weight PLA types [Citation19]. Compared to PHB, PLA has similar manufacturing advantages and can also compete with standard petrochemical polymers [Citation20]. PLA is also a biopolymer which degrades through hydrolyzation and chain cleavage to H2O and CO2 [Citation21,Citation22]. Since the properties of PLA are well-characterized, the polymer is widely used with numerous applications. PLA is commonly used to coat other materials such as textiles or other polymers [Citation21], and other simple products like bottles are made from PLA [Citation20]. PLA has been used in the automotive, medical, and aerospace sector [Citation23,Citation24]. Since both, PLLA and PHB, are biocompatible and they undergo biodegradation to non-toxic byproducts, they have been previously investigated as drug delivery matrices [Citation25]. A drug can be included in the polymer matrix quite easily since these films are made via solvent evaporation methods like dip coating or spray coating, so the drug can be mixed into the initial polymer solution [Citation26]. Those applications are mentioned in the literature, especially as material for scaffolds used in bone tissue engineering [Citation27,Citation28]. Besides PLLA and PHB, blends of both polymers are often used due to the adaptability of particular mechanical properties. There is various literature reporting either on the degradation or drug release kinetics of one or the other polymer [Citation15,Citation21,Citation29]. However, studies differ a lot from each other due to the use of distinct media (pH, ionic strength, additives, etc.) or further conditions (temperature, sampling interval, surface-to-volume ratio, shaking, etc.), so that comparison of the behavior of different materials and correlation of drug release to polymer degradation is often hindered. Within this manuscript we provide an in vitro degradation and drug release study performed under identical rather static conditions, as for example observed in bone with low vascularization, allowing correlation of both mechanisms. Furthermore, PLLA with low and high molecular weight, P(3HB) and blends thereof are investigated using identical conditions to allow for direct comparison of the degradation and drug release behavior.
Materials and methods
Materials
Two different kinds of PLLA were used: Resomer L210 with Mw = 311,000 g/mol (Evonik Industries AG, Essen, Germany) and Biomer with Mw = 65,000 g/mol (Biomer, Krailing, Germany), further in manuscript denoted as PLLA-Resomer L210 and PLLA-Biomer, respectively. The P(3HB) (PHE001, Mw = 132,000 g/mol) was supplied by Lackner Ventures & Consulting GmbH, Wien, Austria. Besides using pure materials, blends with a weight ratio of 3:1 (PLLA:P(3HB)), denoted as PLLA-Resomer L210 blend and PLLA-Biomer blend, were tested. Fluorescein diacetate (FDAc) served as model drug and was purchased from Sigma Aldrich, Munich, Germany.
Film preparation
For the preparation of the samples, the granules of the pure PLLA types or the mixed granules of the blends were solved in CHCl3 (ctotal = 70 mg/ml). Afterwards, the solution (v = 70 ml) was poured into petri dishes (d = 12cm) and covered with foils to slow down evaporation. The solvent was allowed to evaporate from each sample for over 24 hours, and then the samples dried for 1 day at 40°C in a vacuum drying oven. The dried polymer films were 400 to 450 μm thick. It was not possible to dissolve pure P(3HB) in any solvent for film preparation via solution casting. Therefore, the samples were pressed with the Polystat 200T Press, Schwabenthan, Balingen, Germany. Granules as received were laid into the heated press and top and bottom were covered with a polyimide foil (Kapton Type 100HN with a thickness of 25 μm; DuPont, Wilmington, USA) to ease the detachment of the resultant polymer film. The press was heated to 185°C and pressed with a pressure of 100 bar. After 10 minutes, the samples were removed and cooled between two steel plates to hold constant pressure and prevent the film from flowing out of the polyimide foil. After the film cooled down, it was detached from the polyimide foil. Those pressed films were again 400 to 450 μm thick. After the polymer films were produced, they were stamped into 12 mm round specimen for the degradation study.
Preparation of drug release films
For the drug release study, polymer films were prepared with FDAc, which was meant to act as a model drug as previously described in literature [Citation30]. The same solution casting process was followed as above to make polymer films of PLLA Biomer, PLLA Resomer, and their respective 3:1 blends with P(3HB). However, 50 mg of FDAc was added (at a concentration of 100 mg/ml in chloroform) to the polymer solution as well and mixed until homogeneous. These films were also stamped into 12 mm round specimen.
In vitro degradation study
The degradation study was accomplished with a Titramax 1000 with heating module Incubator 100 and a flat incubation cover from Heidolph, Schwabach, Germany. The samples were stored in small glasses with a screw lid to secure the buffer from evaporation. Phosphate buffered saline (PBS, pH = 7.4) was used as degradation medium with a ratio of 10 ml/g sample. The degradation temperature was set to 65°C and shaken at 120 to 150 rpm. No intermediate replacement of the medium was performed to simulate rather static conditions as observed in bone with low vascularization. Polymers samples were removed from solution after increasing time intervals and characterized regarding weight loss, viscosity, water contact angle, chemical composition, and crystallinity.
Characterization methods
Weight loss
Each sample was weighed before the start of the study and after being extracted from the glasses, rinsed with water and dried for 12 hours in a vacuum drying oven at 40°C. They were weighed with a microbalance ACL-110.4 from Aculab, Milton Keynes, United Kingdom.
Viscosimetry
Viscosity measurements were processed with an Ubbelohde type 0c and I from SI Analytics GmbH, Mainz, Germany. The Ubbelohde capillary was set into a water bath at 25°C. Three different concentrations (2.5; 5; 7.5 g/l) were used to calculate the molar mass with the Staudinger-Mark-Houwink Equation 1 with α = 0.82 for all materials and K(PHB) = 0.077 ml/g, K(PLLA) = 0.0129 ml/g and K(blend) = 0.0116 ml/g according to [Citation31]:
FTIR-spectroscopy
FTIR-spectra were recorded with the Avatar 360 FT_IR Nicolet and analyzed with the software OMNIC Spectra 8.3 (Thermo Fischer Scientific, Waltham, USA). Every spectrum, including background and samples, was recorded 32 times. For the measurement, the attenuated total reflection method was used.
Contact angle measurements
Water contact angles were measured with OCA 20 from DataPhysics, Filderstadt, Germany, and analyzed with the SCA 22 Software as a static contact angle with the Young equation. Each sample was tested seven times with the contact angle of water at different extraction points. Deionized water was used at room temperature.
Differential scanning calorimetry
For the DSC-measurement the DSC 823 with the STARe Software (Mettler Toledo, Columbus, USA) was used. All samples were tested with the following program: (i) heating from 25 to 190°C; heat rate: 20 K/min; (ii) holding phase: 2 minutes at 190°C, (iii) cooling from 190 to −20°C; cooling rate: −20 K/min, (iv) holding phase: 2 minutes at −20°C, (v) heating from −20 to 200°C; heat rate: 20 K/min, (vi) holding phase: 2 minutes at 200°C, (vii) cooling from 200 to 25°C; cooling rate: −20 K/min. The polymer samples were weighed in an aluminum crucible during measurements in the DSC oven and nitrogen was used as inert gas with a flow rate of 30 ml/min during the measurement. To calculate the degree of crystallinity a melting enthalpy ΔHf of 146 J/g was assumed for 100% crystalline PHB; 93.6 J/g for 100% crystalline PLLA and 106.7 J/g for the blend according to [Citation32].
pH-value measurements
The pH value of the degradation media was monitored during the in vitro study with the pH 1970i and pH electrode SenTix® 22 Basis pH-electrode (WTW, Dinslaken, Germany).
In vitro drug release study
Conditions (medium, temperature, shaking, etc.) for drug release were identical to those of the degradation study. Briefly, samples of each polymer type were placed into clear glass vials and doused with PBS. Samples were kept at 65°C and shaken constantly at 120–150 rpm. Then, at various timepoints (these had to be varied in comparison to the degradation study due to saturation and instability of the model drug) the solution was completely removed from the vials and fresh buffer was added. 10 µL of the removed solution was mixed with 90 µL of 1.0 M NaOH in a 96-well plate in replicates. Fluorescence intensity (excitation wavelength: 485 nm, emission wavelength: 520 nm) was then measured with a microplate reader (BMG Labtech, Ortenberg, Germany) to estimate the released drug amount.
Results and discussion
Weight loss of polymer samples during in vitro degradation
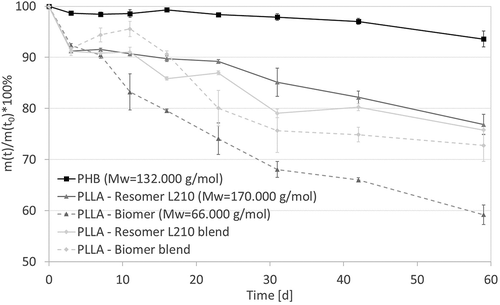
All samples lost mass during the degradation period (). P(3HB) lost the least amount of weight (≈ 5% mass loss) compared to all other samples. There was also a difference in mass loss between both PLLA types. PLLA-Biomer samples lost more weight (≈ 40%) than PLLA-Resomer L210 samples (≈ 25 %), and it is notable that pure PLLA-Biomer lost much more weight compared to the blended samples (PLLA – Resomer L210 blend: ≈25 %, PLLA-Biomer blend: ≈27 %). At all, it is clear to see that mass loss of PHB is slower than pure and blended PLLA. This is caused by the different chemical structure. Because of the added carbon atom at the P(3HB) chain, compared to PLLA, the chain degradation is estimated to be slower [Citation33]. Additionally, one can see the influence of the molecular weight causing faster mass loss of the PLLA-Biomer compared to the PLLA-Resomer L210. The lower mass loss of the PLLA-Biomer blend, compared to the pure biopolymer, results from the more slowly degrading P(3HB) components. However, the PLLA-Resomer L210 blend lost more weight compared to the pure PLLA-Resomer L210. This is probably related to the empty volume between the surface of the P(3HB) and PLLA phases allowing faster medium interpenetration. These both influencing factors compete with other in both blends leading to comparable mass loss kinetics in the presented study.
Figure 1. Weight loss of investigated biopolymers as a function of degradation time (T = 65 ° C, medium: PBS buffer (pH 7.4), 120–150 rpm). Results are represented as mean value ± standard deviation of n ≥ 3 samples.
Molecular weight loss of polymers during in vitro degradation
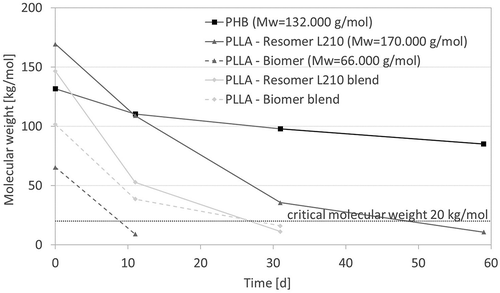
Hydrolysis, which is the main degradation mechanism expected for the investigated polymers under chosen conditions, generally follows three steps: i) uptake of degradation medium, ii) decrease of the average molecular weight associated with loss of mechanical stability, iii) actual weight loss when critical molecular weight is achieved. The critical molecular weight of polymers is in general 20,000 g/mol, for PLLA a more detailed definition was published by Pitt et al., identifying a lower critical molecular weight of 10,000 g/mol [Citation34].
The molecular weight of all samples was determined via Ubbelohde viscosimetry. Besides PHB, all investigated biopolymers crossed the critical molecular weight at 20,000 g/mol during the presented in vitro degradation study, underlining the slow degradation behavior of PHB as mentioned during of mass loss observation (). Both PLLA show faster degradation than PHB. The slope corresponding to degradation velocity seems to be independent of the PLLA chain length, however the time point when the critical molecular weight is reached differs. While the chains of PLLA- Resomer L210 are in the range of the critical molecular weight after 50 days, PLLA-Biomer crosses this barrier already after 11 days. This explains the difference in mass loss described before. Further points of the PLLA-Biomer were not measurable due to nearly similar viscosity to CHCl3. The observation of the molecular weight of corresponding blends, which always present a mean value of contained components, once again underlines mass loss measurements. The blended PLLA-Resomer L210 degrades faster than the pure PLLA-Resomer L210 and reached the area of critical mass after less than 30 days. The faster degradation is caused by the created interfaces between both polymers. The PLLA-Biomer blend shows slower decrease of molecular weight due to the presence of slowly degrading P(3HB) components with higher starting molecular weight.
Figure 2. Molecular weight of biopolymers as a function of degradation time (T = 65 °C, medium: PBS buffer (pH 7.4), 120–150 rpm) as determined via Ubbelohde viscosimetry.
Change of surface composition of polymer samples during in vitro degradation
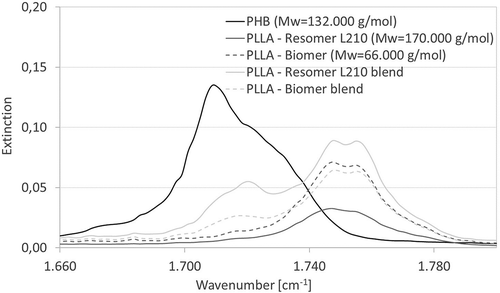
In order to evaluate whether degradation products can be identified on the sample surfaces, FTIR-ATR measurements were performed. shows that PHB has a peak between 1.710 and 1.720 cm−1. PLLA samples have a band at nearly 1.750 cm−1, which is split into two peaks, corresponding to Zhang and Thomas [Citation35]. It is also recognizable, that both blended samples show the peaks of both materials. The peaks of all samples were identical whenever the FTIR-spectrum was measured (data not shown). This indicates that the chemical composition of the surface of any biopolymer sample does not change significantly during degradation. In summary with results for mass loss and molecular weight reduction, one can assume the continuance of surface composition as indice for bulk degradation, which is generally stated for hydrophobic polymers as PLLA and P(3HB) in literature [Citation36].
Figure 3. FTIR-ATR spectra of examined biopolymer samples after three days of degradation in the range of 1660 to 1800 cm−1.
Change of water contact angle of polymer samples during in vitro degradation
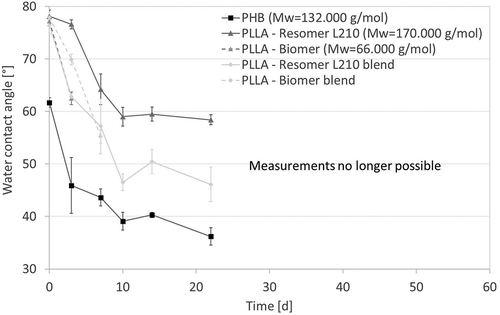
Initially, all samples show similar water contact angles of 78° except P(3HB), which had a much lower water contact angle of 61° (). The contact angle for each polymer decreased over time during the degradation, which could be an indication for the formation of carboxyl- and hydroxyl end groups by cleavage of ester bonds. However, taken together with FTIR spectra showing no alteration of functional groups, the decreasing contact angle is rather associated with surface roughening due to polymer mass erosion. Moreover, it has be stated, that in some cases, samples became too brittle after degradation, making it impossible to measure the water contact angle throughout the entire time course of the study.
Figure 4. Surface water contact angle of biopolymer as function of degradation time (T = 65 °C; Medium: PBS-buffer (pH 7.4); 120–150 rpm). Results are represented as mean +- standard deviation of n ≥ 5 measurements.
Change of crystallinity of polymer samples during in vitro degradation
A higher crystallinity indicates degradation of semi-crystalline polymers since amorphous regions degrade first due to less organization. PHB shows the lowest initial crystallinity of all samples, and it increased less as well with degradation time, adding evidence to support a slower degradation rate than PLLA (). Both investigated PLLA types show a faster increase of crystallinity in the presented in vitro degradation study, while the crystallinity of the blends rose more slowly than the pure PLLA types, a result of the 25 w% of PHB. The increasing extinction of the peak at 1.755 cm−1in recorded FTIR-ATR spectra (), which is attributed to crystalline carbonyl vibration [Citation33], supports the measurements of the DSC. The increase in crystallinity off all samples could be also macroscopically followed, not least by the observed brittleness of samples rendering contact angle measurements impossible ().
Figure 5. Degree of crystallinity of biopolymers as a function of degradation time (T = 65°C, medium: PBS buffer (pH 7.4), 120–150 rpm) as determined via DSC measurements (ΔHf100% (PHB) = 146 J/g; ΔHf100% (PLLA) = 93.6 J/g; ΔHf100% (blend) = 106.7 J/g).
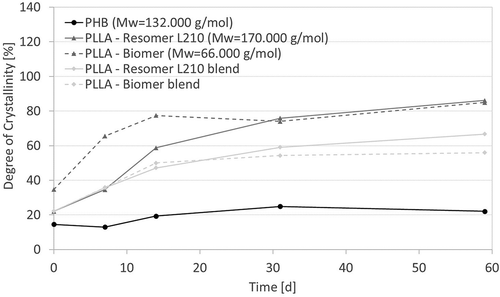
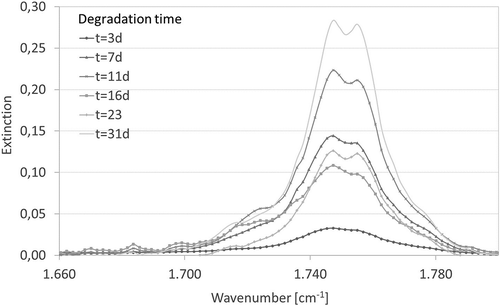
Figure 6. Evolution of FTIR-ATR spectra of PLLA – Resomer L210 samples during in vitro in the range of 1660 to 1800 cm−1.
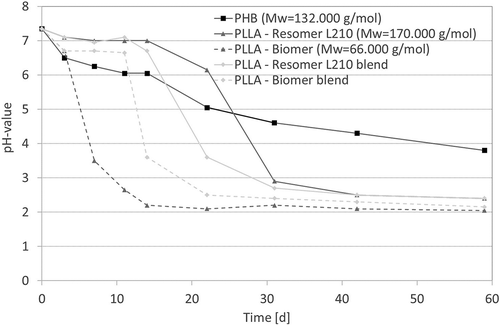
Change of pH-value during in vitro degradation
Degradation of polyesters is accompanied by the formation of alcohols and carboxylic acids, leading to a decrease in pH of the surrounding medium under rather static conditions. This fact could be observed for all investigated samples (). The degradation media of pure PLLA and blended samples show an identical course. After different time intervals (PLLA-Biomer: 7 days; PLLA-Biomer blend: 11 days; PLLA-Resomer L210 blend: 15 days; PLLA Resomer L210: 22 days), the pH of the medium decreases rapidly below three. In contrast, the pH evolvement of the degradation medium of PHB shows a linear course over the whole study. This result corroborates the assumption that PHB shows slower degradation rate while the PLLA-Biomer degrades faster than the PLLA-Resomer L210, and the blends normally degrade faster than the pure materials. The PLLA-Biomer is an exception because of its low molecular mass.
Figure 7. pH-value of the medium as a function of degradation time (T = 65°C, medium: PBS buffer (pH 7.4), 120–150 rpm).
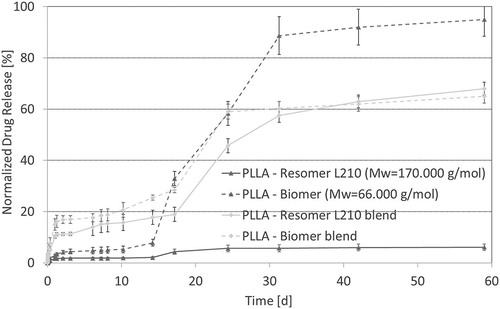
In vitro drug release study
Beyond studying the characteristics of the polymers during degradation, a parallel in vitro drug release study of all polymers except PHB using FDAc as model drug was performed. PHB could not be integrated in the drug release study due to its bad solubility in CHCl3 permitting solution casting of drug-containing polymer films. depicts the normalized release of FDAc from the polymer films. Both blends and the PLLA-Biomer samples evidence a rather four phase release mechanism: (i) Initital burst release, (ii) slow release phase, (iii) secondary burst release at around 20 days and (iv) secondary slow release phase. The first initial burst release is generally triggered by diffusion, which is estimated to be higher in blends due to the presence of interfaces easing the medium interpenetration and thereby release of surface-near drug molecules. The following slow release phase is caused by diffusion of drug molecules from the bulk. The secondary burst release evidenced for the blends and PLLA-Biomer in the presented study is caused by the breakage of samples due to polymer degradation after 20 days. Thereby drug molecules at the boundaries are directly released by a so-called degradation controlled mechanism and the surface to volume ratio increased allowing more rapid diffusion controlled drug release. The intensity of the secondary burst release is in accordance with data from the in vitro degradation study, evidencing a higher degradation rate for PLLA-Biomer and comparable degradation rate for both PLLA blends. At the end of the in vitro drug release study the PLLA-Biomer neared 100% release while the PLLA-Resomer L210 blend and the PLLA-Biomer blend achieved about 60% release. The pure PLLA-Resomer L210 achieved less than 10% release in the same time frame. Comparing again to mass loss measurements we find the same 10% for PLLA, indicating that drug release is mainly degradation controlled, while the diffusion coefficient of FDAc seems to be very low in the higher molecular weight PLLA. In contrast mass losses of blends here are around 20% and for the pure PLLA-Biomer around 30% indicating that diffusion-controlled drug release plays an important role, which is triggered by interphases between both blend components and the lower molecular weight of PLLA-Biomer. Observed mass losses are in general lower compared to the in vitro degradation study discussed above, because of the necessary medium change at removal timepoints to prevent drug saturation and degradation, leading to less decreased pH-values during the in vitro drug release study.
Figure 8. Normalized drug release from each of the polymer films (n = 3) as a function of degradation time (T = 65°C, medium: PBS buffer (pH 7.4), 120–150 rpm).
Conclusion
All investigated polymer samples degraded during the performed in vitro study evidenced by mass loss, molecular weight reduction, decreasing water contact angles, and enhanced crystallinity. Performed FTIR-spectroscopy evidenced no structural/molecular changes at the surface during the study, but it underlined an increasing degree of crystallinity. The rate of observed changes was specific for each polymer tested. PHB degrades slower than all PLLA samples, including blends, which indicates that the chemical structure is the reason for this conduct. The difference between polymers composed of identical monomers (PLLA) with different molecular weight was also clear to see. Lower molecular weight leads to a faster degradation compared to higher molecular weight materials. The blending of PLLA had two effects, lowering degradation rate due to the presence of PHB and at the same time increasing degradation rate by the presence of empty volume in interphases between both blend components.
Based on these differences in degradation rates, it makes sense that investigated biopolyesters would have different drug release profiles as well. One could observe a four phase drug release mechanism, including a diffusion-controlled initial burst release especially elevated for investigated blends due to eased medium interpenetration, and a secondary burst release after 20 days for both blends and the pure PLLA-Biomer with lower molecular weight. The intensity of the secondary burst release corresponded to observed degradation characteristics allowing the conclusion of a degradation controlled drug release here. To the best of our knowledge, this is the first study performing a direct comparison of degradation and drug release profiles for biopolyesters.
Acknowledgments
The authors acknowledge the technical support of Veronika Terveen, Hannelore Schmidt, Marius Barth and Dirk Bröker, who assisted during the analytical and manufacturing process of the study. Moreover, the provision of P(3HB) from Lackner Ventures & Consulting GmbH as well as the PLLA-Biomer from Biomer is appreciated.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Notes on contributors
R. Harting
R. Harting is a Master graduate of Applied Materials Science at the University of Applied Sciences Osnabrück, Collaborator in the Laboratory of Chemistry and Surface Modification at the University of Applied Sciences Osnabrück.
K. Johnston
K. Johnston is a graduate of Biomedical Engineering at the University of Wisconsin-Madison, Research stay in the Laboratory of Chemistry and Surface Modification at the University of Applied Sciences Osnabrück.
S. Petersen
S. Petersen is principal investigator at the University of Applied Sciences Osnabrück, Head of the Laboratory of Chemistry and Surface Modification, and Professor of Chemistry and Adhesive Joints.
Related Research Data
References
- Endres H-J. Biopolymers facts and statistics. Hannover: IfBB - Institute for Bioplastics and Biocomposites; 2017.
- Burk MJ, Osterhout RE, Sun J. Semi-synthetic terephthalic acid via microorganisms that produce muconic acid. Alexandira: USPTO; 2011.
- Cheng Y-T, Jae J, Shi J, et al. Production of renewable aromatic compounds by catalytic fast pyrolysis of lignocellulosic Biomass with bifunctional Ga/ZSM‐5 catalysts. Angew Chem Int Ed. 2012;51:1387–1390.
- Kaiser W. Kunststoffchemie für Ingenieure: von der Synthese bis zur Anwendung. Munich: Carl Hanser Verlag GmbH Co KG; 2015.
- Markl E, Grünbichler H, Lackner M. PHB - bio based and biodegradable replacement for PP. Rev. 4. 2018;2.
- Isikgor FH, Becer RC. Lignocellulosic biomass: a sustainable platform for the production of bio-based chemicals and polymers. Polym Chem. 2015;6:4497–4559.
- Somleva MN, Peoples OP, Snell KD. PHA bioplastics, biochemicals, and energy from crops. Plant Biotechnol J. 2013;11:233–252.
- Endres H-J. Biopolymers facts and statistics. Hannover: IfBB - Institut für Biokunststoffe und Bioverbundwerkstoffe; 2016.
- Kamravamanesh D, Pflügl S, Nischkauer W, et al. Photosynthetic poly-β-hydroxybutyrate accumulation in unicellular cyanobacterium synechocystis sp. PCC 6714. AMB Express. 2017;7:143.
- Kamravamanesh D, Kovacs T, Pflügl S, et al. Increased poly-β-hydroxybutyrate production from carbon dioxide in randomly mutated cells of cyanobacterial strain synechocystis sp. PCC 6714: mutant generation and characterization. Bioresour Technol. 2018;266:34–44.
- Wong JW-C, Tyagi RD, Pandey A. Current developments in biotechnology and bioengineering: solid waste management. Amsterdam: Elsevier; 2016.
- Freier T. Biopolyesters in tissue engineering applications. Polymers for regenerative medicine [Internet]. Berlin, Heidelberg: Springer; p. 1–61. [cited 2017 Aug 31]. Available from: https://link.springer.com/chapter/10.1007/12_073
- Asrar J, Hill JC. Biosynthetic processes for linear polymers. J Appl Polym Sci. 2002;83:457–483.
- Tertyshnaya YV, Khvatov AA, Likhachev AN, et al. Influence of the structural parameters on the biodegradation of poly-3-hydroxybutyrate and composites based on it. Plasticheskie Massy. 2010. p. 62–64.
- Korkusuz F, Korkusuz P, Ekşioĝlu F, et al. In vivo response to biodegradable controlled antibiotic release systems. J Biomed Mater Res. 2001;55:217–228.
- Rettenbacher L. Biokunststoffe Überblick und aktuelle Trends in der Biokunststoffwelt. Wien: OFI; 2014.
- Roch H. Kunststoffe auf Basis nachwachsender Rohstoffe Eigenschaften, Einsatzmöglichkeiten und limitierende Faktoren. Hamburg: Fraunhofer UMSICHT; 2012.
- Sudesh K, Iwata T. Sustainability of biobased and biodegradable plastics. Clean Soil Air Water. 2008;36:433–442.
- Jamshidian M, Tehrany EA, Imran M, et al. Poly-lactic acid: production, applications, nanocomposites, and release studies. Compr Rev Food Sci Food Saf. 2010;9:552–571.
- Baur E, Osswald TA, Rudolph N, Eds. Saechtling Kunststoff Taschenbuch. Vol. 31. München: Hanser; 2016.
- Lv S, Liu X, Gu J, et al. Microstructure analysis of polylactic acid-based composites during degradation in soil. Int Biodeterior Biodegrad. 2017;122:53–60.
- Visakh PM, Arao Y. Thermal degradation of polymer blends, composites and nanocomposites. Schweiz: Springer; 2015.
- Pinto VC, Ramos T, Alves S, et al. Comparative failure analysis of PLA, PLA/GNP and PLA/CNT-COOH biodegradable nanocomposites thin films. Procedia Eng. 2015;114:635–642.
- Bartolo P, Bidanda B. Bio-materials and prototyping applications in medicine. New York (NY): Springer; 2008.
- Yoon S-D, Kwon Y-S, Lee K-S. Biodegradation and biocompatibility of Poly L-lactic acid implantable mesh. Int Neurourol J. 2017;21:S48–54.
- Griesser HJ. Thin film coatings for biomaterials and biomedical applications. Sawston, Cambridge: Woodhead Publishing; 2016.
- Wei G, Ma PX. Structure and properties of nano-hydroxyapatite/polymer composite scaffolds for bone tissue engineering. Biomaterials. 2004;25:4749–4757.
- Rezwan K, Chen QZ, Blaker JJ, et al. Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering. Biomaterials. 2006;27:3413–3431.
- Mainil-Varlet P, Curtis R, Gogolewski S. Effect of in vivo and in vitro degradation on molecular and mechanical properties of various low-molecular-weight polylactides. J Biomed Mater Res. 1997;36:360–380.
- Steele TWJ, Huang CL, Kumar S, et al. High-throughput screening of PLGA thin films utilizing hydrophobic fluorescent dyes for hydrophobic drug compounds. J Pharm Sci. 2011;100:4317–4329.
- Li T. Charakterisierung von Biopolymeren. Darmstadt: Technische Universität Darmstadt; 2013.
- Vogel C. Charakterisierung der thermischen und mechanischen Eigenschaften von Polyhydroxyalkanoat (PHA) Homopolymeren, Copolymeren und Polymermischungen. Essen: Duisburg-Essen; 2008.
- Allen NS, Edge M. Fundamentals of polymer degradation and stabilisation. Manchester: Elsevier Applied Science; 1992.
- Pitt CG, Gratzl MM, Kimmel GL, et al. Aliphatic polyesters II. The degradation of poly (DL-lactide), poly (epsilon-caprolactone), and their copolymers in vivo. Biomaterials. 1981;2:215–220.
- Zhang M, Thomas NL. Blending polylactic acid with polyhydroxybutyrate: the effect on thermal, mechanical, and biodegradation properties. Adv Polym Technol. 2011;30:67–79.
- Lendlein A. Polymere als Implantatwerkstoffe. Chem unserer Zeit. 1999;33:279–295.