
Abstract
Osteoclasts, derived from the monocyte/macrophage line of bone marrow hematopoietic stem cell progenitors, are the sole bone-resorbing cells of the body. Conventional osteoclast differentiation requires macrophage colony-stimulating factor and receptor activator of nuclear factor kappa-B ligand (RANKL) signaling. Rheumatoid arthritis (RA) is the most prevalent systemic autoimmune disease and inflammatory arthritis characterized by bone destruction. Increased levels of proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6), in the serum and joints, cause excessive bone destruction. We have recently reported that stimulation of human peripheral blood monocytes with TNF-α and IL-6 induces the differentiation of osteoclasts with bone resorption activity. This review presents the functional differences between representative osteoclasts, conventional RANKL-induced osteoclasts, and recently identified proinflammatory cytokine (TNF-α and IL-6)-induced osteoclasts in RA patients. We believe novel pathological osteoclasts associated with RA will be identified, and new therapeutic strategies will be developed to target these osteoclasts and prevent the progression of bone destruction.
1. Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease characterized by inflammatory synovitis accompanied by bone destruction [Citation1,Citation2]. Recent advances in therapeutic drugs, including targeted synthetic disease-modifying antirheumatic drugs (tsDMARDs), biologic DMARDs (bDMARDs), and treat-to-target strategies have increased response and remission rates [Citation2]. However, despite treatment according to the current management recommendations, a significant proportion of RA patients remain symptomatic; this group of patients can be designated as having ‘difficult-to-treat RA (D2TRA)’ [Citation3,Citation4]. In addition, a subgroup of D2TRA patients presents rapid bone destruction in plain radiography, regardless of disease activity. Such cases pose a significant challenge to clinical outcomes, quality of life, productivity, prognosis, and medical economics. This rapid progression of bone destruction is believed to be driven primarily by osteolysis involving activated osteoclast formation and bone resorption in bone-adjacent synovial tissue [Citation5]. Therefore, novel therapeutic strategies that target pathological osteoclasts are required to prevent the progression of bone destruction.
This review aims to discuss the differentiation, regulation, and signaling of representative osteoclasts, conventional receptor activator of nuclear factor kappa-B (NF-κB) ligand (RANKL)-induced osteoclasts, and newly discovered proinflammatory cytokine [tumor necrosis factor alpha (TNF-α) or interleukin-6 (IL-6)]-induced osteoclasts, both in vitro and in vivo. We also examined the functional differences between RANKL-, TNF-α and IL-6-induced osteoclasts in RA patients.
2. RANKL-induced osteoclast differentiation
The only cells capable of destroying and resorbing bone tissue in vivo, osteoclasts are multinucleated giant cells with a diameter of approximately 20–100 μm. Histologically, osteoclasts are the only cells that secrete tartrate-resistant acid phosphatase (TRAP). Clinically, serum TRAP levels are indicators of bone resorption marker in osteoporosis patients. In addition, osteoclasts are bone-resorbing cells and primarily located in cancellous bone and differentiate from hematopoietic stem cell-derived monocytes/macrophage progenitor cells (osteoclast precursors). Osteoblasts are bone-building cells differentiated from mesenchymal stem cells, and secrete and express macrophage colony-stimulating factor (M-CSF), RANKL, and osteoclast differentiation inhibitory factor (osteoprotegerin; OPG).
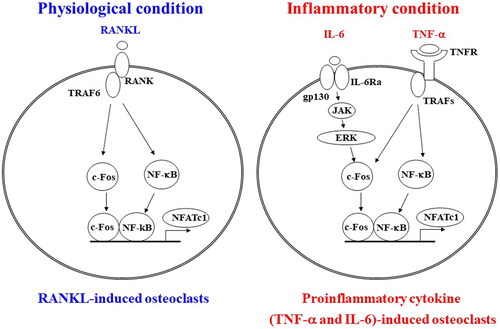
The TNF cytokine superfamily member RANKL is found in both soluble and membrane-bound forms. RANK, the receptor for RANKL, is expressed on the membranes of osteoclast precursor cells (). The RANKL/RANK/OPG signaling pathway regulates osteoclast differentiation and activation [Citation6,Citation7]. An adapter protein, such as TNF receptor-associated factor 6, binds to the intracellular domain of RANK expressed in osteoclast precursors upon RANKL binding. Phosphorylation enzymes such as NF-κB, extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase, and p38 are activated, and c-Fos, nuclear factor of activated T cells, cytoplasmic, and calcineurin-dependent 1 (NFATc1) induce differentiation of osteoclast progenitors into mature multinucleated osteoclasts () [Citation8]. NFATc1 translocates from the cytoplasm into the nucleus and induces osteoclast differentiation following dephosphorylation by calcineurin, a calcium-dependent phosphatase [Citation9]. NFATc1 induces its transcription via a positive feedback loop to promote osteoclast differentiation [Citation10,Citation11].
Figure 1. Comparative mechanisms of osteoclast differentiation in physiological and inflammatory conditions. In physiological conditions, receptor activator of nuclear factor kappa-B ligand (RANKL)-RANK signaling activates the transcription factors c-Fos and nuclear factor of activated T cells, cytoplasmic, calcineurin-dependent 1 (NFATc1), which are essential for osteoclast differentiation in monocyte-macrophage progenitor cells. In inflammatory conditions, tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) signaling synergistically activate c-Fos, and NFATc1 and induce osteoclast differentiation. IL-6 activates c-Fos through its downstream Janus kinase (JAK)-extracellular signal-regulated kinase (ERK) signaling. TNFR: tumor necrosis factor receptor; IL-6Rα: interleukin-6 receptor subunit alpha; TRAFs: TNFR-associated factors; NF-κB: nuclear factor kappa-B.
3. Osteoclast differentiation and bone destruction in RA patients
Bone metabolism results from the remodeling process, in which osteoclasts destroy old bone and osteoblasts form new bone. Consequently, it not only regulates bone remodeling but also control calcium levels in the body. However, excessive osteoclast activity causes numerous diseases, including RA, osteoporosis, periprosthetic osteolysis, bone cancers, and Paget’s disease. Therapeutic agents for osteoporosis include drugs, such as bisphosphonates and anti-RANKL antibodies that target conventional RANKL-induced osteoclasts. Also, RANK knockout arthritic mice showed that the number of osteoclasts was reduced compared with wild-type arthritic models, despite a comparable degree of arthritis [Citation12]. Additionally, anti-RANKL antibodies have been approved for RA in Japan. In detail, anti-RANKL antibodies inhibited the progression of bone erosion, but not joint space narrowing [Citation13]; this comes from the mechanism by which anti-RANKL antibodies inhibits osteoclast differentiation.
On the other hand, it has been reported that osteoclast progenitor cells are induced to differentiate into osteoclasts by the regulation of proinflammatory cytokines other than RANKL [Citation14,Citation15]. In particular, proinflammatory cytokines, including TNF-α and IL-6, directly promote the differentiation of osteoclast precursors into multinucleated osteoclasts [Citation16,Citation17]. In our daily clinical practice, the administration of anti-TNF-α antibodies or anti-IL-6 receptor antibodies in patients with RA not only prevents the progress of bone destruction, but also promotes bone repair. Interestingly, sub-analyses of clinical studies revealed that using anti-TNF-α or anti-IL-6 receptor antibodies significantly inhibited the progression of bone destruction in the patients with poorly controlled disease activity [Citation18,Citation19]. Based on these clinical findings, we hypothesized that strict suppression of circulating TNF-α and IL-6 levels would prevent proinflammatory cytokine-induced osteoclastogenesis, inhibiting osteolytic bone destruction and promoting bone repair.
4. Mouse proinflammatory cytokine (TNF-α and IL-6)-induced-osteoclast-like cell differentiation
Based on the above hypothesis, we stimulated osteoclast precursors with RANKL, TNF-α, and IL-6 to induce osteoclast differentiation. We found that TNF-α and IL-6 induced the differentiation of a significant number of TRAP-positive multinucleated cells in mouse bone marrow macrophages [Citation20]. We also clarified that TRAP-positive multinucleated cells could absorb bone tissue both in vitro and in vivo. Subsequently, OPG, a RANKL decoy receptor, was added to the culture system to determine if RANKL induced by TNF-α and IL-6 on stromal cells contributed to the differentiation of TRAP-positive multinucleated cells. OPG inhibited RANKL-induced TRAP-positive multinucleated cell differentiation, but not TNF-α and IL-6-induced osteoclastogenesis.
We next investigated the molecular mechanisms driving cell differentiation and observed that IL-6 increased the c-Fos protein level while having an insignificant effect on NF-κB. In addition, TNF-α and IL-6 had a synergistic effect on the activity and expression of c-Fos. c-Fos knockdown inhibited the expression of NFATc1 and the differentiation of osteoclast-like cells. Pharmacological inhibitors of Janus kinase (JAK) and extracellular signal-regulated kinase (ERK) blocked the differentiation of osteoclast-like cells but not conventional RANKL-induced osteoclasts in vitro (). Therefore, mouse proinflammatory cytokine (TNF-α and IL-6)-induced osteoclast-like cells have characteristics distinct from those of conventional RANKL-induced osteoclasts () [Citation20].
Table 1. Comparison of TNF-α and IL-6-induced and RANKL-induced osteoclasts.
Notably, O'Brien et al. validated our findings by reporting that TNF-α and IL-6-induced mouse osteoclast-like cell differentiation stimulated RANK-deficient mice-derived bone marrow monocytes. Synovial TRAP-positive multinucleated cells were also found to be responsible for bone erosion in the arthritic joints of RANK-deficient mice with K/BxN serum-transfer arthritis [Citation15].
Furthermore, Hasegawa et al. described murine arthritis-associated osteoclastogenic macrophages (AtoMs) as the osteoclast precursor–containing population in the inflamed synovium, distinct from RANKL-induced osteoclast precursors [Citation21]. These AtoMs differentiated into osteoclasts in a RANKL-dependent manner, and TNF-α induced this differentiation. In addition, the cells were inhibited by OPG. However, rather than relying on RANKL, mouse osteoclast-like cells required TNF-α and IL-6 to differentiate into osteoclasts. Thus, AtoMs were considered to be derived from a distinct developmental lineage from osteoclast-like cells.
5. Human proinflammatory cytokine (TNF-α and IL-6)-induced-osteoclast differentiation
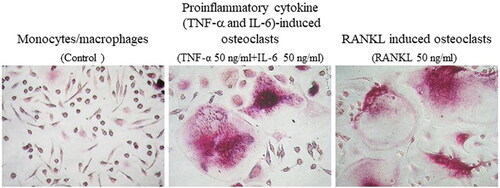
Recently, we found that stimulation of human peripheral blood monocytes with TNF-α and IL-6 induced differentiation of osteoclasts () [Citation22]. By stimulating osteoclast precursors in dentin slices with TNF-α and IL-6, bone resorption pits were identified, indicating that TNF-α and IL-6-induced osteoclasts have bone resorption activity. In addition, the simultaneous addition of OPG suppressed RANKL-induced osteoclast differentiation, but not TNF-α and IL-6-induced differentiation [Citation22]. Notably, TNF-α and IL-6 synergistically stimulated NFATc1 expression in human peripheral blood monocytes. Moreover, NFAT inhibitors inhibited TNF-α and IL-6-induced osteoclast differentiation. Furthermore, TNF-α and IL-6-induced osteoclasts had significantly higher IL-1β mRNA and protein levels than RANKL-induced osteoclasts (). Anti–IL-1β antibody suppressed TNF-α and IL-6–induced but not RANKL-induced osteoclast differentiation. This suggests that IL-1β is a proinflammatory cytokine that promotes TNF-α and IL-6-induced osteoclast differentiation. Expression levels of IL-1β, TNF-α, IL-12p40, and matrix metalloproteinase-3 (MMP-3) were significantly increased in TNF-α and IL-6–induced osteoclasts but not in RANKL-induced osteoclasts () [Citation22]. The functional differences between the two types osteoclast are the following two points: (1) TNF-α and IL-6-induced osteoclasts produce proinflammatory cytokines and chemokines [Citation23] and less expression cathepsin K and bone resorption activity [Citation22]. (2) TNF-α and IL-6-induced osteoclasts cells are immature and partially differentiated from monocytes [Citation24].
Figure 2. Proinflammatory cytokine (TNF-α and IL-6)-induced osteoclasts differentiated from peripheral blood monocytes (PBMs) in patients with rheumatoid arthritis (RA). Photomicrographs of proinflammatory cytokine (TNF-α and IL-6)-induced osteoclasts and RANKL-induced osteoclasts differentiated from PBMs in patients with RA. Original magnification ×200. Refer for other definitions.
6. Effects of JAK inhibitors on the osteoclast differentiation
JAK inhibitors are a unique class of oral tsDMARDs licensed and covered for RA by health insurance in Japan in March 2013. Interestingly, RA patients who continued the combination of methotrexate (MTX) and JAK inhibitors had a significantly slower progression of joint destruction than those who continued the combination of MTX and bDMARDs [Citation25,Citation26]. However, since JAK inhibitors have no direct effect on RANKL-induced osteoclasts, their effects on osteoclasts remain unclear. Therefore, we investigated whether a JAK inhibitor (tofacitinib) could inhibit the differentiation of RANKL- or TNF-α and IL-6-induced osteoclasts using peripheral blood collected from patients with RA. The JAK inhibitor inhibited the differentiation of TNF-α and IL-6-induced osteoclast derived from peripheral blood monocytes of RA patients in a dose-dependent manner [Citation22]. The same concentrations of JAK inhibitor did not inhibit osteoclastogenesis induce by RANKL. Our results suggest that the beneficial effect of JAK inhibitors in RA patients may involve inhibition of TNF-α and IL-6-induced osteoclast differentiation ().
7. RANKL-induced osteoclasts and proinflammatory cytokine (TNF-α and IL-6)-induced osteoclasts in patients with RA
We compared the osteoclastogenic potential of peripheral blood mononuclear cells (PBMCs) from RA patients and healthy donors to better understand the functional differences between RANKL-induced and proinflammatory cytokine (TNF-α and IL-6)-induced osteoclasts in RA [Citation22]. The number of TNF-α and IL-6-induced osteoclasts and RANKL-induced osteoclasts derived from PBMCs in patients with RA was significantly increased compared to that derived from in healthy donors. Interestingly, the number of PBMC-derived TNF-α and IL-6-induced osteoclasts was positively correlated with the structural damage progression (modified total Sharp score). In contrast, there was a negative relationship between the number of PBMCs-derived RANKL-induced osteoclasts and whole body bone mineral density. TNF-α and IL-6-induced osteoclasts expressed MMP-3, and RANKL-induced osteoclasts expressed cathepsin K, indicating that TNF-α and IL-6-induced osteoclasts are involved in joint destruction in RA, and RANKL-induced osteoclasts are involved in osteoporosis (). These results suggest that newly discovered proinflammatory cytokine (TNF−α and IL-6)-induced osteoclasts may be involved in the pathogenic mechanisms of inflammatory bone destruction in RA patients.
8. Conclusion
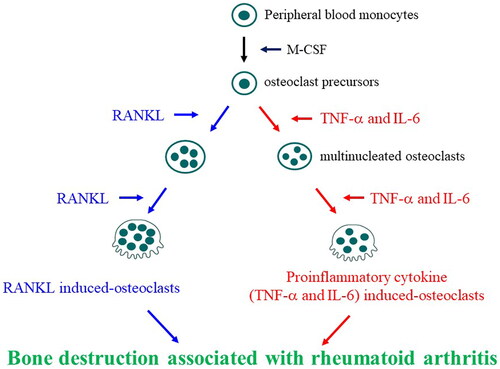
Recent advance of the treatments for RA may be able to reduce the disease activity though it is still difficult to cure the disease and to halt bone destruction. Thus, preventing progression of bone destruction in RA with a poor prognosis has long been a dream for RA patients and rheumatologists, and the new therapeutic strategies and targets urgently need to be developed. Bone-resorbing cells are presumed to be highly functionally heterogeneous and originate from various osteoclastogenesis factors. We clarified a new bone destruction mechanism using a clinical immunological approach. Bone destruction associated with RA involves both conventional RANKL-induced and proinflammatory cytokine (TNF-α and IL-6)-induced osteoclasts (). The development of new therapeutic agents, such as JAK inhibitors, is expected to result from targeting TNF-α and IL-6-induced and RANKL-induced osteoclasts. Furthermore, new therapeutic strategies targeting novel pathological osteoclasts will be developed in the near future, to prevent bone destruction.
Figure 3. Involvement of proinflammatory cytokine (TNF-α and IL-6)-induced osteoclasts and RANKL-induced osteoclasts in bone destruction associated with rheumatoid arthritis. Peripheral blood monocytes differentiate into conventional osteoclasts by macrophage colony-stimulating factor (M-CSF) and RANKL. However, in chronic inflammatory conditions, such as rheumatoid arthritis, monocytes differentiate into inflammatory bone-resorbing cells, such as TNF-α and IL-6-induced osteoclasts, in response to proinflammatory cytokines and may be involved in bone destruction. Refer for other definitions.
Acknowledgments
I would like to express my sincere gratitude to Professor Toshihide Mimura and his entire staff at the Department of Rheumatology and Applied Immunology, Faculty of Medicine, Saitama Medical University, and Professor Kojiro Sato of the Division of Rheumatology and Clinical Immunology, Department of Medicine, Jichi Medical University, for their guidance and encouragement in writing this paper.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Yokota K, Miyazaki T, Hemmatazad H, et al. The pattern-recognition receptor nucleotide-binding oligomerization domain-containing protein 1 promotes production of inflammatory mediators in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2012;64(5):1329–1337. doi: 10.1002/art.34318.
- Schett G, McInnes IB, Neurath MF. Reframing immune-mediated inflammatory diseases through signature cytokine hubs. N Engl J Med. 2021;385(7):628–639. doi: 10.1056/NEJMra1909094.
- Buch MH, Eyre S, McGonagle D. Persistent inflammatory and non-inflammatory mechanisms in refractory rheumatoid arthritis. Nat Rev Rheumatol. 2021;17(1):17–33. doi: 10.1038/s41584-020-00541-7.
- Nagy G, Roodenrijs NMT, Welsing PMJ, et al. EULAR points to consider for the management of difficult-to-treat rheumatoid arthritis. Ann Rheum Dis. 2022;81(1):20–33. doi: 10.1136/annrheumdis-2021-220973.
- Komatsu N, Takayanagi H. Mechanisms of joint destruction in rheumatoid arthritis - immune cell-fibroblast-bone interactions. Nat Rev Rheumatol. 2022;18(7):415–429. doi: 10.1038/s41584-022-00793-5.
- Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4(8):638–649. doi: 10.1038/nrg1122.
- Nakashima T, Hayashi M, Fukunaga T, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17(10):1231–1234. doi: 10.1038/nm.2452.
- Takayanagi H, Kim S, Koga T, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3(6):889–901. doi: 10.1016/s1534-5807(02)00369-6.
- Sato K, Suematsu A, Nakashima T, et al. Regulation of osteoclast differentiation and function by the CaMK-CREB pathway. Nat Med. 2006;12(12):1410–1416. doi: 10.1038/nm1515.
- Asagiri M, Sato K, Usami T, et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med. 2005;202(9):1261–1269. doi: 10.1084/jem.20051150.
- Negishi-Koga T, Takayanagi H. Ca2+-NFATc1 signaling is an essential axis of osteoclast differentiation. Immunol Rev. 2009;231(1):241–256. doi: 10.1111/j.1600-065X.2009.00821.x.
- Pettit AR, Ji H, von Stechow D, et al. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol. 2001;159(5):1689–1699. doi: 10.1016/S0002-9440(10)63016-7.
- Takeuchi T, Tanaka Y, Ishiguro N, et al. Effect of denosumab on Japanese patients with rheumatoid arthritis: a dose-response study of AMG 162 (Denosumab) in patients with RheumatoId arthritis on methotrexate to Validate inhibitory effect on bone Erosion (DRIVE)-a 12-month, multicentre, randomised, double-blind, placebo-controlled, phase II clinical trial. Ann Rheum Dis. 2016;75(6):983–990. doi: 10.1136/annrheumdis-2015-208052.
- Jung YK, Kang YM, Han S. Osteoclasts in the inflammatory arthritis: implications for pathologic osteolysis. Immune Netw. 2019;19(1):e2. doi: 10.4110/in.2019.19.e2.
- O'Brien W, Fissel BM, Maeda Y, et al. RANK-independent osteoclast formation and bone erosion in inflammatory arthritis. Arthritis Rheumatol. 2016;68(12):2889–2900. doi: 10.1002/art.39837.
- Lam J, Takeshita S, Barker JE, et al. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106(12):1481–1488. doi: 10.1172/JCI11176.
- Axmann R, Böhm C, Krönke G, et al. Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis Rheum. 2009;60(9):2747–2756. doi: 10.1002/art.24781.
- Smolen JS, Han C, Bala M, et al. Evidence of radiographic benefit of treatment with infliximab plus methotrexate in rheumatoid arthritis patients who had no clinical improvement: a detailed Sub-analysis of data from the anti-tumor necrosis factor trial in rheumatoid arthritis with concomitant therapy study. Arthritis Rheum. 2005;52(4):1020–1030. doi: 10.1002/art.20982.
- Smolen JS, Avila JCM, Aletaha D. Tocilizumab inhibits progression of joint damage in rheumatoid arthritis irrespective of its anti-inflammatory effects: disassociation of the link between inflammation and destruction. Ann Rheum Dis. 2012;71(5):687–693. doi: 10.1136/annrheumdis-2011-200395.
- Yokota K, Sato K, Miyazaki T, et al. Combination of tumor necrosis factor α and interleukin-6 induces mouse osteoclast-like cells with bone resorption activity both in vitro and in vivo. Arthritis Rheumatol. 2014;66(1):121–129. doi: 10.1002/art.38218.
- Hasegawa T, Kikuta J, Sudo T, et al. Identification of a novel arthritis-associated osteoclast precursor macrophage regulated by FoxM1. Nat Immunol. 2019;20(12):1631–1643. doi: 10.1038/s41590-019-0526-7.
- Yokota K, Sato K, Miyazaki T, et al. Characterization and function of tumor necrosis factor alpha and interleukin-6-Induced osteoclasts in rheumatoid arthritis. Arthritis Rheumatol. 2021;73(7):1145–1154. doi: 10.1002/art.41666.
- Andreev D, Kachler K, Schett G, et al. Rheumatoid arthritis and osteoimmunology: the adverse impact of a deregulated immune system on bone metabolism. Bone. 2022;162:116468. doi: 10.1016/j.bone.2022.116468.
- Iwamoto N, Kawakami A. The monocyte-to-osteoclast transition in rheumatoid arthritis: recent findings. Front Immunol. 2022;13:998554. doi: 10.3389/fimmu.2022.998554.
- Fleischmann RM, Genovese MC, Enejosa JV, et al. Safety and effectiveness of upadacitinib or adalimumab plus methotrexate in patients with rheumatoid arthritis over 48 weeks with switch to alternate therapy in patients with insufficient response. Ann Rheum Dis. 2019;78(11):1454–1462. doi: 10.1136/annrheumdis-2019-215764.
- Combe B, Kivitz A, Tanaka Y, et al. Filgotinib versus placebo or adalimumab in patients with rheumatoid arthritis and inadequate response to methotrexate: a phase III randomised clinical trial. Ann Rheum Dis. 2021;80(7):848–858. doi: 10.1136/annrheumdis-2020-219214.