
Abstract
Lupinus is one of the most geographically widespread genera. It can be divided into New World and Old World Lupinus species according to the geographical distribution of its members. However, there are relatively few studies on their genetic differences, especially regarding the taxonomy and geographical evolutionary history of New World species. Here, we conducted comparative, phylogenetic, and evolution analysis between one New World and five Old World Lupinus species based on their complete chloroplast genomes. Compared with the Old World species, the chloroplast genome of L. westianus in the New World was more variable, showing the deletion of the pseudogene ycf1 and two rps12 gene introns, the 20 kb inversion, the higher number of repeated sequences, the larger genome size, and the lower GC content. Phylogenetic analysis indicated that the genetic relationship between the New World species L. westianus and the Old World species L. albus was closer than to the rest. Evolutionary analysis confirmed that the matK and ycf1 genes were under positive selection, and rpoA, accD, and rpoC2 genes have site-specific selection in L. westianus, which might be related to adaptive evolution. Therefore, our finding would provide genetic information for the evolutionary history and taxonomy of Lupinus species.
Introduction
Fabaceae (or Leguminosae), the third largest angiosperm family, is distributed in very diverse eco-geographic regions around the world (Edwards Citation2007; Bruneau et al. Citation2013). It includes three parallel families, Mimosoideae, Papilionoideae, and Caesalpinioideae, of which the Papilionoideae is the most diverse and contains economically important legumes (Schwarz et al. Citation2017; Cardoso et al. Citation2013; Gepts Citation2005). The Papilionoideae is divided into six major clades: the Genistoids, Dalbergioids, Mirbelioids, Millettioids, and Robinioids, and the inverted repeat-lacking clade (IRLC) (Cronk et al. Citation2006). So far, most of the Papilionoideae plastids that have been sequenced belong to the Millettioids, Robinioids, and IRLC, and only a few are from the Genistoid clade (Cardoso et al. Citation2012). Therefore, it is necessary to further investigate this last clade to better understand plastid evolution within the Papilionoideae. Within this poorly studied lineage, the Lupinus genus is considered a good model system (Keller et al. Citation2017).
Lupinus is one of the most widely distributed genera and is highly diverse. According to their geographical distribution, Lupinus species are usually divided into two main categories: ‘Old World’ species (mainly distributed in the Mediterranean and North and East Africa), including 20 species and subspecies, and ‘New World’ species (mainly distributed in North and South America), including the majority of Lupinus species (Wolko et al. Citation2011). These two geographically independent New-Old World Lupinus species occupy habitats in high mountain areas from sea level to 4000 m, resulting in morphological variations (Wunderlin Citation1982). In addition to morphological differences, several studies have shown that there are genetic separations between the western New World (western North and South America), the eastern New World (including east-central parts of South America and southeastern USA, Florida), and Old World species (Käss and Wink Citation1997; Ainouche and Bayer Citation1999; Maciel and Schifino-Wittmann Citation2002). Phylogenetic evidence has suggested that species from the western New World might be closer to those from the Old World than to those from the eastern New World (Ainouche and Bayer Citation1999; Eastwood and Hughes).
There has been a lack of monographic synthesis for the New World Lupinus species (Wolko et al. Citation2011). Hundreds of species were described only by their morphological characteristics, and many taxa were later considered synonymous (Planchuelo-Ravelo Citation1984). Lupinus westianus, as an eastern New World species, is mainly distributed in subtropical, tropical dry, or coastal dune habitats. It is an endemic species of Florida, United States, and has been classified as a near threatened (NT) species (Xu et al. Citation2019; Bupp et al. Citation2017). In our previous research, next-generation sequencing technology was used to determine the complete chloroplast (cp) genome sequence of L. westianus, which represents the first complete cp genome of a New World Lupinus species (Xu et al. Citation2019). Together with previously reported complete cp genome sequences of other Old World members of this genus, our understanding of the phylogenetic evolution of the Old and New World Lupinus will be enhanced.
In this study, we analyzed the fully assembled complete cp genome of the New World species L. westianus and compared it with five Lupinus cp genomes from the Old World to study the phylogeny and evolutionary characteristic between the New World and Old World Lupinus species. Our findings will not only enhance the understanding of the phylogenetic evolution of the New World and Old World Lupinus species but will also provide valuable genetic information for subsequent taxonomic and phylogenetic identification of members of the genus.
Materials & methods
Genomic materials of Lupinus species
In a previous study (Xu et al. Citation2019), we reconstructed and annotated the complete cp genome of L. westianus, deposited it in GenBank with accession number MG252262. The complete cp genome sequences of the five Old World Lupinus species used in this study (L. albus, L. atlanticus, L. luteus, L. micranthus, and L. princei) were downloaded from the US National Center for Biotechnology Information (NCBI) database, with accession numbers KJ468099, KU726827, NC023090, KU726828, and KU726829, respectively.
Repeated sequences analysis
Repeated sequences in the cp genome were searched including forward, reverse, complement, palindromic using REPuter (Kurtz and Schleiermacher Citation1999) (https://bibiserv.cebitec.uni-bielefeld.de/reputer/), with a similarity percentage for duplicated copies of at least 90%, a minimum repeat size of 30 bp, and a hamming distance of 3. Besides, we used a web-based tandem repeat sequence finder (Benson Citation1999) (TRDB; https://tandem.bu.edu/trf/trf.html) to identify tandem repeated sequences in the genome and set the alignment parameters to match, mismatch, and indel to 2, 7, and 7, respectively. Simple sequence repeats (SSRs) present in the chloroplast genome were identified using the MISA Perl script (Thiel et al. Citation2003). Motif length search parameters were set to > 10 for mononucleotides, > 6 for dinucleotides, and > 5 for trinucleotides, tetranucleotides, pentanucleotides, and hexanucleotides.
Comparative genomic analysis
Blast Ring Image Generator (BRIG) (Alikhan et al. Citation2011) was used to compare the cp genomes of the selected Lupinus species according to Zdravka Ivanova's method (Ivanova et al. Citation2017). Mauve software (Darling et al. Citation2004) was used for genome rearrangement detection, using L. westianus cp genome as a reference. Annotations on inverted repeats (IRs) boundaries within the cp genome of Lupinus were performed using the Plastid Genome Annotator (PGA) (Qu et al. Citation2019). IRs positions were extracted and used as query data for Microsoft Visio professional 2016 to expand and contract these IR areas. For the nucleotide diversity (Pi) comparison, we used a consensus genome for complete alignment according to the method of Njuguna et al. (Citation2019). First, 218 loci shared among all six Lupinus species were extracted, including 77 encoding protein genes, 126 intergenic regions (IGS), and 15 introns. Then, all six cp genomes were compared using MAFFT v7.380 (Katoh and Standley Citation2013). Finally, DnaSP v6.10.04 software (Rozas et al. Citation2017) was used for a sliding window analysis to generate the nucleotide diversity values of the cp genome. The step size was set to 200 bp, and the window length to 600 bp based on Yin's method (Yin et al. Citation2018).
Evolution rate analysis
We tested the synonymous (Ks) and nonsynonymous (Ka) nucleotide substitution rates as well as the Ka/Ks ratios of the coding regions of homologous proteins in all six Lupinus species. After comparing each gene with the ClustalW (Codons) program in MEGA7 (Kumar et al. Citation2016), the Ks, Ka, and Ka/Ks values of each gene were determined based on the model according to Zhang's method (Zhang et al. Citation2018) with the YN00 program in PAML4.9 (Yang Citation2007). We also identified specific selection sites and used the PAML codeML program to analyze these sites for protein-coding gene sequences using four models: M1 (neutral), M2 (selection), M7 (β), and M8 (β & ω). Computational likelihood ratio test (LRTs) was used to confirm the adaptive evolution of genes based on the method of Zhang et al. (Citation2018). Bayes empirical Bayes (BEB) was used to calculate the posterior probability for each site class, and only those sites with a posterior probability > 0.8 were selected.
Phylogenetic analysis
The complete cp genomes of the six Lupinus species and two Sophora species, S. alopecuroides var. alopecu-roides and S. tonkinensis (GenBank accession numbers NC045070 and NC042688, respectively), were used for phylogenetic construction. All data sets were aligned using MAFFT v7.380 (Katoh and Standley Citation2013) under FFT-NS-2 default settings; the comparison results were used for phylogenetic analysis. The phylogenetic tree was constructed using the maximum likelihood (ML) and neighbor-joining (NJ) methods in MEGA7.0 (Kumar et al. Citation2016) with 1,000 bootstrap replication for each matrix, whereas the model finder method was used to find the best model (Kalyaanamoorthy et al. Citation2017).
Results and analysis
Chloroplast genome characteristics of Lupinus species
Like most terrestrial plants, the cp genome of Lupinus species showed a typical quadripartite structure, consisting of a large single copy region (LSC) and a small single copy region (SSC), which separated by two copies of the large inverted repeat (IRa and IRb). As the only one New World Lupinus species whose complete cp genome has been sequenced (Xu et al. Citation2019), the cp genome of L. westianus has similar characteristics to those of L. albus, but slightly different from those of the other four Old World Lupinus species (Table ). The genome size of L. westianus and L. albus are 154,270 bp and 154,140 bp, respectively, which are longer than those of the other Old World Lupinus species (151,808 bp-152,272 bp). However, L. westianus has the same genetic composition as the five Old World Lupinus species, including 77 unique protein-coding genes, 30 tRNAs, and 4 rRNAs. A comparison to determine gene and intron deletions among all six Lupinus species showed that the introns of the rps12 gene were deleted in L. westianus and L. albus. Furthermore, the ycf1 pseudogene of L. westianus was also deleted, whereas it was present in all those Old World Lupinus species studied. Moreover, the genomic GC content of the six Lupinus species did not change much, with a maximum of 36.65% in L. princei and a minimum of 36.47% in L. westianus.
Table 1. Features of six Lupinus species with L. westianus as the reference.
Repeated sequence characteristics of Lupinus species
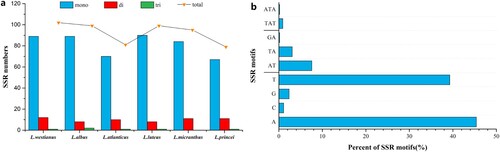
The cp genome of L. westianus contained 102 SSRs, which was higher than that of five Lupinus species in the Old World (ranging from 79 to 99)(Figure (a)). All SSRs of the six Lupinus species were composed of mononucleotides, dinucleotides, and trinucleotides only, and no tetranucleotides, pentanucleotides, or hexanucleotides were present. In addition, SSRs in all six Lupinus cp genomes were AT-rich and rarely contained CG (Figure (b)). The SSRs motifs of mononucleotide repeats were mainly composed of A/T (96.11%), and the SSRs motifs of dinucleotide repeats were almost entirely composed of AT/TA (98.33%).
Figure 1. Analysis of simple sequence repeats (SSRs) in the chloroplast genome of six Lupinus species. (a) The types and numbers of SSRs identified in six Lupinus species; (b) The frequency of SSRs motifs in six Lupinus species.
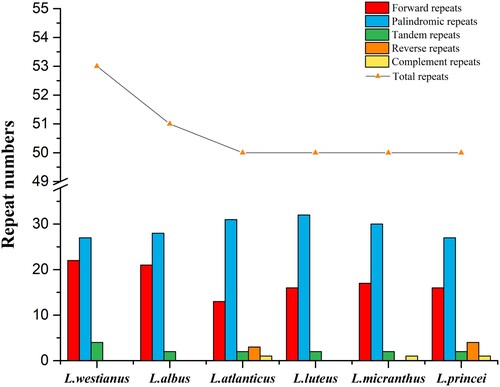
The total number of repeated sequences of L. westianus was greater than that of the five Old World Lupinus species (Figure ). Six Lupinus species contained 13–22 forward repeats, 0–4 reverse repeats, 0–1 complement repeats, 27–32 palindromic repeats, and 2–4 tandem repeats. No reverse repeats or complement repeats were neither found in L. westianus nor L. albus.
Figure 2. Analysis of repeat types and numbers in six Lupinus speices.
Comparison of chloroplast genomes between the Lupinus species
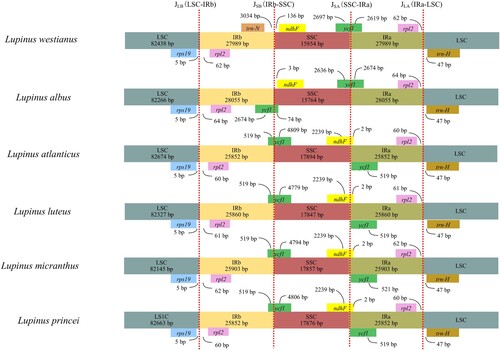
The IR/SC boundary areas of the six Lupinus species were compared (Figure ). Results revealed that the boundaries of the cp genomes of L. westianus and L. albus were similar but slightly different from the other four Old World Lupinus species. Like most angiosperms, the IRa/LSC boundary of the six Lupinus species appeared between the genes rpl2 and trnH-GUG, with 107–111 non-coding nucleotides between the two genes. And the LSC/IRb boundary was found to be between the genes rps19 and rpl2, with 65–69 non-coding nucleotides between the two genes. It is worth noting that the SSC/IRa boundary extended to ycf1, creating a pseudogene in all five Old World Lupinus species that was missing in L. westianus. The ycf1 pseudogene, located at the border of LSC-IRb, was 2748 bp long in L. albus, longer than in other Old World species in which it was 519–521 bp long and was located at the border of SSC-IRa. In addition, compared to other Lupinus species, both the ndhF and ycf1 genes of L. westianus and L. albus were located in different border regions. Overall, compared to four different Old World Lupinus species, the IR in L. westianus and L. albus was extended by 2 kb, and the IR border regions were slightly different.
Figure 3. Comparisons of borders between neighboring genes and junctions of the LSC, SSC and IR regions among the six Lupinus species. Boxes above or below the main line indicate genes adjacent to borders. The figure is not to scale with regard to sequence length and shows only relative changes at or near (inverted repeats/single copy) IR/SC borders. Number above the gene features means the distance between the ends of genes and the borders sites.
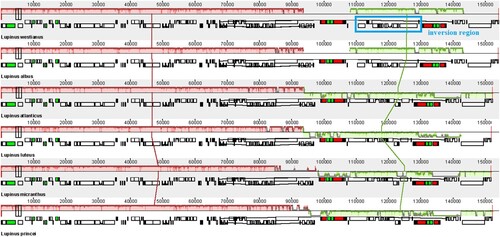
A BRIG-based comparative analysis was performed between the six chloroplast genomes (Figure ). Results showed that the six species were highly similar in genome level, and their genomes were relatively conservative. Besides, the complete cp genome alignment from Mauve was used to detect the rearrangement in six Lupinus species (Figure ). We found that, compared with most of the Old World species, a 20 kb gene inversion (ndhF-ycf1) phenomenon could have had occurred in the New World Lupinus species under study and that this inversion could also be related to the previously described gene deletion.
Figure 4. Comparison of chloroplast genome sequences of six Lupinus species. The outer ring show the coding sequences, tRNA genes, rRNA genes and other genes in the forward and reverse strands. The next six rings show the blast results between the chloroplast genomes of L. westianus and other 5 Old World Lupinus species based on BlastN. The following ring is GC skew curve for the L. westianus chloroplast genome. GC skew+ (green) indicates G > C, GC skew − (purple) indicates G < C. The innermost black ring is the GC content curve for the L. westianus chloroplast genome.
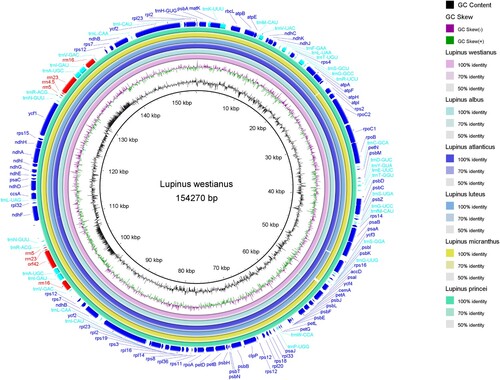
Figure 5. Genomic rearrangement of six Lupinus species. The blue frame represents a 20kb inversion region (ndhF-ycf1).
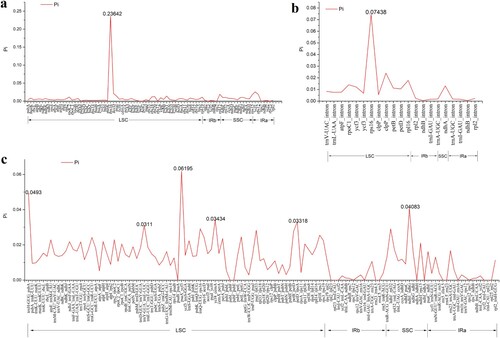
We also made a nucleotide diversity (pi, π) evaluation to determine the hot spots of sequence differences (Figure ). Results showed that the pi values ranged from 0 to 0.23642 and that the IR region showed low nucleotide diversity. Furthermore, eight hypervariable regions were found in the Lupinus chloroplast genome (Pi > 0.03). They were within the rps16, psaA-ycf3, trnH_GUG-psbA, ndhE-ndhG, ycf4-cemA, petD-rpoA, trnY_GUA-trnE_UUC intergenic spacers, and the rps16 intron.
Figure 6. Comparative analysis of nucleotide variability (Pi) values among the chloroplast genome sequences of six Lupinus species.(a) Analysis of the homologous protein-coding genes; (b) Analysis of the intron regions; (c) Analysis of the IGS regions.
Evolution of chloroplast genomes in Lupinus species
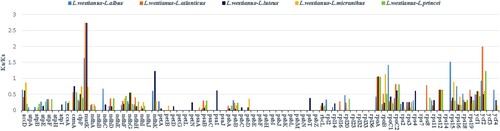
The ratio of nonsynonymous (Ka) to synonymous (Ks) nucleotide substitution rates (expressed as Ka/Ks) is a very important tool for studying the evolution of protein-coding genes; it is used to assess the rate of genetic differences and determine whether a positive, purified, or neutral selection is occurring (Yin et al. Citation2018). If 0.5 < Ka/Ks ratio < 1.0 indicates a relaxed selection, whereas Ka/Ks ratio >1, =1 and <1 (especially less than 0.5) indicate positive, neutral and purifying selection, respectively (Kimura Citation1989). Calculated Ka/Ks ratios between 77 homologous protein-coding genes of the six Lupinus species are shown in Figure . The average Ka/Ks value regarding these 77 shared coding genes was 0.186877. However, the Ka/Ks ratio was 0 for a group of 25 of these genes (i.e. atpH, ndhB, ndhE, ndhJ, petG, petL, psaC, psaJ, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbZ, rpl23, rpl32, rpl33, rpl36, rps7, rps14, and ycf3); >1 for matk and ycf2 genes; and between 0.5 and 1 for a group of 8 genes (accD, rpoA, rpoB, rpoC1, rpoC2, rps12, rps15, ycf1). Noteworthy, the average Ka/Ks of the other genes was between 0 and 0.49, indicating that most genes in the L. westianus cp genome were in a state of purification selection.
Figure 7. The Ka/Ks ratios of homologous protein-coding genes in six Lupinus species, with L. westianus as the reference.
We also investigated site-specific selection events (Table ) and identified five genes with site-specific selection, rpoA, matK, accD, rpoC2, and ycf1 that have 1, 2, 10, 16, and 10 loci under positive selection, respectively.
Table 2. Parameter estimates and log-likelihood values for different models in selectivepressure analysis.
Phylogenetic relationship between Lupinus species
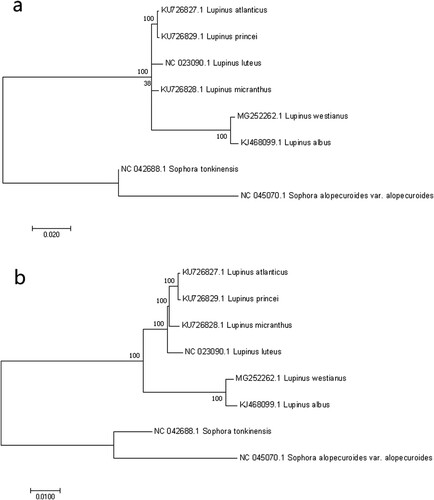
Phylogenetic trees constructed by ML and NJ using the cp genome of the six Lupinus species under study produced the same tree topologies(Figure ), suggesting that the Old World species L. albus was closest to the New World species L. westianus than the other four species. Among the Old World species, L. atlantics and L. princei belong to the same clade. However, it is worth noting that the bootstrap support from ML tree was lower than 50% in L. micranthus node, while was 100% bootstrap support in NJ tree. The reason for the inconsistency might be explained by the small number of taxa.
Figure 8. Phylogenetic relationships based on the chloroplast genomes of six Lupinus species and two Sophera species with the ML and NJ method. (a) ML tree; (b) NJ tree. Sophera species was selected as the outgroup. Numbers on the left side at the branches represent bootstrap values.
Discussion
Chloroplast sequence variation in Lupinus species
The Fabaceae chloroplast genome exhibits significant size variation, cp genome rearrangement, and gene and intron lose (Wang et al. Citation2018) and the events also occurred in the cp genome of Lupinus species. Several factors affect the size of the genome, such as gene deletion (Wakasugi et al. Citation1994), variation in the intergenic region (Tang et al. Citation2004), and expansion or contraction of the IR region (Dugas et al. Citation2015). In this study, the change of cp genomes size in Lupinus from the Old and New world might be related to the variation of the IR / SSC boundary. Firstly, the IR region of L. westianus has expanded by 2 kb compared with other Old World lupines (except L. albus). Secondly, although the pseudogene ycf1 was lost, the length of the intergenic region at the IRb / SSC border becomes longer, which directly leads the size of L. westianus changes. While, the change of the IR / SSC boundary might be related to the inversion event. Hiratsuka's research indicates that inversion causes extensive rearrangement within the rice chloroplast LSC area (Hiratsuka et al. Citation1989). In L. westianus, a 20 kb (ndhF-ycf1) inversion occurred in SSC region, which made the IR / SSC boundary different from other four Old World species, indirectly leading to the change of cp genomes size.
Repeat sequence analysis and molecular marker identification
Regions containing repetitive sequences that cause slip chain mismatches and intramolecular recombination are thought to be responsible for most indel mutations (Kelchner Citation2000). SSRs, also known as microsatellite, are polymorphic (Yin et al. Citation2018) and widely used as a molecular marker to analyze genetic diversity, population structure, and biogeography based on polymorphism (Naydenov et al. Citation2016; Xiao et al. Citation2017). For example, zhang's research shows that SSRs could be used as potential molecular polymorphic markers to reveal the genetic diversity and population structure of Cucurbitaceae (Zhang et al. Citation2018) Jeon et al. find two polymorphic mononucleotide SSRs regions that could differentiate each accession of the Synstylae cp genomes (Jeon and Kim Citation2019). SSRs in L. westianus are consistent with previous reports that SSRs in the cp genomes are usually composed of short polyA or polyT repeats. Other types such as tetra-, penta-, and hexanucleotide repeats are rare in it (Kuang et al. Citation2011; Mehmood et al. Citation2020a, Citation2020b, Citation2020c). These SSRs could contribute to evolutionary and biogeography knowledge in Lupinus species, which warrants further research.
Repeated sequences are thought to cause abnormal replication and repair pathways and have been used extensively for phylogeny, population genetics, genetic mapping, and forensic studies (Huang et al. Citation2018; Bull et al. Citation1999). For example, Li conducted repeated sequence identification of the cp genomes of 7 species of Aristolochiaceae and used them as they can be informative regions for developing genomic markers for phylogenetic analysis (Li et al. Citation2019). Among the complete cp genomes of six Lupinus species, L. westianus had the most repeated sequences. Interestingly, many repetitions occurred in the gene encoding protein ycf2 and the trnN-GUU_ndhF of the intergenic region (Supplementary Table S1), which also exist in Utricularia reniformis and Morella rubra (Silva et al. Citation2016; Liu et al. Citation2017). And these repetitive sequences could be used as information regions to develop genomic markers for phylogenetic analysis of Lupinus.
The highly variable regions would contribute to developing candidate DNA barcodes for future studies (Zhang et al. Citation2018). In this study, 8 highly variable regions (rps16, psaA-ycf3, trnH_GUG-psbA, ndhE-ndhG, ycf4-cemA, petD-rpoA, trnY_GUA-trnE_UUC intergenic spacers, and the rps16 intron) were found in 6 Lupinus species. Except for ndhE-ndhG that was located within the SSC, the other seven areas were all located in the LSC region, which meant the IR region was more conserved than the LSC and SSC regions. And these 8 highly variable regions could be used as phylogenetic markers in L. westianus, which will contribute to the study on the origin of Lupinus in the New World.
Selection events of protein coding genes in Lupinus species
Previous studies have identified the function of the genes contained in most chloroplast DNA by comparing various plant chloroplast DNA sequences and homologous sequences in other organisms (Yan et al. Citation2002). The genes found in L. westianus were divided into three categories according to their functions (Supplementary Table S2). The first category is related to their self-replication, the second category is related to photosynthesis, and the third category is related to other genes. Among these genes, the matK and ycf1 genes were under positive selection, and rpoA, accD, and rpoC2 genes have site-specific selection under positive selection in L. westianus. The plastid accD gene encodes the carboxyl transferase subunit of acetyl-CoA carboxylase and is present in the plastids of most flowering plants (Kode et al. Citation2005). Previous studies have shown that tobacco plastid accD gene is essential for leaf development, which can prolong leaf life and improve seed yield (Kode et al. Citation2005; Yuka et al. Citation2002). The rpoC2 and rpoA gene encode one of the four subunits of RNA polymerase type I (plastid-encoded polymerase, PEP), which is a key enzyme required for transcription of photosynthesis-related genes in the chloroplast (Cummings et al. Citation1994; Yi et al. Citation2018). In the case of rpoA, Kim's research shows that the polycistronic rpoA transcripts increase the stability of chloroplast development in dark-grown plants (Kim et al. Citation1993). As the second largest gene in the plastid genome, ycf1 encodes a protein of approximately 1,800 amino acids. Recent experiments showed that ycf1 is essential for plant viability (Dong et al. Citation2015; Kikuchi et al. Citation2013). The 102 amino acid positions at the carboxyl terminus of matK gene are structurally related to portions of maturase-like polypeptide and might be involved in splicing Group II intron (Yi et al. Citation2018; Georg et al. Citation1993; Neuhaus and Link Citation1987). Therefore, these five genes might be involved in the chloroplast self-replication and plant development of L. westianus.
A study on seed plants shows that genes affected by positive selection always participate in the plant's adaptation process (Zheng et al. Citation2017). Five genes under positive selection might be involved in the adaptation process of L. westianus. In this study, Lupinus from the New-Old World not only have different geographical distribution but also habitats and morphological characteristics. (Supplementary Table S3). For example, L. westianus living in subtropical, tropical dry, or coastal dune in Florida, while the habitats of L. princei is tropical highlands of Kenya, Tanzania, and southern Ethiopia at elevations between 1,700 and 3,000 m. Different habitats lead to changes in the morphology of Lupinus species in the Old and New World. Five Lupinus species from the Old World are all annual herbaceous plants bearing palmate compound leaves; those from the Mediterranean have smooth seed coats (L. albus, L. micranthus, L. luteus), whereas some of those from Africa present rough seed coats (L. princei, L. atlanticus). In contrast, the New World lupine, L. westianus, an endemic species to Florida, is a perennial subshrub plant bearing palmate compound leaves and a smooth seed coat (Ainouche et al. Citation2004). Therefore, the Lupinus species might have produced some corresponding differentiation in morphology to adapt to different habitats during the long geographical evolution process, and these five genes that are under positive selection might be related to it. Our identification of the positively-selected genes in this analysis could lead to a better understanding of the geographical evolution of Lupinus species.
Phylogenetic relationship between Lupinus species
Previous studies have shown that Lupinus plants originated in the Old World and later spread and diversified in the New World (Ainouche and Bayer Citation1999). It is also generally accepted that geographical distribution may be closely related to genetic relationships (Ayele et al. Citation2009). Therefore, the genetic relationship between the Lupinus species from the New World and Old World may be related to the geographical distribution characteristics. This is confirmed by Ainouche, who believes that western New World species may be closer to Old World species than eastern New World species. This is also suggested by the existing relationship between L. albus and L. atlanticus which are widely related to western New World species (Ainouche and Bayer Citation1999; Eastwood and Hughes Citation2008). In this study, the phylogenetic tree showed that L. westianus and L. albus were on the same clade, with 100% bootstraps values, suggesting that L. albus was most closely related to the New World species among the Old world species. Besides, the ML and NJ trees also revealed that the Old World species L. atlanticus and L. princei were on the same branch, which is consistent with the results obtained by Gladstones (Citation1984). And we found their geographical distribution is similar (only distributed in Africa), which is indirectly confirmed that the genetic relationship of Lupinus was related to its geographical distribution.
Except for the geographical distribution, there are a series of evidence of chromosome number, morphology, serology, genetics, isozyme, and interspecific crossing ability, which show that the rough-seeded species from the Old World are very homogeneous and the most supportive clade in the Old World group (Ainouche and Bayer Citation1999; Giovanni Citation1989; Wolko and Weeden Citation1990; Gupta et al. Citation1996). As the rough-seeded species from the Old World, we found L. atlanticus and L. princei have similar morphological traits, the same number of chromosomes (2n = 38). In contrast, the other three species bearing smooth seed coats(including L. albus, L. luteus, and L. micranthus) were located in different clades, which may be because smooth-seeded species are far more heterogeneous than rough-seeded ones (Käss and Wink Citation1997). Furthermore, L. micranthus was located somewhere between smooth-seeded and rough-seeded Old World Lupinus species, consistent with a study by Williams et al (Citation1983; Cristofolini Citation1989; Wolko and Weeden Citation1990). However, there is only one New World Lupinus species whose complete cp genome has been sequenced, which leads to the lower bootstrap values between the New and Old World species. More complete cp genomes data of Lupinus species from the New World should be included in the future. Nevertheless, our phylogenetic studies provided a valuable resource that should contribute to the future taxonomy, phylogeny, and evolutionary history studies of the Lupinus.
Conclusions
In this study, the complete cp genomes of one New world species and five Old World species of Lupinus were selected to compare the genomic variation and evolutionary characteristics of these species. This study provided new insights into the evolutionary dynamics of the poorly studied Genistoid clade. Our results suggested that the New World Lupinus species might have experienced more positive selection in its cp genome and accelerated evolution in its protein-coding genes than the Old World species. The matK and ycf1 genes were under positive selection, and the rpoA, accD, and rpoC2 genes have site-specific under positive selection, which might be related to the adaptive evolution of Lupinus species. Phylogenetic analysis showed that L. westianus and L. albus were included on the same clade and the rough-seeded species from the Old World are very homogeneous. It is expected that this research will attract a high number of researchers toward the New World Lupinus species, leading to the identification of new evidence on the evolution of the chloroplast genome of New World Lupinus species.
Supplemental Material
Download PDF (154.5 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in National Center of Biotechnology Information at https://www.ncbi.nlm.nih.gov/, accession number are MG252262, KJ468099, KU726827, NC023090, KU726828, KU726829, NC045070, NC0-42688.
Additional information
Funding
References
- Ainouche A, Bayer RJ. 1999. Phylogenetic relationships in Lupinus(Fabaceae: Papilionoideae) based on internal transcribed spacer sequences (ITS) of nuclear ribosomal DNA. Am J Bot. 86:590–607. DOI:10.2307/2656820
- Ainouche A, Bayer RJ, Misset MT. 2004. Molecular phylogeny, diversification and character evolution in Lupinus (Fabaceae) with special attention to Mediterranean and African lupines. Plant Syst Evol. 246:211–222. DOI:10.1007/s00606-004-0149-8
- Alikhan N-F, Petty NK, Zakour NLB, Beatson SA. 2011. BLAST ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 12:402. DOI:10.1186/1471-2164-12-402
- Ayele TB, Gailing O, Umer M, Finkeldey R. 2009. Chloroplast DNA haplotype diversity and postglacial recolonization of Hagenia abyssinica (Bruce) JF Gmel. in Ethiopia. Plant Syst Evol. 280:175–185. DOI:10.1007/s00606-009-0177-5
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27:573–580. DOI:10.1093/nar/27.2.573
- Bruneau A, Doyle JJ, Herendeen P, Hughes C, Kenicer G. 2013. Legume phylogeny and classification in the 21st century: progress, prospects and lessons for other species-rich clades. Taxon. 62:217–248. DOI:10.12705/622.8
- Bull LN, Pabón-Peña CR, Freimer NB. 1999. Compound microsatellite repeats: practical and Theoretical features. Genome Res. 9:830–838. DOI:10.1101/gr.9.9.830
- Bupp G, Ricono A, Peterson CL, Pruett CL. 2017. Conservation implications of small population size and habitat fragmentation in an endangered lupine. Conservation Genetics. 18:77–88. DOI:10.1007/s10592-016-0883-9
- Cardoso D, de Queiroz LP, Pennington RT, de Lima HC, Fonty E, Wojciechowski MF, Lavin M. 2012. Revisiting the phylogeny of papilionoid legumes: new insights from comprehensively sampled early-branching lineages. Am J Bot. 99:1991–2013. DOI:10.3732/ajb.1200380
- Cardoso D, Pennington RT, de Queiroz LP, Boatwright JS, Van Wyk BE, Wojciechowski MF, Lavin M. 2013. Reconstructing the deep-branching relationships of the papilionoid legumes. S Afr J Bot. 89:58–75. DOI:10.1016/j.sajb.2013.05.001
- Cristofolini G. 1989. A serological contribution to the systematics of the genus Lupinus (Fabaceae). Plant Syst Evol. 166:265–278. DOI:10.1007/bf00935955
- Cronk Q, Ojeda I, Pennington RT. 2006. Legume comparative genomics: progress in phylogenetics and phylogenomics. Curr Opin Plant Biol. 9:99–103. DOI:10.1016/j.pbi.2006.01.011
- Cummings MP, King LM, Kellogg EA. 1994. Slipped-strand mispairing in a plastid gene: rpoC2 in grasses (Poaceae). Molecular Biology Evolution & Development. 11:1–8.
- Darling AC, Mau B, Blattner FR, Perna NT. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14:1394–1403. DOI:10.1101/gr.2289704
- Dong W, Xu C, Li C, Sun J, Zuo Y, Shi S, Cheng T, Guo J, Zhou S. 2015. Ycf1, the most promising plastid DNA barcode of land plants. Sci Rep. 5:8348–8353. DOI:10.1038/srep08348
- Dugas DV, Hernandez D, Koenen EJM, Schwarz E, Straub S, Hughes CE, Jansen RK, Nageswara-Rao M, Staats M, Trujillo JT. 2015. Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci Rep. 5:16958. DOI:10.1038/srep16958
- Eastwood RJ, Hughes CE. 2008. Origins of domestication of lupinus mutabilis in the Andes. In Proceedings of the 12th International Lupin Conference, Fremantle,Western Australia, 14–18 Sept; pp. 373–379.
- Edwards TJ. 2007. Legumes of the world. S Afr J Bot. 73:272–273. DOI:10.1016/j.sajb.2007.02.187
- Georg M, Perlman PS, Lambowitz AM. 1993. Evolutionary relationships among group II intron-encoded proteins and identification of a conserved domain that may be related to maturase function. Nucleic Acids Res. 21:4991–4997. DOI:10.1093/nar/21.22.4991
- Gepts P. 2005. Legumes as a model plant family. Genomics for food and feed report of the cross-legume advances through genomics conference. Plant Physiol. 137:1228–1235. DOI:10.1104/pp.105.060871
- Gladstones J. 1984. Present situation and potential of Mediterranean/African lupins for crop rotation. In Proceedings of the 3rd International Lupin Conference, La Rochelle, France, 4–8 June; pp. 18–37.
- Gupta S, Buirchell BJ, Cowling WAJPB. 1996. Interspecific reproductive barriers and genomic similarity among the rough-seeded Lupinus species. Plant Breed. 115:123–127. DOI:10.1111/j.1439-0523.1996.tb00886.x
- Hiratsuka J, Shimada H, Whittier R, Ishibashi T, Sakamoto M, Mori M, Kondo C, Honji Y, Sun C-R, Meng B-Y. 1989. The complete sequence of the rice (oryza sativa) chloroplast genome: intermolecular recombination between distinct tRNA genes accounts for a major plastid DNA inversion during the evolution of the cereals. MGG Molecular & General Genetics. 217:185–194. DOI:10.1007/bf02464880
- Huang L-S, Sun Y-Q, Jin Y-Q, Gao Q. 2018. Development of high transferability cpSSR markers for individual identification and genetic investigation in cupressaceae species. Ecol Evol. 8:4967–4977. DOI:10.1002/ece3.4053
- Ivanova Z, Sablok G, Daskalova E, Zahmanova G, Apostolova E, Yahubyan G, Baev V. 2017. Chloroplast genome analysis of resurrection tertiary relict haberlea rhodopensis highlights genes important for desiccation stress response. Front Plant Sci. 8:15. DOI:10.3389/fpls.2017.00204
- Jeon J-H, Kim S-C. 2019. Comparative analysis of the complete chloroplast genome sequences of three closely related East-Asian wild roses (Rosa sect. Synstylae; Rosaceae). Genes (Basel). 10:23.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. Modelfinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14:587–589. DOI:10.1038/nmeth.4285
- Käss E, Wink M. 1997. Molecular phylogeny and phylogeography of Lupinus (Leguminosae) inferred from nucleotide sequences of therbcL gene and ITS 1 + 2 regions of rDNA. Plant Syst Evol. 208:139–167. DOI:10.1007/BF00985439
- Katoh K, Standley DM. 2013. MAFFT multiple Sequence Alignment Software Version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780. DOI:10.1093/molbev/mst010
- Kelchner SA. 2000. The evolution of non-coding chloroplast DNA and its application in plant systematics. Ann Mo Bot Gard. 87:482–498. DOI:10.2307/2666142
- Keller J, Rousseau-Gueutin M, Martin GE, Morice J, Boutte J, Coissac E, Ourari M, Ainouche M, Salmon A, Cabello-Hurtado F, et al. 2017. The evolutionary fate of the chloroplast and nuclear rps16 genes as revealed through the sequencing and comparative analyses of four novel legume chloroplast genomes from Lupinus. DNA Res. 24:343–358. DOI:10.1093/dnares/dsx006
- Kikuchi S, Bedard J, Hirano M, Hirabayashi Y, Oishi M, Imai M, Mai T, Ide T, Nakai M. 2013. Uncovering the protein translocon at the chloroplast inner envelope membrane. Science. 339:571–574. DOI:10.1126/science.1229262
- Kim M, Christopher DA, Mullet JE. 1993. Direct evidence for selective modulation of psbA, rpoA, rbcL and 16S RNA stability during barley chloroplast development. Plant Mol Biol. 22:447–463. DOI:10.1007/BF00015975
- Kimura M. 1989. The neutral theory of molecular evolution and the world view of the neutralists. Genome. 31:24–31. DOI:10.1139/g89-009
- Kode V, Mudd EA, Iamtham S, Day A. 2005. The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 44:237–244. DOI:10.1111/j.1365-313X.2005.02533.x
- Kuang DY, Wu H, Wang YL, Gao LM, Lu L. 2011. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome. 54:663–673. DOI:10.1139/g11-026
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 33:1870–1874. DOI:10.1093/molbev/msw054
- Kurtz S, Schleiermacher C. 1999. REPuter: fast computation of maximal repeats in complete genomes. Bioinformatics. 15:426–427. DOI:10.1093/bioinformatics/15.5.426
- Li X, Zuo Y, Zhu X, Liao S, Ma J. 2019. Complete chloroplast genomes and comparative analysis of sequences evolution among seven Aristolochia (Aristolochiaceae) medicinal species. Int J Mol Sci. 20:1045–1068.
- Liu LX, Li R, Worth JRP, Li X, Li P, Cameron KM, Fu CX. 2017. The complete chloroplast genome of Chinese bayberry (Morella rubra, Myricaceae): implications for understanding the evolution of fagales. Front Plant Sci. 8:968. DOI:10.3389/fpls.2017.00968
- Maciel HS, Schifino-Wittmann MT. 2002. First chromosome number determinations in south-eastern South American species of Lupinus L. (Leguminosae). Bot J Linn Soc. 139:395–400. DOI:10.1046/j.1095-8339.2002.00071.x
- Mehmood F, Abdullah UZ, Bao Y, Poczai P, Mirza B. 2020b. Comparative plastomics of Ashwagandha (Withania, Solanaceae) and identification of mutational hotspots for barcoding medicinal plants. Plants-Basel. 9. DOI:10.3390/plants9060752
- Mehmood F, Abdullah, Shahzadi I, Ahmed I, Waheed MT, Mirza B. 2020a. Characterization of Withania somnifera chloroplast genome and its comparison with other selected species of Solanaceae. Genomics. 112:1522–1530. DOI:10.1016/j.ygeno.2019.08.024
- Mehmood F, Abdullah UZ, Shahzadi I, Ahmed I, Waheed MT, Poczai P, Mirza B. 2020c. Plastid genomics of Nicotiana (Solanaceae): insights into molecular evolution, positive selection and the origin of the maternal genome of Aztec tobacco (Nicotiana rustica). Peerj. 8:30. DOI:10.7717/peerj.9552
- Naydenov KD, Naydenov MK, Alexandrov A, Vasilevski K, Gyuleva V, Matevski V, Nikolic B, Goudiaby V, Bogunic F, Paitaridou D, et al. 2016. Ancient split of major genetic lineages of european black pine: evidence from chloroplast DNA. Tree Genetics & Genomes. 12:61–68. DOI:10.1007/s11295-016-1022-y
- Neuhaus H, Link G. 1987. The chloroplast tRNALys(UUU) gene from mustard (sinapis alba) contains a class II intron potentially coding for a maturase-related polypeptide. Curr Genet. 11:251–257. DOI:10.1007/BF00355398
- Njuguna AW, Li Z-Z, Saina JK, Munywoki JM, Gichira AW, Gituru RW, Wang Q-F, Chen J-M. 2019. Comparative analyses of the complete chloroplast genomes of nymphoides and menyanthes species (menyanthaceae). Aquat Bot. 156:73–81. DOI:10.1016/j.aquabot.2019.05.001
- Planchuelo-Ravelo AM. 1984. Taxonomic studies of Lupinus in South America. In Proceedings of the 3rd International Lupin Conference, La Rochelle, France, 4–8 June; pp. 39–53.
- Qu XJ, Moore MJ, Li DZ, Yi TS. 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15:12. DOI:10.1186/s13007-019-0435-7
- Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sanchez-Gracia A. 2017. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 34:3299–3302. DOI:10.1093/molbev/msx248
- Schwarz EN, Ruhlman TA, Weng ML, Khiyami MA, Sabir JSM, Hajarah NH, Alharbi NS, Rabah SO, Jansen RK. 2017. Plastome-Wide nucleotide substitution rates reveal accelerated rates in Papilionoideae and correlations with genome features across legume subfamilies. J Mol Evol. 84:187–203. DOI:10.1007/s00239-017-9792-x
- Silva SR, Diaz YC, Penha HA, Pinheiro DG, Fernandes CC, Miranda VF, Michael TP, Varani AM. 2016. The chloroplast genome of utricularia reniformis sheds light on the evolution of the ndh gene complex of terrestrial carnivorous plants from the lentibulariaceae family. PLoS One. 11:e0165176. DOI:10.1371/journal.pone.0165176
- Tang J, Xia HA, Cao M, Zhang X, Zeng W, Hu S, Tong W, Wang J, Wang J, Yu J, et al. 2004. A comparison of rice chloroplast genomes. Plant Physiol. 135:412–420. DOI:10.1104/pp.103.031245
- Thiel T, Michalek W, Varshney RK, Graner A. 2003. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet. 106:411–422. DOI:10.1007/s00122-002-1031-0
- Wakasugi T, Tsudzuki J, Ito S, Nakashima K, Tsudzuki T, Sugiura M. 1994. Loss of all ndh genes as determined by sequencing the entire chloroplast genome of the black pine Pinus thunbergii. Proc Natl Acad Sci USA. 91:9794–9798. DOI:10.1073/pnas.91.21.9794
- Wang YH, Susann W, Hong W, Jin JJ, Chen SY, Zhang SD, Li DZ, Yi TSJ. 2018. Plastid Genome evolution in the early-diverging legume subfamily cercidoideae (fabaceae). Front Plant Sci. 9:138–149. DOI:10.3389/fpls.2018.00138
- Williams CA, Demissie A, Harborne JB. 1983. Flavonoids as taxonomic markers in old world Lupinus species. Biochem Syst Ecol. 11:221–231. DOI:10.1016/0305-1978(83)90058-3
- Wolko B, Clements JC, Naganowska B, Nelson MN, Yang H. 2011. Lupinus. In: Kole C., editor. Wild crop relatives: genomic and breeding resources, Legume crops and forages. Germany: Springer Berlin Heidelberg; p. 153–206.
- Wolko B, Weeden NF. 1990. Isozyme number as an indicator of phylogeny in Lupinus. Genetica Polonica. 31:179–187.
- Wunderlin R. 1982. The leguminosae: A source book of characteristics, uses, and nodulation. Econ Bot. 36:224–224. DOI:10.1007/BF02858721
- Xiao Z, Tao Z, Nazish K, Zhao Y, Bai G, Zhao G. 2017. Completion of eight gynostemma BL. (Cucurbitaceae) chloroplast genomes: characterization, comparative analysis, and phylogenetic relationships. Front Plant Sci. 8:1583. DOI:10.3389/fpls.2017.01583
- Xu Z, Zhao Y, Dong M, Dong C, Xue Y, Yang G. 2019. Characterization and phylogenetic analysis of the chloroplast genome of Lupinus westianus, a endemic species to Florida, United States. Conserv Genet Resour. 11:51–54. DOI:10.1007/s12686-017-0965-0
- Yan X, Cui ming L, Shu fang W, Ning ning W, Yong W. 2002. Chloroplast genome and the regulation of chloroplast-encoded gene expression. J Plant Physiol. 38:264–269. DOI:10.13592/j.cnki.ppj.2002.03.030
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24:1586–1591. DOI:10.1093/molbev/msm088
- Yi Z, Cui YL, Zhang XL, Yu QB, Xi W, Yuan XB, Qin XM, He XF, Chao H, Yang ZN. 2018. A nuclear-encoded protein, mTERF6, mediates transcription termination of rpoA polycistron for plastid-encoded RNA polymerase-dependent chloroplast gene expression and chloroplast development. Sci Rep. 8:11929–11941. DOI:10.1038/s41598-018-30166-6
- Yin K, Zhang Y, Li Y, Du FK. 2018. Different natural selection pressures on the atpF gene in evergreen sclerophyllous and deciduous Oak species: evidence from comparative analysis of the complete chloroplast genome of Quercus aquifolioides with other Oak species. Int J Mol Sci. 19. DOI:10.3390/ijms19041042
- Yuka M, Ken-Ichi T, Junya M, Ikuo N, Yukio N, Yukiko S. 2002. Chloroplast transformation with modified accD operon increases acetyl-CoA carboxylase and causes extension of leaf longevity and increase in seed yield in tobacco. Plant Cell Physiology. 43:1518–1525. DOI:10.1093/pcp/pcf172
- Zhang X, Zhou T, Yang J, Sun J, Ju M, Zhao Y, Zhao G. 2018. Comparative analyses of chloroplast genomes of cucurbitaceae species: lights into selective pressures and phylogenetic relationships. Molecules. 23. DOI:10.3390/molecules-23092165
- Zheng XM, Wang J, Li F, Sha L, Pang H, Lan Q, Jing L, Yan S, Qiao W, Zhang LJSR. 2017. Inferring the evolutionary mechanism of the chloroplast genome size by comparing whole-chloroplast genome sequences in seed plants. Sci Rep. 7:1555–1565. DOI:10.1038/s41598-017-01518-5