
ABSTRACT
Forward genetics is the identification and characterization of the gene that is responsible for the mutant phenotype. It starts with the measurement or observation of a phenotype followed by mapping of the causative loci or genes. In addition, forward genetic approaches are, unbiased in terms of gene identification since the phenotype is the primary level of measurement. Reverse genetics, on the other hand, analyzes the phenotype of an organism following the disruption of a known gene. To generate random mutations in an organism, various approaches such as X-rays, ultraviolet irradiation and chemical treatment are used. Genetic variation across the genome is measured using a set of markers that can distinguish between the two or more strains used in an experimental cross. The source of these markers has changed over the years with advances in technology and knowledge of genomic structure. Single nucleotide polymorphisms (SNPs) are single nucleotide changes that are also detectable by PCR, and millions of them exist between inbred strains, providing even greater resolution for genetic mapping. Two of the most widely used mutagens for chemical mutagenesis experiments are ethylmethanesulfonate and ethylnitrosourea. The objective is to review approaches and use of reverse and forward genetics in plant breeding.
KEYWORDS:
Introduction
Forward genetics (or a forward genetic screen) is an approach used to identify genes (or set of genes) responsible for a particular phenotype of an organism. Forward genetics seeks to find the genetic basis of a phenotype or trait, reverse genetics seeks to find what phenotypes arise as a result of particular genetic sequences. Forward and reverse genetics both approaches begin with the development of a suitable mutagenized population. Mutagenesis of multi-cellular tissues such as seeds result in the first generation (termed M1) being a genetic mosaic because each cell carries unique mutations. Traditional or forward genetics involves a phenotypic evaluation and selection of novel plant genotypes. Plants with phenotypic characteristics can be incorporated immediately in to breeding programs and when desired, the mutation causing the phenotype can be cloned to understand the function of genes and to produce a perfect genetic marker.
Reverse genetics is a method that is used to help understand the function of a gene by analyzing the phenotypic effects of specific engineered gene sequences. Reverse genetics usually proceeds in the opposite direction of so-called forward genetic screens of classical genetics. Regular genetics begins with a mutant phenotype, proceeds to show the existence of the relevant gene by progeny ratio analysis, and finally clones and sequences the gene to determine its DNA and protein sequence. However, reverse genetics, a new approach made possible by recombinant DNA technology, works in the opposite direction. Reverse genetics starts from a protein or DNA for which there is no genetic information and then works backward to make a mutant gene, ending up with a mutant phenotype. Important tools for reverse genetics are in vitro mutagenesis and gene disruption, also known as gene knockout. One approach is to insert a selectable marker into the middle of a cloned gene and then to use this construct to transform a wild-type recipient. Selecting for the marker yields some transformants in which the disrupted (mutated) gene has replaced the in situ wild-type allele (Griffiths et al. Citation2000).
Historically, reverse genetics in crops had its origins in the exploitation of biological agents, specifically transposable elements. First discovered in maize by Barbara McClintock, transposons have the ability to move naturally and insert into new regions of a genome (McClintock Citation1950). The first step in forward and reverse genetics is the development of a suitably mutagenized population. This involves the choice of plant material, mutagen and optimizing dosage treatment. For seed propagated crops, seed are typically mutagenized, but examples also exist for pollen and in vitro mutagenesis (Till et al. Citation2004; Serrat et al. Citation2014).
Objective
| • | To review approaches and use of reverse and forward genetics in plant breeding. | ||||
Forward (classical) genetics
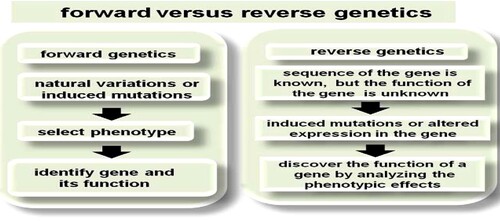
According to Zakhrabekovaa et al. (Citation2013), the process of forward genetics has three steps:
Getting mutants (could be natural variation or induce mutations)
Selecting phenotype, and
Identifying the gene and its function
Mutagens
Mutant phenotypes are known long before their corresponding genes have been identified. These can be phenotypes in model organisms, such as white eyes in Drosophila, or heritable human diseases such as cystic fibrosis or Huntington’s disease (Tarantino and Eisener Citation2012).
To generate random mutations in an organism, various approaches such as X-rays, ultraviolet irradiation and chemical treatment are used. These genes are followed by the selection of mutant phenotypes associated with various traits, such as high yield, early maturity, disease resistance, drought tolerance, cold tolerance, lodging resistance and so on. Following the identification of the mutant, they need to be classified. The aim is to gather mutants into complementation groups by using allelic tests. Such groups of multiple independent mutant alleles can efficiently be used to validate a candidate gene. Scandinavian barley mutant collection can be mentioned as an example. In the mid-1930s, the first viable mutations were observed and notable among the mutant collections are high-yielding, early maturity, dense spike, tillering capacity, straw-stiffness, seed-size and mutants useful for understanding basic agronomical important traits such as photosynthetic capacity and protective outer barrier formation (Lundqvist Citation2005). The following techniques are used to develop mutations in a given population.
X-rays – cause breaks in double-stranded DNA, resulting in large deletions of pieces of chromosome or chromosomal re-arrangements. These mutations are typically easy to map by cytological examination of chromosomes, but are often not limited to single genes. Not good for fine-scale mutagenesis.
Chemical mutation – specified chemicals such as ethylmethanesulfonate (EMS) causes point mutations, which are changes at a single nucleotide position. Mutations may be nonsense (introduce a premature stop codon) or mis-sense (cause an amino acid replacement). They may also be in non-coding sequence, affecting splicing signals or regulatory elements that control gene expression. This approach allows for many different mutations within gene regions.
Insertional (transposon) mutagenesis – Transposable elements (TEs) containing a marker gene(s) are mobilized in the genome. The TE can insert within a coding region and disrupt the amino acid sequence or it may insert into neighboring non-coding DNA and affect intron splicing or gene expression. TE insertion can easily be mapped and the region of genome cloned.
After mutants are identified, they can be separated into complementation groups, mapped to general chromosomal location by linkage with known markers, and eventually cloned and sequenced. A number of mutants have been developed in many crops. Some are mentioned below:
Development of mutant populations
Obtaining a suitably mutagenized population is the first step for both forward and reverse genetics. This involves the choice of plant material, mutagen and optimizing dosage treatment. For seed propagated crops, seeds are typically mutagenized (Serrat et al. Citation2014). Following the mutagenesis procedure, plants are evaluated for heritable traits that are different from the non-mutagenized parental line. When using either physical or chemical mutagenesis, the first generation (referred to as the M 1) is a genetic mosaic owing to the fact that each cell that is treated with mutagen is capable of accumulating different mutations. This chimeric generation is therefore not suitable for phenotypic or genotypic evaluation as different tissues may have different genotypes and thus be expressing different traits. Once potentially interesting traits are recovered, work is undertaken to fix the novel allele(s) in the population and ensure stability and heritability of the desired traits. Typically after 5–10 generations of self-crossing, stable plants are produced and moved forward to multi-location varietal testing before the official release. Crossing is not a requisite for varietal release and many examples can be found in the Mutant Variety Database where plants are released directly after novel traits are fixed and stable.
Tissue-culture mutagenesis for breeding follows the same principles as with seed mutagenesis except the choice of material to mutagenize. These include shoot tips, nodal cuttings, adventitious and lateral shoots, and cell and organ cultures. The exact methods available depend on the species and sometimes even the genotype used (Danso et al. Citation2010). Mutagenesis optimization involves increasing dosage of treatments and measuring the survivability of materials and the ability to generate whole plants (Mba et al. Citation2010).
The ability to screen many thousands of mutant individuals rapidly for traits of interest greatly improves the chances of success. This is because while induced alleles may be produced at a frequency that is orders of magnitude higher than spontaneous mutations, the probability of recovering a mutation generating the desired phenotype is expected to be relatively low (Till et al. Citation2003). Mass screening techniques applied to cultured tissues can be especially powerful as trait development is independent of potentially lengthy field propagation cycles (Lebeda and Svabova Citation2010). There is no intellectual property associated with inducing mutations, and most countries do not require regulation of mutagenized material. This implies that continual and increasing breeding efforts are needed to meet the growing pressures on global food security, suggests that the use of forward genetics will continue to remain important for many years to come. Emerging technologies will help increase the efficiency of forward breeding.
Genetic mapping
The goal of genetic mapping is to identify the locus of the gene responsible for the trait of interest. The first step in all mapping studies is to find markers that are linked with the trait. Physical linkage will lead to co-inheritance of markers, while recombination events will break these associations. The next steps are to develop appropriate mapping populations; screen parents for marker polymorphism and genotype mapping population. Afterwards a linkage analysis is performed to find out recombination frequencies between markers which in turn lead to the fine mapping of the location of the gene of interest. If the genome of the plant of interest is not fully sequenced, the synteny between physical and genetic maps of closely related plants, with sequenced genome enables the assessment of the gene content at the fine mapped locus.
QTL mapping
QTL mapping is a statistical association analysis between phenotype and genotype at specific locations across the genome. In this case, phenotypic and genetic variations are very important things. Phenotypic variation is easily observed among inbred strains (Hamilton and Frankel Citation2001), and experimental crosses between inbred strains provide both phenotypic and genetic variation. Genetic variation across the genome is measured using a set of markers that can distinguish between the two or more strains used in an experimental cross. The source of these markers has changed over the years with advances in technology and knowledge of genomic structure. Genetic markers initially took the form of linked visible characteristics such as coat color and progressed to restriction fragment length polymorphisms (RFLPs). With the advent of the polymerase chain reaction (PCR), scientists had access to a trove of genetic markers called simple sequence length polymorphisms (SSLPs) that were more numerous and provided dense genetic maps. Single nucleotide polymorphisms (SNPs) are single nucleotide changes that are also detectable by PCR, and millions of them exist between inbred strains, providing even greater resolution for genetic mapping. SNPs are now the primary source of genetic variation measured in mapping studies. Genetic markers need not be causative alleles and simply serve as a means of ascertaining allelic state at a genomic location. Once genotypes and phenotypes are collected, association of genotype with phenotype can be performed. This aim can be accomplished by single-marker analysis using basic statistical techniques (ANOVA, marker regression) and comparing marker status at each genomic location with phenotype.
Exome sequencing
is a powerful method to selectively sequence the coding regions of the genome as a less costly alternative to whole genome sequencing (Ng et al. Citation2009). This method can be combined with target-enrichment strategies, which give possibility to selectively capture genomic regions of interest from a DNA sample prior to sequencing (Bashiardes et al. Citation2005). Identification of mutations by this method requires as well as in RNA-seq a number of different mutant alleles to give a trust worthy answer.
Forward genetics vs. reverse genetics
Forward and reverse genetics approaches are used to determine the function of genes. While forward genetics refers to the identification and characterization of the gene that is responsible for the mutant phenotype, the goal of reverse genetics is to examine the effect of induced mutation or altered expression of a particular gene and to understand the gene function.
With the advent of whole genome sequencing many researchers are now in a very different position. They have access to all of the gene sequences within a given organism and would like to know their function. So, instead of going from phenotype to sequence as in forward genetics, reverse genetics works in the opposite direction – a gene sequence is known, but its exact function is uncertain. In reverse genetics, a specific gene or gene product is disrupted or modified and then the phenotype is measured (Figure ).
Figure 1. The difference between forward and reverse genetic techniques is the starting point: the phenotype or the gene.
Advantage and disadvantage of reverse genetics
Advantage:
| • | Less time than classic genetics | ||||
| • | No knowledge of gene function required | ||||
Disadvantage:
Single allele mutations can be misleading
Limited in scopes
Specific reverse genetic approaches
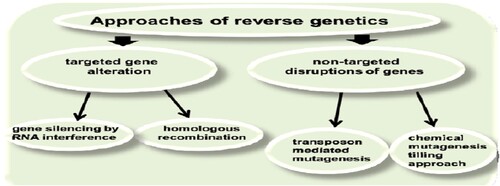
The goal in reverse genetics is to investigate the impact of induced variation within a specific gene and to infer gene function. The process of disruption or alteration can either be targeted specifically as in the case of gene silencing or homologous recombination or can rely on non-targeted random disruptions (e.g. chemical mutagenesis, Transposon mediated mutagenesis) followed by screening a library of individuals for lesions at a specific location.
Gene silencing and homologous recombination are two commonly used approaches used for targeted gene mutation, in contrast to non-targeted disruptions of genes achieved by Transposon mediated and chemical mutagenesis. For such a model plant as Arabidopsis, T-DNA insertion mutants have been produced and are available for researchers (Krysan et al. Citation1999).
two novel approaches of reverse genetics: (1) targeted gene silencing by RNA interference and (2) TILLING (Targeting Induced Local Lesions in Genomes) – a recently developed reverse genetics technique.
Gene silencing
RNA interference (RNAi) is the process by which expression of a target gene is inhibited by antisense and sense RNAs. It works based on the ability of double-stranded sequences to recognize and degrade sequences that are complementary to them (Lewin Citation2004). RNAi was first discovered in Caenorhabditis elegans when the introduction of double-stranded RNA was observed to be an efficient method for silencing gene expression (Fire et al. Citation1998; Kuttenkeuler and Boutros Citation2004). RNAi- based silencing is an exciting strategy for reverse genetics (Waterhouse et al. Citation1998). RNA interference has recently become a powerful tool to silence the expression of genes and analyze their loss-of-function phenotype, allowing analysis of gene function when mutant alleles are not available. Having been shown to work in a similar manner in all metazoans, RNAi has proven to be applicable to many organisms and has been used to generate a wide variety of loss-of-function phenotypes (Kuttenkeuler and Boutros Citation2004). The phenomenon of post-transcriptional gene silencing observed in plants may also be due to a related RNAi mechanism (Waterhouse et al. Citation1998). RNAi-based silencing utilizes the endonuclease Dicer to cleave single-stranded RNAs, abbreviated siRNAs, from double-stranded RNA; the RISC complex then destroys specific target mRNAs based on sequence complementarily with the siRNA (Pattanayak et al. Citation2005).
RNAi has been used for a systematic analysis of gene function in C. elegans by generating loss of function phenotypes, creating a library of worms expressing dsDNA corresponding to different genes (Lewin Citation2004).
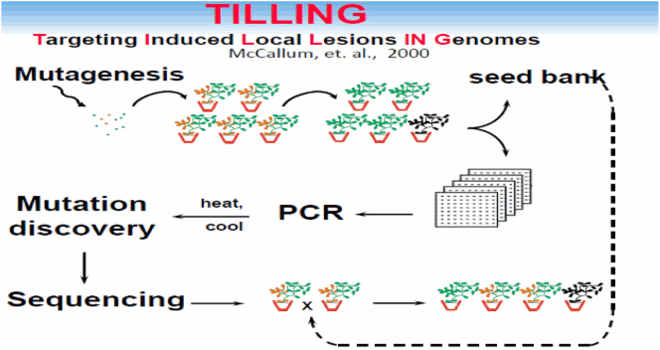
TILLING (Targeting Induced Local Lesions in Genomes)
TILLING (Targeting Induced Local Lesions in Genomes) is a recently developed reverse genetics technique, based on the use of a mismatch-specific endonuclease (CelI), which finds mutations in a target gene containing aheteroduplex formation (Henikoff et al., Citation2004). If the mutation frequency is high and the population size large enough, mutated alleles of most, if not all, genes will be present in the population. The technique involves PCR amplification of the target gene using fluorescently labeled primers, formation of DNA heteroduplex between wild type and mutant alleles (PCR products, corresponding to the mutant and wild type alleles are heated and then slowly cooled), followed by endonuclease digestion specifically cleaving at the site of an EMS-induced mismatch. The sizes of the amplicon cleavage fragments are often analyzed by a Li-COR (McCallum et al. Citation2000).
It is possible to apply TILLING to genetically complicated crops, such as wheat for example (Slade et al. Citation2005). One of the greatest benefits of the TILLING approach is that it does not involve genetic manipulations, that results in Genetically Modified Organisms (GMO), which are not legal for agricultural applications in many countries.
Homologous recombination
Just as homologous recombination has been found to be mainly initiated with a double-strand break, gene targeting by homologous recombination is associated with the repair of double-strand breaks. The double-strand break repair and synthesis-dependent strand-annealing models are the most generally accepted models to explain gene targeting (Iida and Terada Citation2004). A reverse-genetic system using homologous recombination has recently been developed for Drosophila. It is promising, but is a lengthy procedure and requires generation of specific transgenic flies (Stemple Citation2004). Reproducible gene targeting by homologous recombination is now also feasible in rice. With the combination of site-specific recombination systems (such as Cre-lox), the future of gene targeting by homologous recombination as a routine procedure for engineering the genome of rice and presumably other plants is bright (Iida and Terada Citation2004).
Insertional mutagenesis/transposon-mediated mutagenesis
Transposons are mobile genetic elements that can relocate from one genomic location to another (Hayes Citation2003). They are DNA sequences that can insert themselves at a new location in the genome without having any sequence relationship with the target locus (Lewin Citation2004). Transposon-based signature-tagged mutagenesis has been successful in identifying essential genes as well as genes involved in infectivity of a variety of pathogens. Strategies for insertional mutagenesis using transposons have been developed for a number of animal and plant models (Hayes Citation2003). Reverse-genetics is currently being done in Drosophila and C. elegans by utilizing libraries of individuals who carry transposable element insertions, many of which have been mapped, and some of which will disrupt the expression of nearby genes. In Drosophila P-elements, imprecise excision can be driven to generate a mutation in the nearest gene. Transposon-based methods are also being used in Arabidopsis, maize and other plants (Stemple Citation2004). One drawback of insertional mutagenesis is the low frequency of mutations, necessitating the screening of large numbers of individuals to find mutations in any given gene (Gilchrist and Haughn Citation2005). Also, insertions in essential genes will usually cause lethality, and less severe mutations must be generated in these genes in order to understand gene function (Till et al. Citation2003).
The segment of the Ti plasmid of Agrobacterium tumefaciens known as T-DNA that carries genes to transform the plant cell has also been utilized for insertional mutagenesis. T-DNA insertional mutagenesis has been used to obtain gene knockouts for greater than 70 percent of Arabidopsis genes (Alonso et al. Citation2003), but no comparable resources exist for rice or maize even as high-coverage genomic sequence is becoming increasingly available (Henikoff et al. 2004). Unlike other successful gene targeting systems (namely mouse, Physcomitrella, and Drosophila), the precise mechanism of T-DNA integration into the plant genome remains largely unknown (Iida and Terada Citation2004). Like RNA suppression techniques, insertional mutagenesis is limited by its host range and by its limited range of allele types (McCallum et al. Citation2000).
Gene replacement via transformation is a commonly used tool for many filamentous fungi (Fang, Hanau, and Vaillancourt, Citation2002; Lalucque and Silar Citation2004; Takano et al. Citation2000). The transformation can be mediated in many ways, including Agrobacterium and various other transformation vectors (Zhang et al., Citation2003).
Chemical mutagenesis
Two of the most widely used mutagens for chemical mutagenesis experiments are EMS (ethylmethanesulfonate) and ENU (ethylnitrosourea). EMS is a chemical mutagen that alkylates guanine bases. The alkylated guanine will then pair with thymine instead of the preferred cytosine base, ultimately resulting in a G/C to A/T transition. EMS is the most commonly used mutagen in plants. In Arabidopsis, 5 percent of EMS-induced mutations in targeted coding regions result in premature termination of the gene product, while 50 percent result in missense mutations that alter the amino-acid sequence of the encoded protein (Gilchrist and Haughn Citation2005). This high level of missense mutations relative to terminated gene products is very useful in analyzing gene function. ENU also induces point mutations, and is a more potent mutagen than EMS. It is also an alkylating agent, mutagenizing by transferring an ethyl group to oxygen or nitrogen radicals in the DNA molecule, which leads to mispairing and ultimately results in base pair substitutions, and sometimes base pair losses if not repaired (Guénet Citation2004).
Chemical mutagenesis is attractive for reverse genetics because it results in induced point mutations, which create a diverse range of alleles for genetic analysis. It induces a large number of recessive mutations per genome that are randomly distributed (Gilchrist and Haughn Citation2005). Because chemical mutagenesis is already widely used in many organisms for forward genetic screens, it promises to be generally applicable for reverse genetics (Henikoff et al. 2004). The TILLING strategy provides a high throughput strategy to detect single base changes within genetic targets and can be applied to a wide variety of organisms (Figure ).
Figure 2. Approaches of reverse genetics.
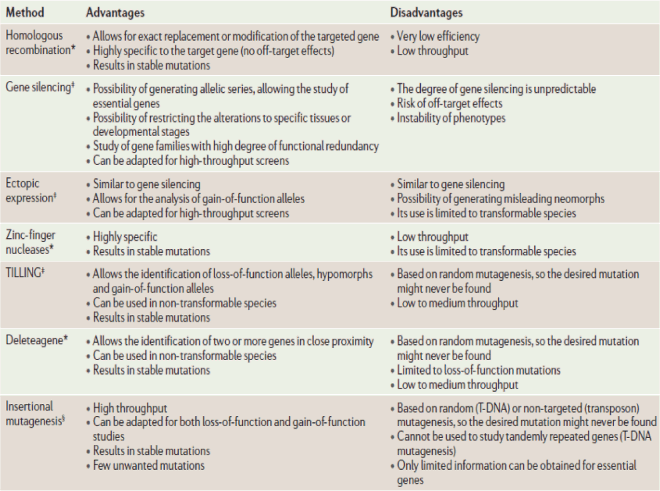
Methods used for reverse genetics in plant
Limitations/roadblocks to completing reverse genetics
Completing reverse genetics is not without its pitfalls, and not all techniques can be applied to all organisms. To be successful, there are several aspects that must be checked. For organisms that do not have efficient transformation systems available techniques such as TILLING that can be applied without transformation may be the only practical choice. In these cases, the rate of mutagenesis is an important factor that can be difficult to determine. The load of mutations must be balanced with the recovery of mutants (Till et al. Citation2003) – in other words, the genome can’t be so riddled with mutations that it is impossible to see a mutant phenotype. Also to be considered is the fertility of the mutagenized organism, especially in the first generation, but also in subsequent generations (Perry et al. Citation2003), both before and after mutagenesis. This is especially true for diploid organisms, because if the sexual machinery is not intact and working properly, then it is impossible to obtain a homozygous mutant. The mutagenized organisms must also be kept alive long enough to screen a mutant population for a specific target. For some organisms, like Arabidopsis or Phytophthora, this is not a problem as the seeds or cultures are relatively easy to store. It presents a challenge for other organisms.
Conclusion
Forward genetics starts with identification of interesting mutant phenotype. Forward genetics seeks to find the genetic basis of a phenotype or trait. Automated DNA sequencing generates large volumes of genomic sequence data relatively rapidly. The main objective of forward genetics is to discover the function of genes defective in mutants.
Reverse genetics (or a reverse genetic screen), on the other hand, analyzes the phenotype of an organism following the disruption of a known gene. Forward genetics seeks to find the genetic basis of a phenotype or trait, reverse genetics seeks to find what phenotypes arise as a result of particular genetic sequences. Reverse genetics starts with a known gene and alters its function by transgenic technology. Reverse genetics however, due to the advent of advances in sequencing is employed to seek out what sequences underlies the phenotype. Reverse genetics starts with known genes e.g. from genomic sequencing. Major goal of reverse genetics is to determine function through targeted modulation of gene activity. Forward and reverse genetics both approaches begin with the development of a suitable mutagenized population. Mutagenesis of multi -cellular tissues such as seeds result in the first generation (termed M1) being a genetic mosaic because each cell carries unique mutations. Traditional or forward genetics involves a phenotypic evaluation and selection of novel plant genotypes. Plants with phenotypic characteristics can be incorporated immediately into breeding programs and when desired, the mutation causing the phenotype can be cloned to understand the function of genes and to produce a perfect genetic marker. Reverse genetic strategies begin with the genotypic screening of the mutant population to identify novel induced mutations in candidate genes. This is followed by phenotypic evaluation of those individuals harboring putative deleterious mutations. A major contributor to both forward and reverse genetics was the discovery in the early twentieth century that mutations can be induced in genomes at frequencies in several orders of magnitude higher than typically observable in nature. The ability to produce novel variation has fueled the development of thousands of new crop cultivars. Examples exist of increased disease resistance, higher yields, tolerance to abiotic stresses such as drought and salinity and improved nutritional quality.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Reference
- Alonso JM, Stepanova AN, Leisse TJ. 2003. Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science. 301:653–657.
- Bashiardes S, Veile R, Helms C, Mardis ER, Bowcock AM, Lovett M. 2005. Direct genomic selection. Nat Methods. 1(2):63–69.
- Danso KE, Elegba W, Oduro V, Kpentey P. 2010. Comparative study of 2,4-D and picloram on friable embryogenic calli and somatic embryos development in cassava (Manihot esculenta Crantz). Int J Integr Biol. 10(2):94–100.
- Fang G-C, Hanau RM, Vaillancourt LJ. 2002. The SOD2 gene, encoding a manganese-type superoxide dismutase, is up-regulated during conidiogenesis in the plant-pathogenic fungus Colletotrichum graminicola. Fungal Gene Biol. 36:155–165.
- Fire A, Siqun XU, Montgomery MK, Kostas SA, Driver SE, Mello CC. 1998. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 391:806–811.
- Gilchrist EJ, Haughn GW. 2005. TILLING without a plough: a new method with applications for reverse genetics. Curr Opin Plant Biol. 8:211–215.
- Griffiths AJF, Miller JH, Suzuki DT, et al. 2000. An introduction to genetic analysis, 7th ed. New York: W. H.
- Guénet J. 2004. Chemical mutagenesis of the mouse genome: an overview. Genetica. 122:9–24.
- Hamilton BA, Frankel WN. 2001. Of mice and genome sequence. Cell. 107:13–16. Hamre.
- Hayes F. 2003. Transposon-based strategies for microbial functional genomics and proteomics. Annu Rev Genet. 37:3–29.
- Henikoff S, Till BJ, Comai L. 2004. TILLING. Traditional mutagenesis meets functional genomics. Plant Physiol. 135(2):630–636. doi:10.1104/pp.104.041061.
- Iida S, Terada R. 2004. A tale of two integrations, transgene and T-DNA: gene targeting by homologous recombination in rice. Curr Opin Biotechnol. 15:132–138.
- Krysan PJ, Young JC, Jester PJ, Monson S, Copenhaver G, Preuss D, Sussman MR. 1999. Characterization of T-DNA insertion sites in arabidopsis thaliana and the implications for saturation mutagenesis. Omics J Integr Biol. 6(2).
- Kuttenkeuler D, Boutros M. 2004. Genome-wide RNAi as a route to gene function in Drosophila. Brief Funct Genom Proteom. 3(2):168–176.
- Lalucque H, Silar P. 2004. Incomplete penetrance and variable expressivity of a growth defect as a consequence of knocking out two K+ transporters in the Euascomycete fungus Podospora anserina. Genetics. 166:125–133.
- Lebeda A, Svabova L. 2010. In vitro screening methods for assessing plant disease resistance. In: Mass screening techniques for selecting crops resistant to disease, 5–46. Vienna: Internal Atomic Energy Agency.
- Lewin B. 2004. Genes VII. Upper Saddle River, NJ: Pearson Prentice Hall.
- Lundqvist U. 2005. The Swedish collection of barley mutants held at the Nordic Genebank. Barley Genetics Newsletter. 35:150–154.
- Mba C, Afza R, Bado S, Jain SH. 2010. Induced mutagenesis in plants using physical and chemical agents. In: Davey MR, Anthony P, editors. Plant cell culture: essential methods. Chichester: John Wiley; p. 111–130.
- McCallum CM, Comai L, Greene EA, Henikoff S. 2000. Targeting induced local lesions in genomes (TILLING) for plant functional genomics. Plant Physiol. 123:439–442.
- McClintock B. 1950. The origin and behavior of mutable loci in maize. Proc Natl Acad Sci U S A. 36(6):344–355.
- Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler EE, et al. 2009. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 461:272–276.
- Pattanayak D, Agarwal S, Sumathi S, Chakrabarti SK, Naik PS, Khurana SM. 2005. Small but mighty RNA-mediated interference in plants. Indian J Exp Biol. 43(1):7–24.
- Perry JA, Wang TL, Welham TJ, Gardner S, Pike JM, Yoshida S, Parniske M. 2003. A TILLILNG reverse genetics tool and a web-accessible collection of mutants of the legume Lotus japonicus. Plant Physiol. 131:866–871.
- Serrat X, Esteban R, Guibourt N, et al. 2014. EMS mutagenesis in mature seed-derived rice calli as a new method for rapidly obtaining TILLING mutant populations. Plant Methods. 10(1):5. doi:10.1186/1746-4811-10-5.
- Slade AJ, Fuerstenberg SI, Loeffler D, et al. 2005. A reverse genetic, nontransgenic approach to wheat crop improvement by TILLING. Nat Biotechnol. 23(1):75–81.
- Stemple DL. 2004. TILLING - a high-throughput harvest for functional genomics. Nat Rev. 5(2): 145–150.
- Takano Y, Kikuchi T, Kubo Y, Hamer JE, Mise K, Furusawa I. 2000. The Colletotrichum lagenarium MAP kinase gene CMK1 regulates diverse aspects of fungal pathogenesis. Mol Plant Microbe Interact. 13(4):374–383.
- Tarantino LM, Eisener-Dorman AF. 2012. Forward genetic approaches to understanding complex behaviors. Behav Neurogenetics. 25–58.
- Till BJ, Reynolds SH, Greene EA, Codomo CA, Enns LC, Johnson JE, Burtner C, Odden AR, Young K, Taylor NE, et al. 2003. Large-scale discovery of induced point mutations with high-througput TILLING. Genome Res. 13:524–530.
- Till BJ, Reynolds SH, Weil C, et al. 2004. Discovery of induced point mutations in maize genes by TILLING. BMC Plant Biol. 4:12.
- Waterhouse PM, Graham MW, Wang M. 1998. Virus resistance and gene silencing in plants can be induced by simultaneous expression of sense and antisense RNA. Proc National Acad Sci. 95:13959–13964.
- Zakhrabekovaa SM, Gougha S, Lundhb L, Hansson M. 2013. Functional genomics and forward and reverse genetics approaches for identification of important QTLs in plants 23–28.
- Zhang A, Lu P, Dahl-Roshak AM, Paress PS, Kennedy S, Tkacz JS, An Z. 2003. Efficient disruption of a polyketide synthase gene (pks1) required for melanin synthesis through Agrobacterium-mediated transformation of Glarea lozoyensis. Mol Genet Genomics. 268:645–655.