
Abstract
The blood-brain barrier (BBB) is the brain-specific endothelial cell barrier that is important for maintaining brain homeostasis and preventing the entry of toxic substances. Pathological BBB dysfunction is a critical step of the disease process in several immuno-mediated neurological diseases, including multiple sclerosis (MS), neuromyelitis optica (NMO), neuropsychiatric systemic lupus erythematosus (NPSLE) and neuro-Behçet diseases. The pathological findings from patients with secondary progressive (SP) MS, NMO and NPSLE showed leaky BBB in the active lesions. NMO is a disease with strong evidence of disease-specific and pathogenic autoantibodies (aquaporin 4 [AQP4] autoantibodies). In the development of NMO, circulating AQP4 autoantibodies need to pass through the BBB in order to reach AQP4 on the astrocyte endfeet. Strong evidence suggests that NPSLE is associated with the disruption of the BBB and NPSLE patients frequently have antibodies bound to endothelial cells in their sera. We recently identified two BBB-reactive autoantibodies in immuno-mediated neurological diseases: galectin-3 autoantibodies in SPMS and GRP78 autoantibodies in NMO. In the present review article, we describe the basic structure and cellular biology of the BBB, discuss recent insights regarding the pathophysiology of the BBB breakdown in the setting of immuno-mediated neurological diseases, and describe our recent findings of autoantibody-mediated BBB breakdown.
1. Introduction
The blood-brain barrier (BBB) functions as a structural and functional barrier that restricts the movement of soluble mediators and leukocytes from the blood to the central nervous system (CNS) [Citation1]. The pathological breakdown of the BBB is associated with various neuro-inflammatory diseases, such as multiple sclerosis (MS), neruomyelitis optica (NMO), neuropsychiatric systemic lupus erythematosus (NPSLE) and neuro-Behçet diseases [Citation2,Citation3]. In this review, we focus on recent findings regarding antibody-mediated BBB dysfunction in neuro-inflammatory diseases.
1.1. Structure and function of the BBB
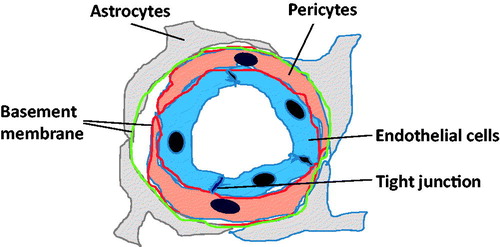
The BBB is a multicellular vascular system that maintains brain homeostasis, regulates the influx and efflux and protects against toxic substances [Citation4]. The main anatomical element of the BBB is the endothelial cells in cerebral blood vessels (). Several factors, such as abundant endothelial tight junction and adhesion junction protein and the inhibition of non-selective fenestration and transcytosis, help from the physical barrier in the BBB [Citation5]. BBB-endothelial cells express a series of specific transporters to deliver nutrients into the brain and excrete toxins [Citation6]. Pericytes and astrocytes are also important components of the BBB. Pericytes cover the abluminal surfaces of cerebral vessels and share the same basement membrane as endothelial cells [Citation7]. Astrocyte endfeet cover most of the surface of brain microvessels [Citation8]. Both astrocytes and pericytes are a source of important regulatory factors, such as fibroblast growth factor (bFGF) and transforming growth factor-β (TGF-β), to strengthen the BBB properties [Citation9].
Figure 1. BBB structure. Endothelial cells have tight junction and form barrier. Pericyte process covers the ablabluminal surface of endothelial cells. Astrocyte endofeet cover the ablabluminal side of the vessel to an extent of nearly 100%. Basement membrane surrounds the pericyte and astrocyte, and cover outside endothelial cells.
1.2. Cell lines for BBB experiments
Immortalized human in vitro BBB-cell lines with in vivo characteristics are a useful tool for understanding the cellular biology of the BBB on a molecular basis. Several human and rodent cell lines consisting of brain microvascular endothelial cells (BMECs) have been established and used to analyze the molecular and cellular characteristics of the BBB [Citation10–13]. Our laboratory established the immortalized human brain microvascular endothelial cell lines (TY08 and TY09) as well as a human brain pericytes cell line [Citation14,Citation15]. These cell lines isolated from human brain tissue harbor a temperature-sensitive SV40 large T-antigen (tsA58) protein. The gene product of tsA58 is in an active conformation and binds to p53 at 33 °C, leading to the immortalization of the cells. However, the conformation of the gene product has changed when the cells are grown at 37 °C and p53 is degraded and released. As a result, these cells are conditionally immortal, retaining BBB-like differentiated characteristics. Both TY08 and TY09 expressed all key tight junctional proteins, such as occludin, claudin-5, ZO-1 and ZO-2, and have a low permeability to inulin across monolayers and a high expression of influx and efflux transporters. We previously showed that the brain pericytes secreted several growth factors, including bFGF, TGF-β, Ang-1, VEGF, GDNF and BDNF, and that the barrier function in TY08 was increased through the upregulation of claudin-5 by soluble factors released from brain pericytes [Citation16,Citation17]. These cell lines are useful tools for understanding the BBB function under both physiological and pathological conditions.
1.3. Clinical biomarkers of BBB alterations under condition of neuroinflammation
Disruption of the BBB gives rise to the intrusion of serum pathogenic T cells, immunoglobulin G and inflammatory cytokines into the CNS parenchyma. Magnetic resonance imaging (MRI) is a reliable method of detecting BBB breakdown in several neuroinflammatory diseases. Gadolinium enhancement of the brain parenchyma on T1-weighted MRI is occasionally observed in neuroinflammatory diseases, including MS, NMO and immune-mediated encephalitis. This change is interpreted as a hallmark of BBB breakdown [Citation18]. Another standard measure of BBB disruption in clinical practice is the cerebrospinal fluid (CSF)/serum albumin ratio [Citation19]. As the BBB limits the movement of albumin from blood to the CSF and albumin is not produced in the CNS, the CSF/serum albumin ratio can be used to assess the integrity of the BBB.
1.4. Molecular basis of the BBB breakdown
Pathological BBB disruption leads to two events: (1) increased paracellular leakage of soluble mediators into the CNS via the disruption of tight junctions and (2) increased transcellular entry of inflammatory T cells across brain endothelial cells via the upregulation of adhesional molecules [Citation9]. Decreases in claudin-5 protein, which is one of the most important components of tight junction proteins, leads to increased paracellular permeability across the BBB [Citation20,Citation21]. VCAM-1 on the endothelial cells plays an important role in the ‘adhesion process’ for the transmigration of T cells across the BBB [Citation22,Citation23]. The VCAM-1 expression was found to be upregulated on CNS endothelial cells in autopsy cases of multiple sclerosis and NPSLE [Citation24]. Several studies have provided evidence that T lymphocytes bind through α4-integrin to the endothelial ligand VCAM-1 on inflamed cerebral vessels and the blockade of interactions between VCAM-1 and α4-integrin prevent the recruitment of circulating T cells into the CNS.
1.5. Treatment for BBB disruption in neuroinflammatory diseases
Some studies have shown that corticosteroids increase the expression of claudin-5 and strengthen the barrier property [Citation25]. Our data demonstrated that fingolimod or the active form of vitamin D, which are used to treat multiple sclerosis (MS), enhance barrier function via the upregulation of claudin-5 and reduction in the VCAM-1 expression [Citation26,Citation27].
Natalizumab, a humanized monoclonal antibody against VLA-4, is widely used to treat the relapsing-remitting MS (RRMS) [Citation28]. This drug strongly interferes with the ‘adhesion process’ by blocking the interaction of VLA-4 with the VCAM-1 expressed on the BMEC surface and subsequently inhibiting the entry of T cells into the CNS. As of 31 May 2018, approximately 184,900 MS patients have received natalizumab worldwide (Biogen Idec, 20 Jun 2018), which has reportedly shown greater efficacy than any other drug for MS. However, the use of natalizumab for the treatment of MS patients has been associated with the development of progressive multifocal leukoencephalopathy (PML), with 781 PML cases reported as of 1 June 2018 (Biogen Idec, 2018).
1.6. Multiple sclerosis
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the CNS, characterized pathologically by the presence of focal demyelinated plaques within the white matter [Citation29]. MS is classified into three clinical phenotypes: relapse-remitting MS (RRMS), primary progressive MS (PPMS) and secondary progressive MS (SPMS) [Citation30]. Most MS patients start with a phase of RRMS, characterized by alternating episodes of neurological disability and recovery [Citation31]. After this course (within 20–25 years), 60–70% of RRMS patients shifted into a phase of SPMS, characterized by a progressive neurological decline [Citation32,Citation33]. Our understanding regarding the mechanisms underlying multiple sclerosis is limited, but it is believed that multiple genetic, environmental and endogenous factors lead to inflammation and ultimately neurodegeneration. Axonal loss and grey matter damage have been considered major causes of irreversible neurological disability in the progressive stages [Citation34]. Disease modifying therapies (DMTs) for RRMS that interfere with a variety of immunological mechanisms have reduced the relapse rate and decline in the neurological function in several randomized controlled trials [Citation35]. However, current DMTs fail to exert any benefit on the SPMS phase as these agents cannot stop neurodegenerative mechanisms.
1.7. Blood-brain barrier disruption in MS
The pathological changes in RRMS are associated with the inflammatory process: newly formed lesions within the CNS can be detected by contrast gadolinium (Gd)-enhancement of the brain parenchyma in MRI. Multifocal inflammation, demyelination, oligodendrocyte loss, reactive gliosis and axonal degeneration are observed in MS lesion. In addition, anti-inflammatory or immunomodulatory therapies have shown a beneficial effect in this phase. However, SPMS is less associated with the inflammatory process than RRMS: new Gd-enhancing lesions are rare, but progressive loss of brain volume is observed on MRI. A hypocellular and gliotic core without active inflammation or axon swelling was observed in the brains of SPMS patients.
Pathological changes in the BBB are a critical features of the disease process in all clinical subtypes of MS [Citation36–Citation38]. Acute MS lesions have demonstrated the BBB disruption, as evidenced by in vivo Gd-enhancement on MRI and post-mortem evidence of focal micro-vascular leakage and the accumulation of inflammatory cells around the vessels [Citation39]. In both PPMS and SPMS, the persistent loss of BBB integrity, as indicated by vascular leakage and the decreased expression of tight junction protein, has been clearly observed in both active and inactive lesions [Citation40].
1.8. Galectin-3 antibodies is associated with BBB disruption in SPMS
Gadolinium-enhanced lesions on brain MRI are rarely observed, but some pathological studies have described minor but persistent BBB damage in SPMS patients. We, therefore, hypothesized that serum autoantibodies in SPMS can disrupt the BBB, and that the phenotypic discrepancy between the remitting phase of RRMS and SPMS may be derived from the presence of persistent BBB breakdown. To examine this hypothesis, we designed a series of studies with the following findings [Citation41,Citation42]: (1) IgG from SPMS sera (SPMS-IgG) induced BBB damage through claudin-5 degradation and VCAM-1 upregulation in BBB-endothelial cells; (2) the target molecule of SPMS-IgG responsible for BBB disruption was galectin-3 according to a proteomic approach; (3) most SPMS patients (10 of 11 SPMS patients) were positive for this antibody, although the patients with other neurological diseases did not have these antibodies. (4) Galectin-3 autoantibodies in the SPMS sera influenced the NF-κB p65 signaling and induced the upregulation of the ICAM-1 expression.
Based on these findings, we reported the anti-galectin-3 antibodies were the contributing factors responsible for persistent BBB damage in progressive MS.
Galectin-3 is a β-galactoside-binding lectin that has been expressed extra- and intracellularly in several cell types [Citation43]. The activation of intracellular galectin-3 stimulates the NFκB p65 pathways [Citation44]. The upregulation of galectin-3 in the glial cells (astrocytes, oligodendrocytes and microglia) has been observed in both the acute and progressive stage in MS brain sections and experimental autoimmune encephalomyelitis (EAE) mice [Citation45,Citation46]. We therefore speculate that the upregulation of galectin-3 in glial cells by inflammatory stimulation may result in the production of anti-galectin-3 antibodies. Interestingly, galectin-3 autoantibodies also may have an additional effect of inhibiting remyelination, as exposure to these antibodies causes the morphological and functional differentiation of in vitro rat oligodendrocyte progenitor cells [Citation47]. Anti-galectin-3 antibodies were reportedly detected in SLE patients [Citation48]. The association between these antibodies and the pathophysiology of NPSLE has now been examined in our laboratory.
1.9. Neuromyelitis optica
Neuromyelitis optica (NMO) is a severe autoimmune CNS disease, and its main clinical manifestations are recurrent optic neuritis and longitudinal transverse myelitis [Citation49]. Limited forms of the disease are known as NMO spectrum disorder (NMOSD) [Citation50]. Most patients with NMO and many with NMOSD have autoantibodies against the water channel aquaporin-4 (AQP4) expressed on astrocyte endfeet (known as NMO-IgG or AQP4-Ab), which are thought to have pathogenic effects on astrocytes with complement- and antibody-dependent cellular cytotoxicity [Citation51,Citation52]. Following the discovery of AQP4 autoantibodies, NMO has been considered a distinct clinical entity from MS. The detection of serum AQP4-IgG has been used for the clinical diagnosis and early treatment of NMO patients worldwide [Citation53]. NMO is therefore considered to be the first CNS autoimmune disease for which a specific tissue target molecule has been identified.
1.10. BBB breakdown in NMO
Gadolinium-enhanced lesions on MRI and/or increased albumin leakage in the CSF were observed clinically in the acute phase of NMO [Citation54]. More AQP4-Abs is produced by peripheral plasmablasts than intrathecal plasmablasts, suggesting that these antibodies are produced in the serum in NMO [Citation55,Citation56]. The peripheral injection of AQP4-IgG alone is not sufficient to induce NMO-like histopathology when injected into normal mice [Citation57]. However, these antibodies can cause the disease in experimental animals with pre-existing T-cell mediated CNS inflammation and BBB disruption [Citation58]. Thus, BBB breakdown and subsequent entry of AQP4-IgG into the brain are critical steps for the development of NMO.
1.11. GRP78 autoantibodies are associated with BBB disruption in NMO
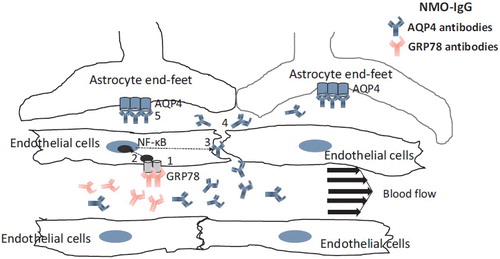
To determine how circulating AQP4-Abs reach the targeted AQP4 on the astrocyte endfeet behind the BBB, we designed a set of experiments. As the BBB-endothelial cells do not express the AQP4 protein and serum AQP4-Abs itself cannot influence the BBB, we hypothesized that another endothelial cell-specific Ab may be presented in sera from NMO patients and promote transit of AQP4-Abs across the BBB. We obtained the following findings: (1) Sera from the acute phase of NMO patients decreased the BBB function via a claudin-5 decrease; (2) the decrease in claudin-5 in the BBB after exposure to NMO sera was induced by the secretion of VEGF and MMP-2/9 from endothelial cells in an autocrine manner; (3) IgG from 50 AQP4 Ab-positive NMO sera samples was able to bind and activate BBB-endothelial cells via the NF-κB p65 pathway in vitro; (4) two AQP4-nonreactive monoclonal recombinant Abs (rAbs) prepared from CSF plasmablasts were present in NMO, and able to strongly activate BBB-endothelial cells; (5) glucose-regulated protein 78 (GRP78) as the target antigen of NMO-rAb was identified using proteome technology; (6) peripheral injection of a GRP78-specific NMO-rAb gave rise to increased BBB leakage in vivo; (7) the absorption of GRP78-specific Abs from NMO-IgG pooled from 50 patients resulted in a decreased effect on BBB-endothelial cell activation [Citation59–62].
Taken together, the finding from our series of studies suggest that GRP78 autoantibodies directly induce increased BBB leakage and are associated with NMO attacks in at least some NMO patients ().
Figure 2. Mechanism of BBB disruption induced by GRP78 autoantibodies in NMO. NMO-IgG involves 2 autoantibodies (AQP4 antibodies and GRP78 antibodies). Binding of GRP78 antibodies to GRP78 on BBB-endothelial cells (1) causes the activation of NF-κB signal (2). Enhancement of BBB permeability (3) via the disruption of tight junction leads to diffusion of AQP4 antibodies into CNS (4) and binding of this Abs to AQP4 on astrocytes (5).
1.12. GRP78 antibodies in cancer and RA patients
GRP78 (alternatively, HSPA5) is a stress protein of the heat shock protein 70 family, expressed in all CNS cells as an endoplasmic reticulum (ER) chaperone [Citation63]. GRP78 as an ER-localized chaperone protein prevents the accumulation of unfolded protein and protects cells from apoptosis. In contrast, cell surface of GRP78 is involved in NF-κB signal transduction [Citation64]. As expression of GRP78 is abundant on cell surface of the BBB-endothelial cells, serum GRP78 autoantibodies can bind to and influence BBB-endothelial cells [Citation62].
GRP78 is highly expressed on the cell surface of cancer cells in vitro and in vivo, suggesting that this molecule may be the target for cancer-specific therapy [Citation65]. GRP78 autoantibodies have been observed in sera from patients with cancer [Citation66]. These antibodies were also detected in sera from RA patients (60%) [Citation67]. GRP78 is reported to be highly abundant in the synovial fluids of patients with rheumatoid arthritis (RA). GRP78 autoantibodies may be produced in reaction to the pathological overexpression of GRP78 in patients with cancer and RA.
1.13. NPSLE
Systemic lupus erythematosus (SLE) is a chronic, systemic, relapsing-remitting autoimmune inflammatory disease [Citation68]. About 40–75% of patients with SLE have experienced neuropsychiatric symptoms, including seizures, cerebrovascular events, cognitive changes, mood and anxiety disorders, acute confusional state and psychosis (named neuropsychiatric SLE [NPSLE]) [Citation68]. The diagnosis of NPSLE is challenging, as it is not easy to distinguish a variety of NPSLE symptoms from other neuropsychiatric diseases with different etiologies [Citation69]. Further complicating matters is the lack of accurate and reliable biomarkers for the diagnosis of NPSLE. The pathogenesis of NPSLE is complex and the precise mechanisms remain unclear, but complicated etiologies are involved, including BBB dysfunction, vascular occlusion, neuro-inflammation, tissue and neuronal damage mediated by autoantibodies and proinflammatory cytokines (IL-1, IL-6, IL-8, IL-17, tumor necrosis factor [TNF]-α) [Citation70,Citation71]. Autoantibodies against anti-DNA antibodies, anti-phospholipid, ribosomal P protein and N-methyl-D-aspartate (NMDA) receptor have been reported as possible biomarkers in SLE [Citation72,Citation73]. A subset of anti-DNA antibodies can cross-react with NMDAR in neurons, which functions as a major excitatory neurotransmitter in the brain and is involved in the memory function [Citation72]. In addition, autoantibodies against the NR1 subunit of NMDAR are also found in patients with NMDAR encephalitis, who presents with acute behavior changes, seizure, movement disorders and memory loss, suggesting that the cross-reaction of anti-DNA antibodies in SLE with NMDAR may cause neuropsychiatric symptoms [Citation73]. However, peripheral increases in anti-DNA antibodies are not associated with neuronal damage without BBB damage. The direct injection of NMDAR autoantibodies into the cerebral cortices of healthy mice causes CNS diseases. BALB/c mice immunized with the DWEYS peptide overproduces anti-DNA/NMDAR antibodies in the sera, but binding of these antibodies was not observed in the brain without the stimulation of LPS, suggesting that BBB breakdown is necessary for these antibodies to induce NPSLE symptoms [Citation74,Citation75]. Increased levels of immunoglobulins, albumin and proinflammatory cytokines have been observed in CSF from both NPSLE patients and MRLlpr/lpr lupus-prone mice [Citation76]. In particular, increased albumin leakage (Q Alb) is considered to reflect BBB damage in NPSLE [Citation77].
1.14. Endothelial cell autoantibodies in SLE
Endothelial cell antibodies are common in SLE: 64.7% of NPSLE patients were found to be positive for endothelial cell antibodies in their sera, compared with only 29.4% of non-NPSLE patients [Citation78]. Endothelial cell autoantibodies were reported to induce the upregulation of major cellular adhesion molecules, such as ICAM-1, VCAM-1 and E-selectin, in the endothelial cells, along with the production of cytokines and chemokines, including interleukin (IL)-1b and IL-8 and monocyte-chemotactic protein-1 (MCP-1) [Citation79]. Endothelial cells autoantibodies also contribute to the direct cytotoxic effect due to complement- or antibody-dependent cytotoxicity and induce coagulation on endothelial cells [Citation80]. Our recent data demonstrated that IgG from SLE patients induce NF-κB p65 nuclear translocation and increased ICAM-1 expression in BBB-endothelial cells [Citation62]. The target molecules of endothelial cell autoantibodies in NPSLE are still unclear, but we speculate that GRP78 may be a target of endothelial cell autoantibodies in SLE.
1.15. Neuro-Behçet disease
Behçet’s disease is a multi-systemic, recurrent, auto-inflammatory disorder affecting the skin, eyes, joint, and even the nervous system [Citation81]. The etiology is considered to involve vascular inflammation in the large/small veins [Citation82]. CNS involvement, usually called neuro-Behçet disease (NBD), can present as an acute or chronic progressive type [Citation83,Citation84]. Acute NBD is characterized by acute meningoencephalitis with focal lesions on MRI and increased neutrophil cell counts in the CSF. In contrast, chronic progressive NBD is characterized by intractable slowly progressive dementia, ataxia and dysarthria with persistent elevation of the CSF IL-6 and progressive brain stem atrophy on MRI [Citation85]. Acute NBD responds well to high doses of corticosteroid, while chronic progressive NBD does not respond to steroid, but dose respond to low doses of methotrexate [Citation86]. The histopathological findings of NBD from biopsied and autopsied brain sections have shown the infiltration of inflammatory cells around small vessels with neuron loss, particular in the brainstem [Citation82]. BBB breakdown may be associated with NBD, but no studies to prove this hypothesis have yet been reported.
2. Conclusions
Our understanding of the role of autoantibodies that activate BBB-endothelial cells in the pathology and pathophysiology of BBB dysfunction in immuno-mediated neurological disease has expanded. Further studies to identify disease-specific and pathogenic autoantibodies may lead to the development of novel molecular-targeted drugs for several immune-mediated neurological diseases.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53.
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201.
- Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19:1584–1596.
- Man S, Ubogu EE, Ransohoff RM. Inflammatory cell migration into the central nervous system: a few new twists on an old tale. Brain Pathol. 2007;17:243–250.
- Kniesel U, Wolburg H. Tight junctions of the blood-brain barrier. Cell Mol Neurobiol. 2000;20:57–76.
- Zhao Z, Nelson AR, Betsholtz C, et al. Establishment and dysfunction of the blood-brain barrier. Cell. 2015;163:1064–1078.
- Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14:1398–1405.
- Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133–150.
- Takeshita Y, Ransohoff RM. Inflammatory cell trafficking across the blood-brain barrier: chemokine regulation and in vitro models. Immunol Rev. 2012;248:228–239.
- Bernas MJ, Cardoso FL, Daley SK, et al. Establishment of primary cultures of human brain microvascular endothelial cells to provide an in vitro cellular model of the blood-brain barrier. Nat Protoc. 2010;5:1265–1272.
- Nakagawa S, Deli MA, Kawaguchi H, et al. A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem Int. 2009;54:253–263.
- Weksler BB, Subileau EA, Perrière N, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19:1872–1874.
- Poller B, Gutmann H, Krähenbühl S, et al. The human brain endothelial cell line hCMEC/D3 as a human blood-brain barrier model for drug transport studies. J Neurochem. 2008;107:1358–1368.
- Sano Y, Shimizu F, Abe M, et al. Establishment of a new conditionally immortalized human brain microvascular endothelial cell line retaining an in vivo blood-brain barrier function. J Cell Physiol. 2010;225:519–528.
- Sano Y, Kashiwamura Y, Abe M, et al. Stable human brain microvascular endothelial cell line retaining its barrier-specific nature independent of the passage number. Clin Exp Neuroimmunol. 2013;4:92–103.
- Shimizu F, Sano Y, Maeda T, et al. Peripheral nerve pericytes originating from the blood-nerve barrier expresses tight junctional molecules and transporters as barrier-forming cells. J Cell Physiol. 2008;217:388–399.
- Shimizu F, Sano Y, Abe M, et al. Peripheralnerve pericytes modify the blood-nerve barrier function and tight junctional molecules through the secretion of various soluble factors. J Cell Physiol. 2011;226:255–266.
- Waubant E. Biomarkers indicative of blood-brain barrier disruption in multiple sclerosis. Dis Markers. 2006;22:235–244.
- Lefvert AK, Link H. IgG production within the central nervous system: a critical review of proposed formulae. Ann Neurol. 1985;17:13–20.
- Wolburg H, Lippoldt A. Tight junctions of the blood-brain barrier: development, composition and regulation. Vasc Pharmacol. 2002;38:323–337.
- Nitta T, Hata M, Gotoh S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–660.
- Engelhardt B. T cell migration into the central nervous system during health and disease: different molecular keys allow access to different central nervous system compartments. Clin Exp Neuroimmunol. 2010;1:79–93.
- Shimizu F, Kanda T. The blood-brain barrier in neuroinflammation. In: Suzumura A, Ikenaka K, editors. Neuron-glia interaction in neuroinflammation. New York, NY: Springer Science + Business Media New York; 2013. p. 157–179.
- Alvarez JI, Saint-Laurent O, Godschalk A, et al. Focal disturbances in the blood-brain barrier are associated with formation of neuroinflammatory lesions. Neurobiol Dis. 2015;74:14–24.
- Förster C, Burek M, Romero IA, et al. Differential effects of hydrocortisone and TNF alpha on tight junction proteins in an in vitro model of the human blood-brain barrier. J Physiol. 2008;586:1937–1949.
- Nishihara H, Shimizu F, Sano Y, et al. Fingolimod prevents blood-brain barrier disruption induced by the sera from patients with multiple sclerosis. PLoS One. 2015;10:e0121488.
- Takahashi S, Maeda T, Sano Y, et al. Active form of vitamin D directly protects the blood–brain barrier in multiple sclerosis. Clin Exp Neuroimmunol. 2017;8:244–254.
- Miller DH, Soon D, Fernando KT, et al. MRI outcomes in a placebo-controlled trial of natalizumab in relapsing MS. Neurology. 2007;68:1390–1401.
- Noseworthy JH, Lucchinetti C, Rodriguez M, et al. Multiple sclerosis. N Engl J Med. 2000;343:938–952.
- Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology. 1996;46:907–911.
- Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14:183–193.
- Ontaneda D, Fox RJ, Chataway J. Clinical trials in progressive multiple sclerosis: lessons learned and future perspectives. Lancet Neurol. 2015;14:208–223.
- Dutta R, Trapp BD. Relapsing and progressive forms of multiple sclerosis: insights from pathology. Curr Opin Neurol. 2014;27:271–278.
- Leray E, Yaouanq J, Le Page E, et al. Evidence for a two-stage disability progression in multiple sclerosis. Brain. 2010;133:1900–1913.
- Oh J, O’Connor PW. Established disease-modifying treatments in relapsing-remitting multiple sclerosis. Curr Opin Neurol. 2015;28:220–229.
- Alvarez JI, Cayrol R, Prat A. Disruption of central nervous system barriers in multiple sclerosis. Biochim Biophys Acta. 2011;1812:252–264.
- Larochelle C, Alvarez JI, Prat A. How do immune cells overcome the blood-brain barrier in multiple sclerosis? FEBS Lett. 2011;585:3770–3780.
- Kirk J, Plumb J, Mirakhur M, et al. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood-brain barrier leakage and active demyelination. J Pathol. 2003;201:319–327.
- Plumb J, McQuaid S, Mirakhur M, et al. Abnormal endothelial tight junctions in active lesions and normal-appearing white matter in multiple sclerosis. Brain Pathol. 2006;12:154–169.
- Leech S, Kirk J, Plumb J, et al. Persistent endothelial abnormalities and blood-brain barrier leak in primary and secondary progressive multiple sclerosis. Neuropathol Appl Neurobiol. 2007;33:86–98.
- Shimizu F, Tasaki A, Sano Y, et al. Sera from remitting and secondary progressive multiple sclerosis patients disrupt the blood-brain barrier. PLoS One. 2014;9:e92872.
- Nishihara H, Shimizu F, Kitagawa T, et al. Identification of galectin-3 as a possible antibody target for secondary progressive multiple sclerosis. Mult Scler. 2017;23:382–394.
- Li LC, Li J, Gao J. Functions of galectin-3 and its role in fibrotic diseases. J Pharmacol Exp Ther. 2014;351:336–343.
- Dumic J, Dabelic S, Flögel M. Galectin-3: an open-ended story. Biochim Biophys Acta. 2006;1760:616–635.
- Radosavljevic G, Volarevic V, Jovanovic I, et al. The roles of galectin-3 in autoimmunity and tumor progression. Immunol Res. 2012;52:100–110.
- Jeon SB, Yoon HJ, Chang CY, et al. Galectin-3 exerts cytokine-like regulatory actions through the JAK-STAT pathway. J Immunol. 2010;185:7037–7046.
- Cristofanilli M, Rosenthal H, Cymring B, et al. Progressive multiple sclerosis cerebrospinal fluid induces inflammatory demyelination, axonal loss, and astrogliosis in mice. Exp Neurol. 2014;261:620–632.
- Shi ZR, Tan GZ, Meng Z, et al. Association of anti-acidic ribosomal protein P0 and anti-galectin 3 antibodies with the development of skin lesions in systemic lupus erythematosus. Arthritis Rheumatol. 2015;67:193–203.
- Wingerchuk DM, Lennon VA, Pittock SJ, et al. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485–1489.
- Yanagawa K, Kawachi I, Toyoshima Y, et al. Pathologic and immunologic profiles of a limited form of neuromyelitis optica with myelitis. Neurology. 2009;73:1628–1637.
- Lennon VA, Kryzer TJ, Pittock SJ, et al. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–477.
- Hinson SR, Romero MF, Popescu BF, et al. Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proc Natl Acad Sci USA. 2012;109:1245–1250.
- Waters P, Reindl M, Saiz A, et al. Multicentre comparison of a diagnostic assay: aquaporin-4 antibodies in neuromyelitis optica. J Neurol Neurosurg Psychiatry. 2016;87:1005–1015.
- Kim SM, Waters P, Vincent A, et al. Cerebrospinal fluid/serum gradient of IgG is associated with disability at acute attacks of neuromyelitis optica. J Neurol. 2011;258:2176–2180.
- Jarius S, Franciotta D, Paul F, et al. Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: frequency, origin, and diagnostic relevance. J Neuroinflammation. 2010;7:52.
- Kowarik MC, Dzieciatkowska M, Wemlinger S, et al. The cerebrospinal fluid immunoglobulin transcriptome and proteome in neuromyelitis optica reveals central nervous system-specific B cell populations. J Neuroinflammation. 2015;12:19.
- Bradl M, Misu T, Takahashi T, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol. 2009;66:630–643.
- Sharma R, Fischer MT, Bauer J, et al. Inflammation induced by innate immunity in the central nervous system leads to primary astrocyte dysfunction followed by demyelination. Acta Neuropathol. 2010;120:223–236.
- Shimizu F, Sano Y, Takahashi T, et al. Sera from neuromyelitis optica patients disrupt the blood-brain barrier. J Neurol Neurosurg Psychiatry. 2012;83:288–297.
- Tasaki A, Shimizu F, Sano Y, et al. Autocrine MMP-2/9 secretion increases the BBB permeability in neuromyelitis optica. J Neurol Neurosurg Psychiatry. 2014;85:419–430.
- Shimizu F, Nishihara H, Sano Y, et al. Markedly increased IP-10 production by blood-brain barrier in neuromyelitis optica. PLoS One. 2015;10:e0122000.
- Shimizu F, Schaller KL, Owens GP, et al. Glucose-regulated protein 78 autoantibody associates with blood-brain barrier disruption in neuromyelitis optica. Sci Transl Med. 2017;9. DOI:10.1126/scitranslmed.aai9111
- Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014;14:263–276.
- Misra UK, Deedwania R, Pizzo SV. Activation and cross-talk between Akt, NF-kappaB, and unfolded protein response signaling in 1-LN prostate cancer cells consequent to ligation of cell surface-associated GRP78. J Biol Chem. 2006;281:13694–13707.
- Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2013;32:805–818.
- Raiter A, Vilkin A, Gingold R, et al. The presence of anti-GRP78 antibodies in the serum of patients with colorectal carcinoma: a potential biomarker for early cancer detection. Int J Biol Markers. 2014;29:e431.
- Bläss S, Union A, Raymackers J, et al. The stress protein BiP is overexpressed and is a major B and T cell target in rheumatoid arthritis. Arthritis Rheum. 2001;44:761–771.
- Jeltsch-David H, Muller S. Neuropsychiatric systemic lupus erythematosus: pathogenesis and biomarkers. Nat Rev Neurol. 2014;10:579–596.
- Sciascia S, Bertolaccini ML, Baldovino S, et al. Central nervous system involvement in systemic lupus erythematosus: overview on classification criteria. Autoimmun Rev. 2013;12:426–429.
- Stock AD, Wen J, Putterman C. Neuropsychiatric lupus, the blood brain barrier, and the TWEAK/Fn14 pathway. Front Immunol. 2013;4:484.
- Bertsias GK, Boumpas DT. Pathogenesis, diagnosis and management of neuropsychiatric SLE manifestations. Nat Rev Rheumatol. 2010;6:358–367.
- Brimberg L, Mader S, Fujieda Y, et al. Antibodies as mediators of brain pathology. Trends Immunol. 2015;36:709–724.
- Mader S, Brimberg L, Diamond B. The role of brain-reactive autoantibodies in brain pathology and cognitive impairment. Front Immunol. 2017;8:1101.
- Kowal C, Degiorgio LA, Lee JY, et al. Human lupus autoantibodies against NMDA receptors mediate cognitive impairment. Proc Natl Acad Sci USA. 2006;103:19854–19859.
- Kowal C, DeGiorgio LA, Nakaoka T, et al. Cognition and immunity; antibody impairs memory. Immunity. 2004;21:179–188.
- McLean BN, Miller D, Thompson EJ. Oligoclonal banding of IgG in CSF, blood-brain barrier function, and MRI findings in patients with sarcoidosis, systemic lupus erythematosus, and Behçet's disease involving the nervous system. J Neurol Neurosurg Psychiatry. 1995;58:548–554.
- Hirohata S, Arinuma Y, Yanagida T, et al. Blood-brain barrier damages and intrathecal synthesis of anti-N-methyl-D-aspartate receptor NR2 antibodies in diffuse psychiatric/neuropsychological syndromes in systemic lupus erythematosus. Arthritis Res Ther. 2014;16:R77.
- Conti F, Alessandri C, Bompane D, et al. Autoantibody profile in systemic lupus erythematosus with psychiatric manifestations: a role for anti-endothelial-cell antibodies. Arthritis Res Ther. 2004;6:R366–R372.
- Carvalho D, Savage CO, Isenberg D, et al. IgG anti-endothelial cell autoantibodies from patients with systemic lupus erythematosus or systemic vasculitis stimulate the release of two endothelial cell-derived mediators, which enhance adhesion molecule expression and leukocyte adhesion in an autocrine manner. Arthritis Rheum. 1999;42:631–640.
- Renaudineau Y, Dugué C, Dueymes M, et al. Antiendothelial cell antibodies in systemic lupus erythematosus. Autoimmun Rev. 2002;1:365–372.
- Kalra S, Silman A, Akman-Demir G, et al. Diagnosis and management of Neuro-Behçet's disease: international consensus recommendations. J Neurol. 2014;261:1662–1676.
- Hirohata S. Histopathology of central nervous system lesions in Behçet's disease. J Neurol Sci. 2008;267:41–47.
- Hirohata S, Kikuchi H, Sawada T, et al. Clinical characteristics of neuro-Behcet's disease in Japan: a multicenter retrospective analysis. Mod Rheumatol. 2012;22:405–413.
- Ishido M, Horita N, Takeuchi M, et al. Distinct clinical features between acute and chronic progressive parenchymal neuro-Behçet disease: meta-analysis. Sci Rep. 2017;7:10196.
- Kikuchi H, Takayama M, Hirohata S. Quantitative analysis of brainstem atrophy on magnetic resonance imaging in chronic progressive neuro-Behçet's disease. J Neurol Sci. 2014;337:80–85.
- Hirohata S, Kikuchi H, Sawada T, et al. Retrospective analysis of long-term outcome of chronic progressive neurological manifestations in Behcet's disease. J Neurol Sci. 2015;349:143–148.