
Abstract
CD8+T cells are important in protective immunity against intracellular pathogens and tumors. In chronic infections or cancer, CD8+T cells are constantly exposed to antigens and inflammatory signals. Such excessive and constitutive signals lead to the deterioration of T cell function, called ‘exhaustion’. Exhausted T cells are characterized by low proliferation in response to antigen stimulation, progressive loss of effector function (cytokine production and killing function), expression of multiple inhibitory receptors such as PD-1, Tim3, and LAG3, and metabolic alterations from oxidative phosphorylation to glycolysis. These dysfunctions are associated with altered transcriptional programs and epigenetic regulations and recent studies suggested that NR4a and TOX transcription factors are deeply involved in exhaustion phenotypes. However, an increase the early memory T cells including stem cell memory T (TSCM) cells is critical for T cell persistence and efficient tumor killing especially for adoptive cancer immunotherapy such as CAR-T cell therapy. An increasing amount of evidence supports the therapeutic potential of targeting exhausted T cells and TSCM cells. We have begun to understand the molecular mechanisms of T cell exhaustion and early memory formation, and the clinical application of converting exhausted T cells to rejuvenated early memory T cells is the goal of our study.
1. Introduction
Currently, the immune checkpoint blockade is the major anti-tumor immunotherapy and its basic mechanism has been intensively studied. T cells from patients who did or did not respond to checkpoint blockade therapy such as anti-PD-1 or anti-CTLA-4 antibodies were analyzed using single cell RNA sequencing (scRNAseq). In addition to neo-antigen presentation, these studies suggested that TCF-1 (also called TCF-7)+ T cells and exhausted T cells play a key role in anti-tumor immunity. Initially, immune checkpoint inhibition was thought to reactivate exhausted T cells, but recent studies in humans and mice showed that T cells that were reactivated by immune checkpoint inhibition with CTLA4 or that PD-1 antibodies were in pre-exhausted states. In chronic inflammatory conditions such as cancer and chronic infection, sustained T cell receptor (TCR) stimulation results in a phenomenon known as the exhaustion of cytotoxic CD8+ T cells (CTL) [Citation1]. These exhausted T cells express large amounts of inhibitory molecules such as PD-1 (CD279), cytotoxic T lymphocyte antigen-4 (CTLA-4, CD152), lymphocyte-activation gene 3 (LAG3), T cell immunoglobulin domain and mucin domain 3 (Tim-3), CD244/2B4, CD160, and T cell immunoreceptor with Ig and ITIM domain (TIGIT) [Citation1]. They do not respond to TCR stimulation or secrete anti-tumor cytokines such as interferon γ (IFN γ) and Tumor Necrosis Factor-α (TNFα), and they have a reduced ability to kill tumors or infected cells. However, TCF-1+ T cells, which represent early memory T cells including stem cell memory T (TSCM) cells, have been shown to play important roles in T cell persistence and efficacy of tumor immunotherapy. In this short review, we will discuss TSCM cells, T cell exhaustion, regulatory T cells (Tregs), and tumor immunity.
T cell dysfunction or exhaustion may differ for specific pathogens or tumor types, and this state of T cell differentiation prevents optimal control of infections and tumors, and a better understanding of the molecular mechanism of CD8+ T cell differentiation and exhaustion should lead to new clinical opportunities. The emerging results support the notion that reversal of T cell exhaustion and reprograming to early memory T cells in humans is ideal to improve the efficacy of antitumor immune therapy.
2. T cell memory
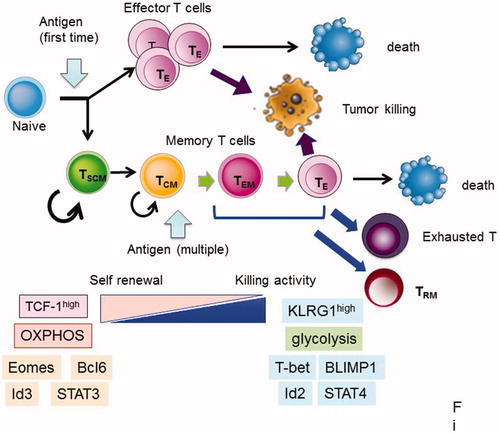
T cell populations have been classified by several surface markers and distinguished by their functions and residency, along with their effector cytokine production. T cell subsets were originally classified into naïve, effector, and several memory cell populations [Citation2]. Naïve T cells, which have not been exposed to antigens, possess strong proliferative potential after antigen stimulation, and resist terminal differentiation and exhaustion when compared with memory T cells. Memory T cells are typically classified into discreet subsets based on phenotypic definitions, and each subset has distinct roles in immunity [Citation3,Citation4]. Effector memory T (TEM) cells and central memory T (TCM) cells circulate in the blood and target the secondary lymphoid tissues. Resident memory T (TRM) cells stay at local sites to respond immediately to secondary infection. These cells can rapidly produce multiple functional molecules after restimulation to control the invasion and spread of pathogens. However, TEM and TRM cells have a limited potential for population expansion, and tend to become terminally differentiated and subsequently die or become exhausted (). Memory stem cells (TSCM) or memory cells with naïve phenotypes (TMNP) have been detected in T cell populations that were previously characterized as being naïve [Citation5,Citation6]. Both TSCM and TMNP cells, which express naïve T cell markers (CD45RA+CD45RO-CCR7+CD62L+), respond rapidly to antigens, express multiple effector molecules, and produce memory and effector cells. TSCM is also related to TCF-1high memory T cells with naïve-like surface markers or memory-precursor-like T cells [Citation7,Citation8]. These subsets have greater proliferative potential than naïve cells, and produce larger amount of effector T cells compared with other memory subsets. The heterogeneous differentiation states of memory/effector CD8+ T cells can be distinguished by the expression of several surface markers, such as killer cell lectin-like receptor G1 (KLRG1), IL-7 receptor subunit-α (IL-7Rα), CD27, CXC-chemokine receptor 3 (CXCR3) and CD62L, and differentiation from TSCM to terminal effector T cells is regulated by the graded expression or activity of certain sets of transcription factors, as shown in [Citation9].
Figure 1. Fate of CD8+ T cells after antigen stimulation. After naïve T cells are activated by the corresponding antigens, they become effector T (TE) cells and die after attack to tumor cells or infected cells. Part of the activated T cells became memory T cells. Memory T cells further differentiated into several subsets after re-stimulation, as follows: stem cell memory (TSCM), central memory T (TCM), effector memory T (TEM) cells, and resident memory T (TRM) cells. TRM stay at local sites to respond immediately to secondary infection. TSCM and TCM cells have a high potential for self-renewal and produce TEM and TE cells after re-stimulation, while killing activity for target cells is high in TEM and TE cells compared with TSCM and TCM cells. TEM, TE, and TRM cells have a limited potential for population expansion, and tend to become terminally differentiated and subsequently die or exhausted (). Genes, markers, and metabolism are listed in this figure, and they are characteristic in early and late memory status.
3. Memory T cell subsets related to efficacy of tumor immunotherapy
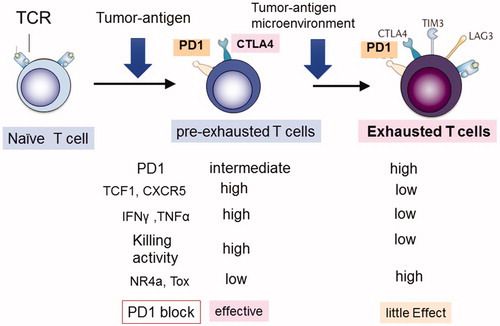
Several scRNAseq studies have been performed to understand the characteristics of tumor infiltrating lymphocytes (TILs) that respond to immune checkpoint blockade therapy [Citation8,Citation10–16]. Although some variations exist depending on tumor type and therapy, most studies agree that the TILs in patients responding to checkpoint-blockade therapy contain more TCF1+ early memory T cells, while fewer exhausted T cells compared with TILs in non-responders (). For example, Kurtulus et al. revealed that checkpoint blockade induced a shift from naïve-like to memory precursor-like subsets, which are maintained by the transcriptional regulator TCF-1 [Citation8]. The effectiveness of CAR-T therapy has also been reported to depend on the degree of early memory T cells and T cell exhaustion [Citation17]. However, Li et al. reported that dysfunctional T cells are the major intratumoral proliferating immune cells and that the intensity of the dysfunctional signature is associated with tumor reactivity [Citation11]. Miller et al. suggested that exhausted CD8+ cells include a subpopulation of ‘progenitor exhausted’ (pre-exhausted) T cells that retain polyfunctionality, persist for a long time and differentiate into ‘terminally exhausted’ TILs [Citation16,Citation18]. Pre-exhausted CD8+ T cells can be defined by intermediate expression of PD-1 and expression of TCF-1 and the chemokine receptor CXCR5 [Citation19,Citation20]. Thus, PD-1 blockade acts mostly on progenitor exhausted CD8+ TILs, which can suppress tumor growth rather than on terminally exhausted T cells. Melanoma patients with a higher percentage of progenitor exhausted T cells showed a longer duration of response to checkpoint-blockade therapy [Citation16]. Approaches to expand the population of pre-exhausted CD8+ T cells might be important for improving the response to checkpoint blockade. Pre-exhausted CD8+ T cells probably contain effector T cells and TCF-1+ early memory T cells.
Figure 2. Two types of TIL T cells. Activated tumor infiltrating lymphocytes (TILs) can be divided into two stages: pre-exhausted (or effector) T cells and exhausted T cells. Pre-exhausted T cells respond to checkpoint blockade therapy, while terminally exhausted T cells do not respond to this therapy. Pre-exhausted T cells express higher levels of TCF-1, CXCR5, cytokines including IFNγ, and TNFα, and possess higher killing activity compared with terminally exhausted T cells.
4. Mechanism of exhaustion
During persistent antigen stimulation, CD8+ T cells show a gradual decrease in effector function, which is referred to as exhaustion, and which impairs responses in tumors and infections. As shown in , terminally exhausted T cells have high levels of inhibitory molecules including PD-1, CTLA4, Tim-3, and Lag-3, and thus, they do not respond well to antigen-mediated TCR stimulation. Exhausted CD8+ T cells do not proliferate well because they lack killing activity, and they secrete low amounts of effector cytokines such as IFNγ and TNFα. T cell exhaustion has been studied using the following three models: chronic viral infections such as lymphocytic choriomeningitis virus (LCMV) [Citation21], tumor models, and overexpression of NFAT1 (nuclear factor of activated T cells-1) without AP1-binding site [Citation22]. The last NFAT lacking AP-1 mimics continuous TCR signaling without co-stimulatory signals, which induces ‘T cell anergy’. NFAT1 is unable to interact with AP-1 transcription factors that increase the expression of inhibitory cell surface receptors including PD-1, which interferes with the ability of CD8+ T cells to protect against infection and attenuate tumor growth; they also exhibited similar gene expression as the exhausted T cells [Citation23]. ATAC-seq analysis revealed that the regions that were differentially accessible between functional subsets were enriched for binding sites of transcription factors that are known to regulate T cell exhaustion, including E2A, BATF, IRF4, T-bet, and TCF1 [Citation24–28]. Exhaustion-specific accessible regions were enriched with NFAT and Nr4a family member consensus binding sites [Citation22,Citation29]. The accessible chromatin analysis (ATA-seq) revealed that the 23-kb upstream enhancer of the Pdcd1 gene locus encoding PD-1 plays an essential role in stable, high PD-1 expression in exhausted T cells [Citation28]. This enhancer contains three Nr4a and two NFAT binding sites [Citation23]. Therefore, NR4a has been implicated as a master regulator of T cell exhaustion.
5. The NR4a family is a nuclear transcription factor that is involved in T cell anergy, exhaustion, and regulatory T cells
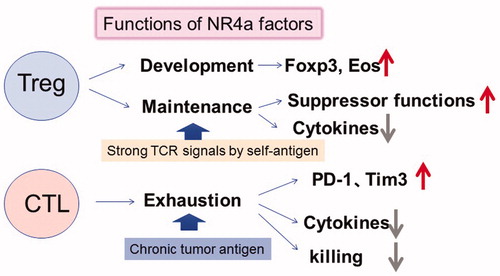
Nuclear receptor transcription factors NR4A1 (also known as NUR77), NR4A2 (NURR1), and NR4A3 (NOR1) are up-regulated by TCR stimulation, and these are targets of NFAT. We have cloned NR4a family proteins as an activator of the Foxp3 promoter [Citation30]. Expression of Nr4a factors is greatly increased in Tregs compared with other T-cell subsets [Citation31–34]. Subsequently, we demonstrated that Nr4a family members are redundantly essential for Treg development in the thymus, as revealed by the nearly complete loss of Tregs in T cell-specific conditional triple knockout (TKO) mice lacking all three Nr4a factors in T cells [Citation35]. Nr4a factors are crucial mediators of TCR signaling for Foxp3 induction and for Il2r and Ikzf4 expression. Nr4a-deficient Tregs easily lost Foxp3 expression and became Th2 and T follicular helper cells, and NR4a factors repressed cytokine expression, including IL-4, IL-5, IL-21, and IFNγ [Citation30,Citation36]. Thus, NR4a is important for the Treg function and for anergic phenotypes of Tregs ().
Figure 3. Functions of NR4a factor in Tregs and CTLs (CD8+ T cells). NR4a factors are upregulated by strong or chronic TCR signals in Tregs and CTLs. NR4a factors are essential for Treg development, suppressive functions, maintenance of Foxp3 and Eos expression, and suppression of cytokine expression. While NR4a factors in CTLs induce exhaustion phenotypes including high expression of PD-1 and Tim3, lower expression of cytokines results in lower tumor killing activity.
NR4a has been shown to be expressed in Tregs and in both activated CD4+ and CD8+ T cells. Nr4a1 has been shown to regulate the expansion, differentiation, and function of CD8+ T cells through direct transcriptional repression of IRF4 or Runx1 [Citation37,Citation38]. NR4a1 is highly expressed in tolerant (i.e., anergic) CD4+ and CD8+ T cells that are stimulated by strong TCR without co-stimulatory signals [Citation39]. Liu et al. showed that NR4A1 overexpression inhibits effector T cell differentiation, whereas NR4A1 deletion overcomes T cell tolerance and exaggerates effector function, as well as enhances immunity against tumors and chronic viruses [Citation39]. They showed that a genome wide-ChIP assay revealed that NR4A1 preferentially binds to AP-1 binding sites, suggesting that NR4a represses effector-gene expression by inhibiting AP-1.
In a recent Nature paper, Chen et al. demonstrated that NR4a factors are deeply involved in T cell exhaustion (). NR4a family factors are highly expressed in exhausted T cells in the tumor compared with the pre-exhausted stage in both humans and mice (). NR4a up-regulates the inhibitory receptors PD-1 and TIM3, while it represses the IFNγ and TNFα expression. Overexpression of each NR4a factor exhibited gene expression profiles similar to those of exhausted T cells. T cells that are lacking all three NR4a transcription factors showed a reduced exhausted fraction, promoted tumor regression, and prolonged survival of tumor-bearing mice. ATAC-seq analysis defined that NR4a directly opens the chromatin of up-regulated genes by exhaustion, such as PD-1 and Tim3, while closes that of repressed genes that contain the binding motifs for NF-κB and AP-1 (). Thus, NR4a transcription factors could be an important regulator of exhaustion and anergy, which are induced by chronic TCR signaling.
Figure 4. Effect of NR4a factors on PD-1 gene chromatin status and target sequences. (A) The upper two panels show ATAC-seq analysis of the PD-1 gene (pdcd1) in WT and Nr4a-triple KO (TKO) T cells in the tumor. The lower 4 panels show activated T cells in which each NR4a gene or control cDNA (empty) is introduced. The 23 kbp upstream enhancer is shown. (B) The left panels show the comparison of ATAT-seq density between WT and NR4a3-TKO CAR T cells. Concentrated transcription factor binding sites in the region that are differentially activated are shown on the right. Data are from reference [Citation77].
![Figure 4. Effect of NR4a factors on PD-1 gene chromatin status and target sequences. (A) The upper two panels show ATAC-seq analysis of the PD-1 gene (pdcd1) in WT and Nr4a-triple KO (TKO) T cells in the tumor. The lower 4 panels show activated T cells in which each NR4a gene or control cDNA (empty) is introduced. The 23 kbp upstream enhancer is shown. (B) The left panels show the comparison of ATAT-seq density between WT and NR4a3-TKO CAR T cells. Concentrated transcription factor binding sites in the region that are differentially activated are shown on the right. Data are from reference [Citation77].](/cms/asset/596853c1-e4a0-4933-99bc-fdc0e0b90a5f/timm_a_1698261_f0004_c.jpg)
Thus, NR4a inhibition could be a promising strategy for cancer immunotherapy because deletion of NR4a increases functional CD8+ T cells while reducing Tregs that suppress anti-tumor immunity. We have shown that mice lacking Nr4a1 and Nr4a2 genes specifically in Tregs showed resistance to tumor growth in transplantation models [Citation40]. The chemotherapeutic agent camptothecin and a common cyclooxygenase-2 inhibitor were found to inhibit transcriptional activity and NR4a factor induction, and they synergistically exerted antitumor effects, unleashed effector activities of CD8+ cytotoxic T cells, and reduced the number of intratumor Tregs. These findings demonstrate that pharmacologic modulation of NR4a activity or expression may be a novel cancer treatment strategy targeting the immunosuppressive tumor microenvironment.
6. TOX and exhaustion
Five recent papers [Citation41–45] showed that the nuclear factor TOX mediates transcriptional and epigenetic changes of exhausted T cells during chronic infection and in cancer [Citation46]. TOX and TOX2 are highly induced in PD-1high TIM3high exhausted TILs (). The proteins TOX and TOX2 induce epigenetic changes and activate the transcription of genes that encode transcription factors (including Tcf7, Nr4a2 and Tox itself) and co-inhibitory receptors (including Pdcd1, Lag3, Cd244 and Havcr2) that are hallmarks of CD8+ T cell exhaustion, but they are not required for functional effector or memory cells. TOX overexpression promotes TCF1+PD-1int progenitor cell survival while increasing the inhibitory receptor expression and suppressing effector functions. Deleting Tox and Tox2 increased IFN-γ and TNFα production and enhanced anti-tumor immunity [Citation44]. Heterozygous deletion of Tox also strengthened the anti-tumor T cell responses [Citation42]. Thus, TOX seems to be necessary and sufficient for the induction of most features of exhausted T cells. However, Tox-deficient CD8+ T cells do not become exhausted but instead adopt the KLRG1+ terminal effector state and die by apoptosis induced by restimulation via the TCR. This vulnerability underscores the need to activate a persistence program and to repress the KLRG1+ differentiation pathway. Thus, TOX may fine-tune CD8+ T cell differentiation by promoting exhaustion and by preventing KLRG1+ effector differentiation to elicit a more prolonged response.
Tox is also suggested to be a downstream of NFAT, and the expression and functions of NR4a and TOX are strongly linked. Similar to Nr4a-TKO T cells, Seo et al. reported that Tox and Tox2 double KO TILs show increased accessibility of chromatin regions that are enriched for motifs that bind to NF-κB and AP-1 (44). NR4a expression is reduced in Tox double KO T cells, while Tox expression is low in Nr4a TKO T cells. Thus, both Tox and Nr4a seem to be critical for the transcriptional program of CD8+ T cell exhaustion downstream of NFAT.
However, the story may not be so simple. Yao et al. reported CD8+ T cell transcriptomes in LCVM acute and chronic infections using scRNA-seq [Citation45]. They found that progenitor-like CD8+ T cells become distinct from memory precursor cells before the peak of the T cell response. Tox-coexpression gene module containing Tox that exhibited higher transcriptional activity associated with more abundant active histone marks in progenitor-like cells than memory precursor cells. Overexpression of Tox in CD8+ T cells induced a high expression of NR4a genes in progenitor-like (TIM3loLy108hi) cells rather than in terminally exhausted (TIM3hiLy108lo) cells [Citation45]. Tox is required for promoted the persistence of antiviral CD8+ T cells and for the programming of progenitor-like CD8+ T cells. Thus, the role of TOX and NR4a factors may also be deeply involved in transition sate of CD8+ T cells from progenitor to terminal exhaustion.
7. Application of TSCM for cancer immunotherapy
To improve the efficacy of adoptive T cell immunotherapy including CAR-T cell therapy, various methods have been tried for adjusting T-cell differentiation and memory formation [Citation47–50]. The strategy of converting from terminally exhausted T cells into pre-exhausted T cells or further younger early memory T cells is important for cancer immunotherapy. Recent studies have challenged the rejuvenation of differentiated T cells by various approaches. Clinical trials evaluating rejuvenation of T cells using iPS technology from antigen-specific T cells (T-iPS cells) are on going [Citation51–54]. However, a significant length of time is needed to establish iPS cells from patient T cells, and the T cells generated from iPS cells must be expanded by TCR stimulation in vitro, which may still induce T cell exhaustion. One method is conversion of effector T cells to early memory T cell subsets with naïve markers and another method is TSCM because TSCM cells possess stronger proliferative potential, long-term survival, and in vivo durability compared with naïve and other memory T cells [Citation5,Citation55]. Compared with other differentiated T cell populations, TSCM cells provide a long-term antitumor response and the TSCM phenotype is, thus, considered to be fit for adoptive T cell immunotherapy [Citation56].
Although remarkable responses have been demonstrated in CAR-T cell immunotherapy in patients with relapsed or chemo-refractory B-lymphoblastic leukemia, a low response or relapse was reported in a subset of patients [Citation57]. Recent studies have revealed two major problems that are associated with a low response to, or relapse during, CAR-T therapy [Citation58–60]. The first is the loss of CAR target molecules on tumor cells [Citation61]. The second is the reduced in vivo persistence of transferred CAR-T cells, mostly because of T cell exhaustion and dysfunction by continuous TCR and cytokine stimulation [Citation60,Citation62]. Sustained remission was associated with an elevated frequency of early memory (CD27+CD45RO-CD8+) T cells before CAR-T cell generation [Citation17]. Therefore, generation of CAR-TSCM cells is expected to improve the quality of CAR-T cells and antitumor activity.
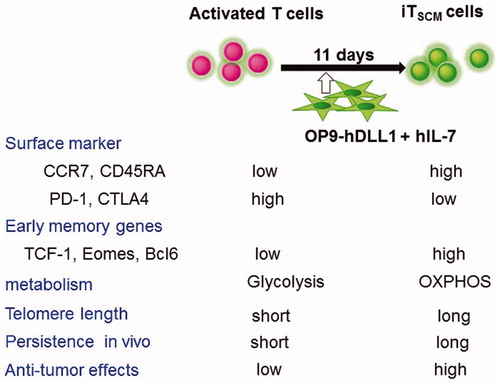
Several in vitro methods for TSCM generation have been reported, but most of them generate TSCM cells from naïve T cells [Citation47,Citation50,Citation55,Citation63–66]. We recently reported that co-culture with NOTCH ligand-expressing feeder cells converts fully activated or more differentiated memory cells into induced TSCM-like (iTSCM) cells [Citation67,Citation68] (). These ‘functional’ iTSCM cells expanded more efficiently in response to antigen re-stimulation compared to any other T cell subsets. iTSCM cells also showed a long-lived and self-renewing potential, were resistant to cell cycle arrest and apoptosis after TCR stimulation, and exhibited a superior antitumor ability compared to naïve and other memory T cell subsets (). We recently reported that the NOTCH/OP9 system efficiently converts conventional human CAR-T cells into TSCM-like CAR-T, which are called ‘CAR-iTSCM’ cells [Citation69].
Figure 5. Summary of phenotypes and anti-tumor activity of activated T cells and iTSCM cells. Antigen-specific activated human CD8+ T cells were co-cultured with OP9-hDLL1 cells in the presence of IL-7 for 11 days. Surface markers, memory genes, metabolism, and anti-tumor effects in NOG mice are shown.
The potent antitumor activity of early memory T cells including TSCM cells appears to be associated with early memory phenotypes and with fatty acid oxidation (FAO)-dependent oxidative phosphorylation (OXPHOS) [Citation70]. Fraietta et al. reported that early memory-related genes with reduced glycolysis signatures were enriched in CAR-T cells from complete-responding patients against chronic lymphocytic leukemia (CLL) [Citation17]. Less-differentiated CAR-T induction was obtained by inhibiting the Akt-mTOR pathway, enhancing FOXO1 activity, and potentiating mitochondrial function [Citation68,Citation71–75]. Additionally, IL-21 in combination with rapamycin has been reported to enhance generation of tumor antigen-specific TSCM-like cells [Citation64], and IL-21 enhances TCM cell formation in a FAO-dependent manner [Citation76]. We found that FOXM1 plays an important role of the induction of the FAO-dependent OXPHOS-related genes in NOTCH-induced iTSCM cells [Citation69]. These reports suggest that the metabolic fitness regulates T cell differentiation, memory formation, and exhaustion, and, therefore, maintains the antitumor ability of T cells.
8. Conclusion
We have just begun to understand the differentiation of memory T cells, and there are several stages of T cell exhaustion that show different responses to checkpoint blockade. If we can discover the method for converting exhausted T cells to rejuvenated early memory T cells, it may be an ideal method to promote successful cancer immunotherapy. However, the goal has not yet been reached, and many difficulties need to be overcome.
Disclosure statement
The authors declare no competing financial interests.
Additional information
Funding
References
- Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–499.
- Maecker HT, McCoy JP, Nussenblatt R. Standardizing immunophenotyping for the Human Immunology Project. Nat Rev Immunol. 2012;12(3):191–200.
- Mueller SN, Gebhardt T, Carbone FR, et al. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol. 2013;31(1):137–161.
- Jameson SC, Masopust D. Understanding subset diversity in T cell memory. Immunity. 2018;48(2):214–226.
- Gattinoni L, Lugli E, Ji Y, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17(10):1290–1297.
- Pulko V, Davies JS, Martinez C, et al. Human memory T cells with a naive phenotype accumulate with aging and respond to persistent viruses. Nat Immunol. 2016;17(8):966–975.
- Miron M, Kumar BV, Meng W, et al. Human lymph nodes maintain TCF-1(hi) memory T cells with high functional potential and clonal diversity throughout life. J Immunol. 2018;201(7):2132–2140.
- Kurtulus S, Madi A, Escobar G, et al. Checkpoint blockade immunotherapy induces dynamic changes in PD-1(-)CD8(+) tumor-infiltrating T cells. Immunity. 2019;50(1):181–194.e6.
- Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12(11):749–761.
- Siddiqui I, Schaeuble K, Chennupati V, et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. 2019;50(1):195–211.e10.
- Li H, van der Leun AM, Yofe I, et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. 2019;176(4):775–789.e18.
- Clarke J, Panwar B, Madrigal A, et al. Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J Exp Med. 2019;216(9):2128–2149.
- Jerby-Arnon L, Shah P, Cuoco MS, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175(4):984–997.e24.
- Wang B, Zhang W, Jankovic V, et al. Combination cancer immunotherapy targeting PD-1 and GITR can rescue CD8(+) T cell dysfunction and maintain memory phenotype. Sci Immunol. 2018;3:eaat7061.
- Yost KE, Satpathy AT, Wells DK, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med. 2019;25(8):1251–1259.
- Miller BC, Sen DR, Al Abosy R, et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20(3):326–336.
- Fraietta JA, Lacey SF, Orlando EJ, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24(5):563–571.
- Paley MA, Kroy DC, Odorizzi PM, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. 2012;338(6111):1220–1225.
- He R, Hou S, Liu C, et al. Follicular CXCR5- expressing CD8(+) T cells curtail chronic viral infection. Nature. 2016;537(7620):412–428.
- Im SJ, Hashimoto M, Gerner MY, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537(7620):417–421.
- Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682–687.
- Martinez GJ, Pereira RM, Aijo T, et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity. 2015;42(2):265–278.
- Mognol GP, Spreafico R, Wong V, et al. Exhaustion-associated regulatory regions in CD8(+) tumor-infiltrating T cells. Proc Natl Acad Sci U S A. 2017;114(13):E2776–e85.
- Kao C, Oestreich KJ, Paley MA, et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat Immunol. 2011;12(7):663–671.
- Doering TA, Crawford A, Angelosanto JM, et al. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37(6):1130–1144.
- Quigley M, Pereyra F, Nilsson B, et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat Med. 2010;16(10):1147–1151.
- Man K, Gabriel SS, Liao Y, et al. Transcription factor IRF4 promotes CD8(+) T cell exhaustion and limits the development of memory-like T cells during chronic infection. Immunity. 2017;47(6):1129–1141.e5.
- Sen DR, Kaminski J, Barnitz RA, et al. The epigenetic landscape of T cell exhaustion. Science. 2016;354(6316):1165–1169.
- Scott-Browne JP, Lopez-Moyado IF, Trifari S, et al. Dynamic changes in chromatin accessibility occur in CD8(+) T cells responding to viral infection. Immunity. 2016;45(6):1327–1340.
- Sekiya T, Kashiwagi I, Inoue N, et al. The nuclear orphan receptor Nr4a2 induces Foxp3 and regulates differentiation of CD4+ T cells. Nat Commun. 2011;2(1):269.
- Hill JA, Feuerer M, Tash K, et al. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. 2007;27(5):786–800.
- Lin W, Haribhai D, Relland LM, et al. Regulatory T cell development in the absence of functional Foxp3. Nat Immunol . 2007;8(4):359–368.
- Moran AE, Holzapfel KL, Xing Y, et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med. 2011;208(6):1279–1289.
- Wei G, Wei L, Zhu J, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30(1):155–167.
- Sekiya T, Kashiwagi I, Yoshida R, et al. Nr4a receptors are essential for thymic regulatory T cell development and immune homeostasis. Nat Immunol. 2013;14(3):230–237.
- Sekiya T, Kondo T, Shichita T, et al. Suppression of Th2 and Tfh immune reactions by Nr4a receptors in mature T reg cells. J Exp Med. 2015;212(10):1623–1640.
- Nowyhed HN, Huynh TR, Thomas GD, et al. Cutting edge: the orphan nuclear receptor Nr4a1 regulates CD8+ T cell expansion and effector function through direct repression of Irf4. J Immunol. 2015;195(8):3515–3519.
- Nowyhed HN, Huynh TR, Blatchley A, et al. The nuclear receptor nr4a1 controls CD8 T cell development through transcriptional suppression of runx3. Sci Rep. 2015;5(1):9059.
- Liu X, Wang Y, Lu H, et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature. 2019;567(7749):525–529.
- Hibino S, Chikuma S, Kondo T, et al. Inhibition of Nr4a receptors enhances antitumor immunity by breaking Treg-mediated immune tolerance. Cancer Res. 2018;78(11):3027–3040.
- Alfei F, Kanev K, Hofmann M, et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature. 2019;571(7764):265–269.
- Khan O, Giles JR, McDonald S, Manne S, et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature. 2019;571(7764):211–218.
- Scott AC, Dundar F, Zumbo P, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019;571(7764):270–274.
- Seo H, Chen J, Gonzalez-Avalos E, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci U S A. 2019;116(25):12410–12415.
- Yao C, Sun HW, Lacey NE, et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8(+) T cell persistence in chronic infection. Nat Immunol. 2019;20(7):890–901.
- Mann TH, Kaech SM. Tick-TOX, it’s time for T cell exhaustion. Nat Immunol. 2019;20(9):1092–1094.
- Kagoya Y, Nakatsugawa M, Ochi T, Cen Y, et al. Transient stimulation expands superior antitumor T cells for adoptive therapy. JCI Insight. 2017;2(2):e89580.
- Kagoya Y, Nakatsugawa M, Yamashita Y, et al. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J Clin Invest. 2016;126(9):3479–3494.
- Kagoya Y, Tanaka S, Guo T, et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat Med. 2018;24(3):352–359.
- Alvarez-Fernandez C, Escriba-Garcia L, Vidal S, et al. A short CD3/CD28 costimulation combined with IL-21 enhance the generation of human memory stem T cells for adoptive immunotherapy. J Transl Med. 2016;14:214.
- Nishimura T, Kaneko S, Kawana-Tachikawa A, et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12(1):114–126.
- Vizcardo R, Masuda K, Yamada D, et al. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8+ T cells. Cell Stem Cell. 2013;12(1):31–36.
- Kawamoto H, Masuda K, Nagano S, et al. Cloning and expansion of antigen-specific T cells using iPS cell technology: development of “off-the-shelf” T cells for the use in allogeneic transfusion settings. Int J Hematol. 2018;107(3):271–277.
- Minagawa A, Yoshikawa T, Yasukawa M, et al. Enhancing T cell receptor stability in rejuvenated iPSC-derived T cells improves their use in cancer immunotherapy. Cell Stem Cell. 2018;23(6):850–858.e4.
- Sabatino M, Hu J, Sommariva M, et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood. 2016;128(4):519–528.
- Gattinoni L, Speiser DE, Lichterfeld M, et al. T memory stem cells in health and disease. Nat Med. 2017;23(1):18–27.
- Park JH, Riviere I, Gonen M, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449–459.
- Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448.
- Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12(20):6106–6115.
- Jensen MC, Popplewell L, Cooper LJ, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16(9):1245–1256.
- Lamers CH, Gratama JW, Pouw NM, et al. Parallel detection of transduced T lymphocytes after immunogene therapy of renal cell cancer by flow cytometry and real-time polymerase chain reaction: implications for loss of transgene expression. Hum Gene Ther. 2005;16(12):1452–1462.
- Levine BL, Miskin J, Wonnacott K, et al. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. 2017;4:92–101.
- Gattinoni L, Zhong X-S, Palmer DC, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15(7):808–813.
- Jeza VT, Li X, Chen J, et al. IL-21 augments rapamycin in expansion of alpha fetoprotein antigen specific stem-cell-like memory T cells in vitro. Pan Afr Med J. 2017;27:163.
- van der Waart AB, van de Weem NM, Maas F, et al. Inhibition of Akt signaling promotes the generation of superior tumor-reactive T cells for adoptive immunotherapy. Blood. 2014;124(23):3490–3500.
- Scholz G, Jandus C, Zhang L, et al. Modulation of mTOR signalling triggers the formation of stem cell-like memory T cells. EBioMedicine. 2016;4:50–61.
- Kondo T, Imura Y, Chikuma S, et al. Generation and application of human induced-stem cell memory T cells for adoptive immunotherapy. Cancer Sci. 2018;109(7):2130–2140.
- Kondo T, Morita R, Okuzono Y, et al. Notch-mediated conversion of activated T cells into stem cell memory-like T cells for adoptive immunotherapy. Nat Commun. 2017;8(1):15338.
- Kondo T, Ando M, Nagai N, et al. The NOTCH-FOXM1 axis plays a key role in mitochondrial biogenesis in the induction of human stem cell memory-like CAR-T cells. Cancer Res. 2019. pii: canres.1196.2019. DOI:10.1158/0008-5472.CAN-19-1196.
- O'Sullivan D. The metabolic spectrum of memory T cells. Immunol Cell Biol. 2019;97:636–646.
- Sukumar M, Liu J, Ji Y, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. 2013;123(10):4479–4488.
- Crompton JG, Sukumar M, Roychoudhuri R, et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res. 2015;75(2):296–305.
- Klebanoff CA, Crompton JG, Leonardi AJ, et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight. 2017;2(23):e95103.
- Mousset CM, Hobo W, Ji Y, et al. Ex vivo AKT-inhibition facilitates generation of polyfunctional stem cell memory-like CD8(+) T cells for adoptive immunotherapy. Oncoimmunology. 2018;7(10):e1488565.
- Pilipow K, Scamardella E, Puccio S, et al. Antioxidant metabolism regulates CD8+ T memory stem cell formation and antitumor immunity. JCI Insight. 2018;3(18):e122299.
- Loschinski R, Bottcher M, Stoll A, et al. IL-21 modulates memory and exhaustion phenotype of T-cells in a fatty acid oxidation-dependent manner. Oncotarget. 2018;9(17):13125–13138.
- Chen J, Lopez-Moyado IF, Seo H, et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature. 2019;567(7749):530–534.