
Abstract
Systemic Lupus Erythematosus (SLE) is an autoimmune disease characterized by inflammation in multiple organs. A few treatments for SLE currently exist, including antimalarials, glucocorticoids, immunosuppressants, and two recently approved antibody agents; however, an unmet medical need remains for SLE. In addition, developing new drugs targeting SLE is a challenge since no specific biomarkers exist for the prediction of disease progression or drug response. A new drug candidate, E6742, is a specific antagonist of the toll-like receptors 7/8. To address the challenges for drug development in SLE, the process of developing E6742 utilizes a unique system of the Japan Agency for Medical Research and Development (AMED), the Cyclic Innovation for Clinical Empowerment (CiCLE) program. In the CiCLE program, a Phase 1 study in healthy adults was completed (NCT04683185) and a Phase 1/2 study in patients with SLE is on-going (NCT05278663). One of the potential benefits of this program is to conduct academia-led clinical research to identify specific biomarkers for E6742 in parallel with clinical studies (UMIN000042037). The aim of this review is to present current progress within the strategic collaboration of the AMED CiCLE program that optimize clinical development for patients with SLE.
1. Introduction
Systemic Lupus Erythematosus (SLE) is an autoimmune disease characterized by inflammation in multiple organs such as skin, joints, connective tissues, kidneys, lungs, and the central nervous systems. Like most other autoimmune diseases, SLE is more common in young female adults. The prevalence of SLE varies considerably by studies, the global prevalence ranges from 3.2 to 517.5 per 100,000 individuals, from 42.0 to 300 per 100,000 in the U.S, and 3.7 to 37.7 per 100,000 in Japan [Citation1–3]. The 2019 European League Against Rheumatism (EULAR)/American College of Rheumatology classification criteria for SLE is generally used for diagnosis, and a treatment plan is determined by comprehensive evaluation to include disease activity, organ damage, classification of disease type, and complications such as infections [Citation4]. The goal of treatment is remission without systemic symptoms or organ damage. For the treatment of SLE, hydroxychloroquine, the antimalarial drug, is recommended as a standard drug for all patients with fewer contraindications. If there is severe organ involvement or disease activity assessed by comprehensive index, high-dose glucocorticoids and immunosuppressants are promptly initiated. On the other hand, these drugs are the main cause of some serious side effects such as infections due to suppression of immune function. Therefore, the development of more targeted therapies with high efficacy and less side effects has been expected [Citation5–7]. In SLE, B cells can differentiate into autoantibody-producing cells and form immune complexes. These immune complexes activate the complement system, leading to inflammation and tissue damage in various organs including the kidneys, joints, skin, and central nervous system. Therefore, the development of drugs targeting B cells has been driven by the central role of B cells and autoantibodies in the pathogenesis of SLE. B-cell targeting drugs like rituximab had indeed shown mixed results in late-phase clinical studies for SLE or lupus nephritis (LN). While some studies suggested potential benefits in the subgroups, others did not demonstrate statistically significant efficacy [Citation8,Citation9]. However, EULAR/European Renal Association-European Dialysis and Transplant Association (ERA-EDTA) recommends rituximab for induction of remission in treatment-resistant lupus nephritis based on subsequent evidence [Citation10]. Very recently, the additional indication of rituximab for ‘lupus nephritis that has not responded sufficiently to existing therapies’ based on a public knowledge-based application was officially approved in Japan. In addition, obinutuzumab, a type-II anti CD20 antibody, has also been in phase 3 studies targeting SLE and LN with positive result in phase 2 study [Citation11].
Genome-wide association studies (GWAS) have identified numerous genetic variants associated with the risk of SLE, including variants in genes involved in both the innate and adaptive immune responses. While the adaptive immune response, specifically the role of B cells and autoantibodies, has been traditionally thought to be central to SLE pathogenesis, recent evidence has highlighted the importance of the innate immune response in disease development and progression [Citation5]. Various aspects of the innate immune response, such as toll-like receptors (TLRs), interferon regulatory factors (IRFs), and complement pathways are genetic risk factors for SLE. These genetic variants may contribute to dysregulated innate immune activation and increased production of type I interferons (IFNs) and proinflammatory cytokines, leading to tissue damage and inflammation [Citation5,Citation12,Citation13]. Belimumab, which targets soluble B lymphocyte stimulator (BLyS) and is also known as B cell-activating factor (BAFF), as well as anifrolumab, which targets the type I interferon receptor 1 (IFNAR1), highlight the growing recognition of the importance of the innate immune system in the pathogenesis of SLE. Both belimumab and anifrolumab target molecules produced by the innate immune system that play a key role in the activation of adaptive immune response in SLE. Therefore, the role of innate immune system, centered on dendritic cells, has been the focus of new therapeutic targets for SLE. These next generation targeted therapies, which are currently under development, are expected to be the future promise of more efficient and effective treatment of SLE with reduced dependence on glucocorticoids [Citation5,Citation14].
2. The relationship between SLE and TLR7/8
Although the pathophysiology of SLE is not fully understood, there is much evidence indicating that autoimmune response via type I IFNs and activation of IFN gene signatures are important in the pathogenesis. Plasmacytoid dendritic cells (pDCs) are specialized subset of dendritic cells that can produce large amounts of type I IFNs in response to viral or bacterial pathogens. In SLE, pDCs are thought to play a central role in the pathogenesis of the disease by promoting the activation of autoreactive T and B cells through the production of type I IFNs, suggesting the importance of TLR signaling in SLE. TLR7/8 are endosomal receptors for single-stranded RNA; the resulting complexes recruit MyD88 and activate signal cascade via IFN regulatory factor 7 and nuclear factor-kappa B (NF-κB) and trigger the induction of IFN-α. These are the fundamental defense reactions against viral infection [Citation15–17], but dysregulation of these IFNs can drive autoimmunity. TLR7, which recognizes single-stranded RNA, is highly expressed on pDCs and thought to be major triggers of type I IFN production in SLE [Citation18–21]. With regards to the genetic evidence supporting the association between TLR7 and SLE, single nucleotide polymorphisms (SNPs) in the TLR7 gene have been identified as risk factors associated with SLE. Furthermore, a recent study demonstrated that a rare missense variant in the TLR7 gene, that was identified in patients with childhood-onset SLE, leads to a gain-of-function of the TLR7 signaling, resulting in increased activation of the immune system. These examples provide stronger evidence for the genetic contribution to the disease, leading to increased attention to the importance of TLR7 in the pathogenesis of SLE [Citation22,Citation23]. TLR8 is more abundant in neutrophils, monocytes, and myeloid dendritic cells, inducing strong production of inflammatory cytokines by the activation of NF-κB signaling [Citation24]. Growing evidence demonstrates that dual inhibition of TLR7/8 may have stronger effects than inhibiting either receptor alone, for the treatment of SLE, through modulation of innate immune responses and suppressing the production of many inflammatory cytokines including IFN-α. Therefore, the development of a selective TLR7/8 inhibitor may be valuable to achieving disease control and preventing organ damage.
3. A selective dual TLR7/8 antagonist, E6742
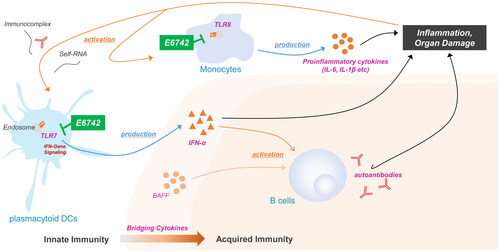
E6742 is a dual antagonist for TLR7/8, and blocks activation by either synthetic RNA or small molecule ligands. The targeted mechanism of action is illustrated in . E6742 was discovered and optimized using cell-based assays in Eisai Co., Ltd. In-vitro studies demonstrated that E6742 inhibited TLR7/8-mediated NF-κB activation in HEK-293 human embryonic kidney cells expressing human or murine TLR7 (hTLR7 and mTLR7, respectively), or human TLR8 (hTLR8). In our preclinical data, E6742 inhibited IL-6 and IFN-α production in response to TLR7/8 agonists with nanomolar potency, but did not block stimulation by TLR9 agonists. In contrast, hydroxychloroquine, a standard of care of SLE and its part of mechanism are known as the inhibition of TLR 7/8/9, was able to inhibit IL-6 and IFN-α production in response to TLR9 agonists with micromolar potency, but it had less potent activity than E6742 in inhibiting TLR7/8 agonist-induced IL-6 and IFN-α production. Thus, E6742 has a distinctly different drug profile from hydroxychloroquine in terms of selectivity for TLR7/8 and potency of activity, and could have clinical benefit for SLE, either as a replacement or an add-on therapy for patients with inadequate response to hydroxychloroquine. The crystal structure of the extracellular domain of human TLR7/8 in complex with E6742 showed that E6742 binds specifically and noncovalently to the interface of each TLR7 and TLR8 dimer. In vivo studies showed therapeutic effects in two SLE model mice. In NZB/W model mice, which spontaneously developed SLE-like autoimmune disease, E6742 reduced mortality and proteinuria, and suppressed weight loss and exacerbation of renal pathology score after 5 months of administration, compared to the vehicle control group. In pristane-induced SLE model mice, which induced human SLE-like pathology (especially in autoimmune arthritis by administering pristane to DBA/1J mice), E6742 decreased arthritis scores (hind paw swelling and erythema) and suppressed the increase of anti-ribosomal P protein in plasma after 3 months of administration, compared with vehicle-treated control mice. Furthermore, it was confirmed that the increase in plasma anti-small nuclear ribonucleoprotein and anti-double stranded DNA antibodies was suppressed after 3 months of administration.
Figure 1. Mechanism of E6742 inhibition in SLE pathology. TLR7 and 8 recognize single-stranded RNA. TLR7 and TLR8 are selectively expressed on plasmacytoid dendritic cells (pDCs), monocytes. TLR 7/8 are also involved in the production of inflammatory cytokines such as IFN-α and the production of autoantibodies. Simultaneous inhibition of TLR7/8 by E6742 may enable SLE disease control, to include the prevention of organ damage. BAFF: B cell-activating factor; IFN: interferon; IL: interleukin; TLR: toll-like receptor.
In humans, E6742 was evaluated in phase 1 studies consisting of a single ascending dose (SAD) study and a multiple ascending dose (MAD) study in healthy adult subjects. After single and multiple oral administrations of E6742, it was confirmed that E6742 was well tolerated within the established dose range, 10 mg to 800 mg in the SAD study and 100 mg to 400 mg twice a day for a week in the MAD study. The safety results from the SAD and MAD studies demonstrated that E6742 has an adequate single and multiple dose safety and tolerability profile, with no clinically significant drug-related laboratory, vital sign, electrocardiogram, or physical examination safety findings, or dose limiting adverse events across the evaluated cohorts [Citation25].
4. Challenges in drug development in SLE
Although many clinical studies have been conducted to date, it is difficult to develop new drugs targeting SLE. The barriers to accelerate SLE drug development include the following: 1) the pathophysiology of SLE is heterogeneous and multiple physiological pathways are involved in the disease development process, 2) there are no specific biomarkers that can predict disease progression or drug response, and 3) the placebo response rate tends to be high due to the concomitant oral glucocorticoids in the clinical studies. These factors make it more difficult to precisely assess the potential clinical efficacy of investigational drugs [Citation26]. In fact, several investigational drugs have shown promising results in the phase 2 clinical studies but failed to achieve the primary endpoints in the phase 3 clinical studies. In hindsight, one of the possible reasons for failures in the confirmatory studies was the overestimation of the effect size between the active drug and placebo based on the results from phase 2 studies. Also, the inadequate sensitivity of efficacy endpoints or the limitations in study design (e.g., insufficient restriction of concomitant drugs, setting of rescue arms which may lead to discontinuation from clinical studies, etc.) may lead to higher failure rate in clinical studies in patients with SLE [Citation27].
One of the solutions to these challenges is to identify drug-specific biomarkers for setting optimal efficacy endpoints. In the case of E6742, a deep understanding of the structure, biological functions of TLR7/8, and target engagement of E6742 are essential for discovering such biomarkers. From the point of view of preclinical research, many key studies to clarify the intracellular signaling and the relationship between TLRs and SLE have been led by academia in Japan [Citation28,Citation29]. While such abundant pre-clinical research exists in academia, there are limits to research and development (R&D) funding and clinical experiences conducted by academia in Japan [Citation25,Citation30]. Such situations lead to the importance of collaboration between academia and industries [Citation31].
Japan Agency for Medical Research and Development (AMED) is a national research and development agency established in Japan in 2015 to promote R&D in medical field. One of the AMED programs, the Cyclic Innovation for Clinical Empowerment (CiCLE), accelerates the processes in R&D by harmonizing intelligence through collaboration among stakeholders in Japan. Under the CiCLE program, academia and industries can work together with government funding and oversight. Through the CiCLE program it is possible to consistently conduct academic research in observational studies and clinical studies, and clinical experience can be reverse translated into preclinical research efficiently, leading to the identification of optimal biomarkers, since the structure of the CiCLE program is suitable for resolving some challenges in drug development targeting SLE in Japan.
5. Future of SLE treatments and E6742
The preclinical and clinical data of E6742 demonstrated compelling justifications to proceed with the clinical development for patients with SLE. On the other hand, considering the difficulty of developing new drugs for SLE, pharmaceutical company-centered drug development is insufficient to ensure the successful development of E6742. Therefore, the authors have established an academia-industry collaboration with basic researchers, clinicians, and a pharmaceutical company in AMED CiCLE program to overcome some difficulties in drug development to create a new solution for Japan. Thus, a favorable situation was established for academic physicians, pharmaceutical scientists, and regulatory authorities to embark on clinical studies under the solid research framework connecting three stakeholders. In the AMED CiCLE program, academia-led clinical research in patients with SLE was initiated in 2020 in parallel with E6742 clinical studies (UMIN000042037). Academia-led clinical research is a long-term observational study in Japan to investigate the pathogenesis of SLE and to seek prognostic biomarkers which reflect the disease activity or progression of SLE. This clinical research includes a comprehensive analysis of genetic, immunophenotypic, and proteomic factors to investigate the relationship between TLR7/8-related signaling and disease activity or organ damage. Additionally, it aims to examine the biological responses following treatment with existing therapies for SLE. The evidence generated from this academia-led clinical research will be utilized to identify specific biomarkers for E6742 and guide the implementation of clinical studies supported by an optimal biomarker strategy. This strategy will involve precise patient selection and the establishment of objective endpoints based on biomarkers.
In recent years, the development of new drugs targeting TLR7/8 has remained competitive, with several compounds already undergoing clinical studies (). Thus, it is imperative to differentiate E6742 from competitive products through the utilization of comprehensive biomarker data generated from academia-led clinical research, and accelerate the clinical program into global clinical studies in collaboration with regulatory authorities. By overcoming challenges to move this program forward, the authors aim to fulfill the unmet medical needs of SLE patients by developing E6742 as a game-changing drug that can potentially have huge impacts on the therapeutic area of SLE.
Table 1. Development status of TLR7/8 antagonists on clinical stage in SLE.
Acknowledgements
The authors thank all the physicians involved in the E6742 academia-industry collaboration program. We also thank AMED for its financial support of the clinical development of E6742.
Disclosure statement
Yoshiya Tanaka has received speaker fees and/or honoraria from Eli Lilly, AstraZeneca, Abbvie, Gilead, Chugai, Behringer-Ingelheim, GlaxoSmithKline, Eisai, Taisho, Bristol-Myers, Pfizer, Taiho, received research grants from Mitsubishi-Tanabe, Eisai, Chugai, Taisho. Shizuo Akira has received consultant fees from Eisai. Fumitoshi Tago, Naoto Yamakawa, and Mari Aoki are employees of Eisai Co., Ltd. Takuya Yagi is an employee of Eisai Inc.
Additional information
Funding
References
- Barber MRW, Drenkard C, Falasinnu T, et al. Global epidemiology of systemic lupus erythematosus. Nat Rev Rheumatol. 2021;17(9):515–532.
- Carter EE, Barr SG, Clarke AE. The global burden of SLE: prevalence, health disparities and socioeconomic impact. Nat Rev Rheumatol. 2016;12(10):605–620.
- Tanaka Y, Mizukami A, Kobayashi A, et al. Disease severity and economic burden in japanese patients with systemic lupus erythematosus: a retrospective, observational study. Int J Rheum Dis. 2018;21(8):1609–1618.
- Aringer M, Costenbader K, Daikh D, et al. 2019 European league against rheumatism/American college of rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. 2019;71(9):1400–1412.
- Tanaka Y. State-of-the-art treatment of systemic lupus erythematosus. Int J Rheum Dis. 2020;23(4):465–471.
- Tanaka Y, O'Neill S, Li M, et al. Systemic lupus erythematosus: targeted literature review of the epidemiology, current treatment, and disease burden in the asia pacific region. Arthritis Care Res (Hoboken). 2022;74(2):187–198.
- Mok CC, Teng YKO, Saxena R, et al. Treatment of lupus nephritis: consensus, evidence and perspectives. Nat Rev Rheumatol. 2023;19(4):227–238.
- Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62(1):222–233.
- Rovin BH, Furie R, Latinis K, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the lupus nephritis assessment with rituximab study. Arthritis Rheum. 2012;64(4):1215–1226.
- Fanouriakis A, Kostopoulou M, Cheema K, et al. 2019 Update of the joint european league against rheumatism and european renal Association-European dialysis and transplant association (EULAR/ERA-EDTA) recommendations for the management of lupus nephritis. Ann Rheum Dis. 2020;79(6):713–723.
- Furie RA, Aroca G, Cascino MD, et al. B-cell depletion with obinutuzumab for the treatment of proliferative lupus nephritis: a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2022;81(1):100–107.
- Bentham J, Morris DL, Graham DSC, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet. 2015;47(12):1457–1464.
- Liu Z, Davidson A. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med. 2012;18(6):871–882.
- Tanaka Y. Systemic lupus erythematosus. Best Pract Res Clin Rheumatol. 2022;36(4):101814.
- Lund J, Sato A, Akira S, et al. Toll-like receptor 9-mediated recognition of herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med. 2003;198(3):513–520.
- Heil F, Hemmi H, Hochrein H, et al. Species-Specific recognition of Single-Stranded RNA via toll-like receptor 7 and 8. Science. 2004;303(5663):1526–1529.
- El-Zayat SR, Sibaii H, Mannaa FA. Toll-like receptors activation, signaling, and targeting: an overview. Bull Natl Res Cent. 2019;43(1):187.
- Barrat FJ, Meeker T, Gregorio J, et al. Nucleic acids of mammalian origin can act as endogenous ligands for toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005;202(8):1131–1139.
- Sakata K, Nakayamada S, Miyazaki Y, et al. Up-Regulation of TLR7-Mediated IFN-alpha production by plasmacytoid dendritic cells in patients with systemic lupus erythematosus. Front Immunol. 2018;9:1957.
- Ronnblom L, Leonard D. Interferon pathway in SLE: one key to unlocking the mystery of the disease. Lupus Sci Med. 2019;6(1):e000270.
- Postal M, Vivaldo JF, Fernandez-Ruiz R, et al. Type I interferon in the pathogenesis of systemic lupus erythematosus. Curr Opin Immunol. 2020;67:87–94.
- Shen N, Fu Q, Deng Y, et al. Sex-specific association of X-linked toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2010;107(36):15838–15843.
- Brown GJ, Canete PF, Wang H, et al. TLR7 gain-of-function genetic variation causes human lupus. Nature. 2022;605(7909):349–356.
- Bender AT, Tzvetkov E, Pereira A, et al. TLR7 and TLR8 differentially activate the IRF and NF-kappaB pathways in specific cell types to promote inflammation. Immunohorizons. 2020;4(2):93–107.
- Yamakawa N, Tago F, Nakai K, et al. First-in-Human study of the safety, tolerability, pharmacokinetics, and pharmacodynamics of E6742, a dual antagonist of toll-like receptors 7 and 8, in healthy volunteers. Clin Pharmacol Drug Dev. 2023;12(4):363–375.
- Touma Z, Gladman DD. Current and future therapies for SLE: obstacles and recommendations for the development of novel treatments. Lupus Sci Med. 2017;4(1):e000239.
- A. L-V, DA. I. Clinical trials in systemic lupus erythematosus: the dilemma—why have phase III trials failed to confirm the promising results of phase II trials? Ann Rheum Dis. 2022;0(1):6.
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511.
- Santiago-Raber ML, Dunand-Sauthier I, Wu T, et al. Critical role of TLR7 in the acceleration of systemic lupus erythematosus in TLR9-deficient mice. J Autoimmun. 2010;34(4):339–348.
- Kneller R. The importance of new companies for drug discovery: origins of a decade of new drugs. Nat Rev Drug Discov. 2010;9(11):867–882.
- Tsuruya N, Kawashima T, Shiozuka M, et al. Academia-industry cooperation in the medical field: matching opportunities in Japan. Clin Ther. 2018;40(11):1807–1812.