
Abstract
Acetyl xylan esterases are among the key enzymes in the xylan degradation enzyme system. However, acetyl xylan esterases from natural microorganisms have low expression and low enzyme activity and are impure. In this study, a new xylanase gene, est1051, from the metagenomic library, was expressed in the prokaryotic system. Its enzymatic properties were explored, including optimum temperature and pH, thermal and pH stability, and tolerance against organic solvents, metal ions and salt solutions. Then the fermentation conditions of EST1051 were optimized by the response surface method, and the maximum enzyme yield reached 1909.32 U/L. Finally, the synergism with cellulase on straw degradation was evaluated. EST1051 displays high homology with acetylxylan esterases in terms of amino acid sequences and conserved active sites. EST1051 shows high stability across a broad temperature range, and retains more than 60% of its enzymatic activity between 4 and 60°C after 24 h of incubation. Single-factor analysis and orthogonal design were conducted to determine the optimal conditions for the maximizing the saccharification rate of wheat straws. Interestingly, the synergism of EST1051 with cellulase contributes to the efficient transformation of wheat straws. These findings may open the door to significant industrial applications of this novel acetylxylan esterase.
Introduction
Acetyl xylan esterases (AXE, EC 3.1.1.72) are among the least known hemicellulose degrading enzymes. The acetyl group hinders the hydrolysis of xylanase to xylan. Deacetylation of acetylated xylan can improve the hydrolysis efficiency, and therefore has been widely used as a carbon source of microbial growth for long time. However, the ability of the microorganisms to produce AXEs is inhibited under this condition. Moreover, the discovery and research of AXEs are later than other lignocellulose degrading enzymes due to the lack of suitable substrates and the low contents. AXEs were first reported by Biely et al. (Citation1985). They also studied the enzymatic properties, fermentation expression and purification of acetylxylanase (Biely et al. Citation1988a, Citation1986, Citation1988b). Subsequently, more reports about acetylxylanase show that AXEs come from a wide range of sources, mostly from fungi (Trichoderma reesei [Biely et al. Citation1997], Coriolus versicolor [Tsujiyama and Nakano Citation1996], Schizophyllum commune [Halgasová et al. Citation1994], Aspergillus versicolor [Prashant et al. Citation2016], rumen fungi [Cybinski et al. Citation1999], Streptomyces lividans [Dupont et al. Citation1996], Streptomyces albicans [Wenjing et al. Citation2012]) and bacteria (Albus [Weina et al. Citation2015], Streptomyces griseus [Krastanova et al. Citation2005], Bacillus pumilus [Adesioye et al. Citation2018]). The CAZy database divides carbohydrate enzymes into 16 families, among which acetylxylanase belongs to 9 families according to sequence similarity, namely CE 1-7, CE 12 and CE 15. For example, AXEs from Aspergillus sp. & Volvariella volvacea, Vibrio japonicus, Talaromyces Cellulolyticus (Watanabe and Ishikawa Citation2014), Bacillus thermophilus (Aysenur et al. Citation2015) and Trichoderma reesei belongs to CE 1, CE 2, CE 3, CE 4 and CE 5 respectively. Some AXEs from bacteria have activity towards cephalosporin C and belong to CE 7. Xylanase is structurally similar to other glycohydrolase family proteins, and generally consists of a functional domain, a nonfunctional domain and a junction domain. The functional domain refers to a catalytic domain (a key region of endoxylanase and hydrolyzing the substrate) and a substrate binding domain (Fujimoto et al. Citation2000). The substrate binding domain refers to a cellulose binding domain and an xylan binding domain (XBD) (Kuno et al. Citation2000). The function of XBD is to combine endoxylanase with the xylan substrate. AXEs have the same conserved sequence, G-X-S-X-G (Hea et al. Citation2002), as the serine esterase family. The substrate binding site of AXEs is very small and only contains one acetyl group. Thus, the specificity of the enzyme is maintained (Hakulinen et al. Citation2000, Citation1998). As far as we know, many acetyl xylanases (Ding et al. Citation2007), (Koseki et al. Citation2006; Huy et al. Citation2013) reported so far can be hardly used industrially due to their lack of stability and activity. Thermostable AXEs with excellent properties still need to be discovered.
Nonrenewable resources are gradually exhausted, which has caused serious environmental pollution (Aiguo and Hao Citation2010). The demand for better development and utilization of renewable resources is increasing worldwide (Kirtay Citation2009). With the continuous progress of Chinese agriculture, the total output of crop straws is also rising year by year. However, only a small part of crop straws is returned to the field or used in feeding and industrial applications (Miao et al. Citation2020), while the rest is directly burned. Straws are mainly composed of cellulose, lignin and hemicellulose, of which hemicellulose accounts for about 30% (Huang et al. Citation2010). Hemicellulose is the second largest renewable biomass resource after cellulose in plant cell walls (Prade Citation1996). The original soluble sugar and hydrolyzed small- molecular sugar in hemicellulose can be further fermented into ethanol (Ghysel et al. Citation2010). Hemicellulose also contains a variety of side chains and various side chains of lignin. Cellulose is wrapped inside, which reduces the accessibility of cellulose and enzymes. Therefore, degradation of hemicellulose can also improve the utilization of cellulose. Synergism between acetyl xylanase and cellulase can effectively improve the conversion rate of biomass sugar, which lowers the costs of biofuels (Agrawal et al. Citation2015a, Citation2015b, Citation2018). Treatment of hemicelluloses by organic solvents or other nonbiological methods will result in large loss of hemicellulose, high energy consumption and high equipmental cost. Therefore, enzymatic treatment of hemicellulose is a feasible, green and reasonable way. Despite some studies on enzymatic degradation of straws (Tamilvendan et al. Citation2017; Shohreh et al. Citation2020), the AXE with efficient transformation of wheat straw is rarely reported. In this work, the saccharification rate reached 76.12% when EST1051 and cellulase reacted with alkali-pretreated wheat straw for 8 h.
In this study, EST1051 was processed by sequencing and multiple sequence alignment. Results indicate it is a novel AXE. EST1051 shows excellent thermal stability and tolerance against metal ions, organic solvents and salt solutions, and has excellent wheat straw degrading ability and great potential for industrial application.
Materials and methods
Strains, materials, and chemicals
EST1051 and Cel1029 were collected from the soil on the northern margin of the Junggar Basin in Altai Mountains, Xinjiang Province, China (44°30′ N, 86°05′ E). The strains with esterase and cellulase properties were screened in our laboratory. The strain with AXE properties was preliminarily identified through 16S rDNA sequencing and morphological, physiological and biochemical characterizations as Clostridium saccharobutylicum and stored at – 80 °C. E. coli BL21 (DE3) and E. coli DH5α purchased from TSINGKE Biological Technology (Guangzhou, China) were used as the expression host and cloning host, respectively. The pET-28a (+) was used for protein expression. T4 DNA ligases, restriction endonucleases, and DNA and protein markers were bought from TaKaRa (Dalian, China). The DNA extraction kit and plasmid extraction kit were purchased from OMEGA (San Diego, CA, USA). Kanamycins (Kan), 5-bromo-4-chloro-3-indolyl β-D-galactopyranoside (X-Gal), IPTG and ρ-nitropheno (ρNP)-C2 were provided by Sigma. Tryptone and yeast extract powder were offered by British Oxoid, and agar powder was purchased from Coolable. Electrophoretic agarose and Tris were produced by PUBO Biological Technology (Beijing, China). Commercial acetylxylanase (Item: EAXEAO; Orpinomyces sp.) was purchased from Shanghai Jingke Chemical Technology Co., Ltd. Other chemical reagents were domestic and analytically pure. Gene sequencing and primer synthesis were entrusted to Guangzhou TSINGKE Biological Technology.
Preparation of wheat straw biomass (WSB)
Wheat straw was collected from rural areas in Heze, Shandong Province, China. Then the wheat straw was chopped, dried at 105°C, and finally ground into 0.1-1 mm powder. A certain amount of WSB was taken, and added with a dilute NaOH solution (2%) at the ratio of 1:10 (g/ml) under soaking for 1 h at 40°C. Then the dilute NaOH was drained, and the WSB was neutralized by washing with distilled water (Coniglio et al. Citation2020).
Construction of recombinant plasmid and transformation
In PCR amplification, pUC118-est1051 plasmid was adopted as the template, and est1051-F and est1051-R as primers. The amplified product and vector, pET-28a (+), was digested with EcoR I and Xhol I, and then linked by T4 DNA ligase. Finally, the constructed vector, pET28a-est1051, was transferred into E.coli BL21 (DE3). PCR primers for est1051 amplification were as follows: est1051-F, 5’-CCGGAATTCCCC GTTGCGGTGTCATTATACTGAC-3’; est1051-R, 5’-CCGCTCGAGGCTCCCGAAGAACC TATGAAACCAATTG-3’ (the EcoR I and Xhol I restriction sites were italicized). The PCR is as follows: denaturation for 2 min at 98°C; 30 cycles of 10 sec at 98°C, 5 sec at 64°C, and 5 sec at 72°C; elongation for 8 min at 72°C. The expression vector pET-28a (+) was digested using EcoR I and Xhol I at 37°C for 20 min and then ligated to the PCR products, which treated with the same restriction endonuclease. The double-digested expression vector pET-28a (+) and the PCR products were ligated by TaKaRa T4 DNA ligase at 16°C for 14 h. This recombinant plasmid was transformed into E. coli BL21 (DE3) by a heat shock method.
Expression, protein purification, and electrophoresis
The protein was expressed by growing the E. coli BL21(DE3) cells in an LB medium (50 µg/mL Kan). For purification of the EST1051 expressed by E. coli, the sediments were collected by centrifugation at 8000 rpm and 4°C for 10 min. Then the sediments were washed with sterile water twice and broken using an ultrasonicator with amplitude at 30 W for 15 min. The recombinant AXE EST1051 was purified using His-tag protein purification kit (Novagen). Molecular weight of the denatured protein was determined by SDS-PAGE and based on the protein markers (TaKaRa) as the standards. According to the predicted molecular weight of the protein and its relationship with different concentrations of polyacrylamide gels, 12% separation gels and 5% concentration gels were selected. The protein was stained with Coomassie brilliant blue G-250 for 10-12 h, and then decolorized with eluent.
AXE activity assay and substrate specificity
AXE activity was measured using ρ-nitrophenol esters as the substrates (Bahiru et al. Citation2020), since AXE can decompose the ester bond in ρ-nitrophenol esters to form yellow ρNP. Moreover, the absorption value of ρNP at 405 nm (A405) is linearly related with the ρNP content. First, a standard curve was drawn using ρNP content as the X axis, and A405 as the Y axis. Then ρNP content was determined through the standard curve by measuring the A405 of the solution. One unit is defined as the amount of an enzyme to release 1 μmol of ρNP per minute under the assayed conditions (Cheeseman et al. Citation2001). Seven substrates at 1 mM were prepared, including ρ-nitrophenol acetate (ρNP-C2), ρ-nitrophenol butyrate (ρNP-C4), ρ-nitrophenol hexanoate (ρNP-C6), ρ-nitrophenol octanoate (ρNP-C8), ρ-nitrophenol decanoate (ρNP-C10), ρ-nitrophenol laurate (pNP-C12) and ρ-nitrophenol myristate (ρNP-C14). Then 190 μL of each substrate prepared with the 40 mM Britton–Robinson (BR) buffer at pH 7 was taken and added with 10 μL of the purified enzyme solution for 20 min of reaction. After that, the supernatant was collected to determine A405. Finally, the optimal substrate can be determined. The maximum enzyme activity was set at 100%. All assays were performed in triplicate.
Evaluation of enzymatic properties
AXE activity was measured using ρ-nitrophenol esters as the substrates from pH 2–10 and at 40°C. Firstly, 190 μL of 1 μmol/L ρNP-C2 prepared with the 40 mM BR buffer of different pHs was taken, and added with 10 μL of the purified enzyme solution for 20 min of reaction. Then the supernatant was collected to determine the A405 and obtain the optimal pH. The optimal temperature was investigated from 4 to 80°C at the optimal pH. A crude enzyme solution (2 mL) was prepared with the BR buffer solution of different pHs, and stored at 4°C for 24 h. Then in 10 μL of the crude enzyme solution was taken, the residual enzyme activity was detected under the optimum temperature and pH, and the maximum enzyme activity was set as 100%. After the enzyme solution was treated at different temperatures (4-80°C) for 24 h, the residual enzyme activity at the optimum temperature and pH was measured according to the method above.
The effects of various metal ions (Ni2+, Na+, K+, Mg2+, Zn2+, Fe2+, Mn2+, Li+, Ag+, Co2+, Cu2+) and chemicals (methanol, ethanol, isopropanol, DMSO, Triton X-100) on EST1051 activity were investigated by preincubating the enzyme for 20 min at the optimum temperature and pH. The enzyme activity without any reagent addition was set as 100%. NaCl was used to study the salt tolerance of the recombinant enzyme. The enzyme activity was determined with salt solutions from 0 to 3 mM at the optimal temperature and pH. The solution with no agent added was used as a control.
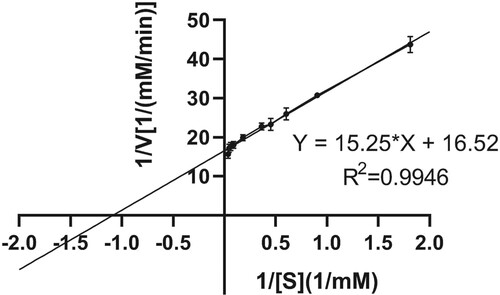
The kinetic constants Km and Vmax of AXE were determined using ρNP-C2 as the substrate. Then ρNP-C2 solutions with the concentration from 0.0–5.0 mg/mL were prepared with the optimal pH buffer solution. Firstly, 190 μL of the ρNP-C2 solution with different concentrations was taken and each added with 10 μL of the 0.62 mg/ml enzyme solution for 20 min of reaction under the optimal temperature. Then the reaction solutions were taken out to measure A405. The ρNP-C2 concentration was determined with the absorption value at 0.0 mM as the negative control. According to the substrate concentration [S] and the measured initial reaction rate V, the Linear Weaver Burk double reciprocal plot (V = Vmax [S]/(Km + [S])) was drawn for 1/V, and the enzymatic kinetic parameters Km and Vmax of AXE were calculated.
Design and optimization of EST1051 fermentation conditions
Firstly, the factors that have significant influence on the EST1051 yield were determined by the Plackett-Burman design (N = 12, 11 factors). The 11 factors included 7 actual factors (triglyceride, yeast powder, tryptone, inoculation quantity, liquid volume, Na2HPO4 and MgSO4) and 4 virtual factors (Table ). Then Box–Behnken design with N = 17 on Design-Expert 10.0 was used (Table ). Finally, the experimental results were statistically analyzed to study the interaction among the three factors and the optimal value, and the optimal enzyme production conditions were predicted. The enzyme activity was taken as the standard, and all assays were performed in triplicate.
Table 1. Plackett-Burmen level range of experimental design.
Table 2. Experimental ranges and levels with Box–Behnken design.
Enzymatic hydrolysis of WSB
The absorption value is linearly related to the content of reducing sugar. A standard curve was drawn using the glucose concentration as the X axis, and A540 as the Y axis. Reducing sugar content can be obtained through the standard curve by measuring the A570 of the solution.
The optimal conditions were tested with WSB. The wheat straw was rich in lignin, cellulose, and hemicellulose that were converted to a reducing sugar by cellulase and AXE. First, single-factor analysis was used to study WSB degradation by changing the pH, temperature, enzyme dosage, and enzymatic hydrolysis time. Then an L9(34) orthogonal design (Table ) was used to determine the optimal degradation combination. The reaction mixtures contained the WSB (0.2 g), BR buffer (40 mM), cellulase Cel1029 and EST1051 enzyme solution (v/v, 1:1) in a 20-mL system at 200 rpm. A 0.01 mg/mL commercial AXE solution was prepared with the BR buffer (40 mM), and the mixed enzyme solution of Cel1029 and commercial AXE was used as a positive control. Enzymatic extraction of WSB was determined by spectrophotometry. The supernatant was centrifuged to measure the absorbance at 570 nm. All reactions were performed in triplicate.
Table 3. Orthogonal test factors at levels 1, 2 and 3 from low to high.
Nucleotide sequence accession number
This nucleotide sequence was declared to the GenBank with the registration number as MK111431.
Results
Genetic characterization
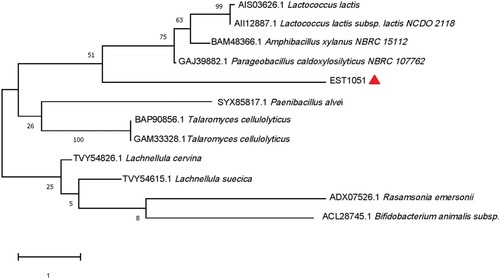
The est1051 was sequenced and analyzed with ORFfinder of the National Center for Biotechnology Information (NCBI). Results show the full length of the ORF sequence is 1051 bp containing AXE gene named est1051. The est1051 encodes a 35.07 kDa protein containing 307 amino acids. The basic properties of EST1051 were predicted by the online program ProtParam from ExPASy. The isoelectric point, instability index and average hydrophilicity are 5.04, 38.71 and −0.481 respectively. EST1051 is an amphoteric stable protein. Multiple sequence alignment and protein BLAST search by BioEdiet, ESPript 3.0 and NCBI demonstrate that EST1051 is similar to acetylxylan esterase (81.40%, NCBI accession: WP_189603113.1), acetylxylan esterase (67.24%, WP_089683842.1), acetylxylan esterase (66.55%, OGP96767.1), and acetylxylan esterase (64.69%, WP_074225788.1). These sequences are strictly conserved with G-X-S-X-G and catalytic triad Ser22-His145-Asp218 (Figure ). The catalytic triad forms the catalytic active center of AXE. A phylogenetic tree was constructed by the neighbor joining method on MEGA-X to verify the evolutionary relationship of EST1051 with 11 known AXEs. Results show that EST1051 has low affinity to the 11 known AXEs (Figure ). Multiple sequence alignment and phylogenetic tree analysis suggest that EST1051 is a new AXE (EC3.1.1.72).
Figure 1. Multiple amino acid sequence alignment of EST1051 with its closest homologues. Sequences of all closest homologues were obtained from NCBI database. WP_189603113.1, WP_089683842.1, OGP96767.1, and WP_074225788.1: AXEs from Salinimicrobium marinum, Catalinimonas alkaloidigena, Deltaproteobacteria bacterium and Algoriphagus halophilus, respectively. Similar sequences were indicated by colored backgrounds. The six residue sites Ser22, Glu52, Ser54, Glu55, His145, and Asp218 labeled as * were strictly conserved in amino acid sequences of AXEs.
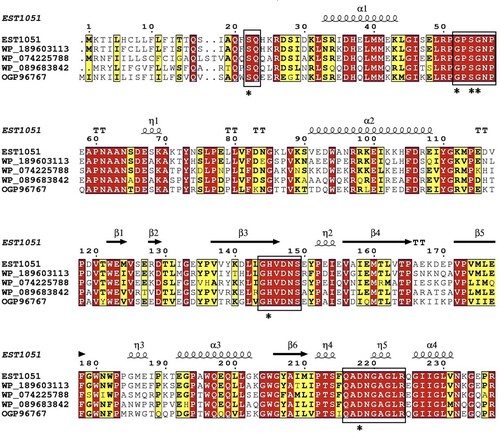
Figure 2. Phylogenetic tree of EST1051 and 11 known AXEs built by neighbor-joining. Evolutionary history was inferred using neighbor-joining by MEGA-X. Sequences of all AXEs were cited from the NCBI. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches
Cloning and overexpression of the AXE Gene in E. coli and purification of the recombinant protein
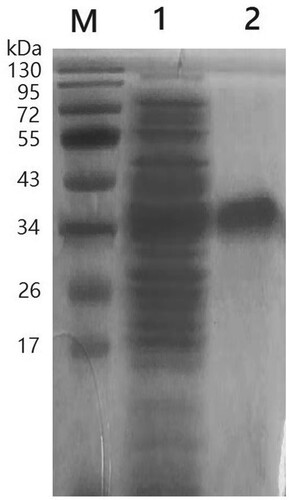
PCR was conducted by using plasmids containing pUC118-Est1051 as the template, and Est1051-F and Est1051-R as primers. The amplified fragment length is consistent with the predicted target DNA fragment length of 1051 bp. The pET28a-est1051 was identified by enzyme digestion verification (Figure ) and DNA sequencing confirmation. EST1051 was expressed in E. coli BL21 (DE3). According to the optimal induction temperature (37°C) and time (10 h), the optimal induction concentration of isopropyl β-D-1thiogalactopyranoside (IPTG) is 0.8 mM. The expression strain was induced by IPTG and then broken by ultrasound. The crude enzyme was purified and then analyzed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) (Wang et al. Citation2004). The enzyme was purified with a specific activity of 1046 ± 17 U/L. Figure shows the approximate target band (38.4 kDa), including theoretical calculation of cellulose molecular weight (35.07 kDa) and protein molecular tag weight (3.33 kDa). Results show the recombinant protein is highly soluble and expressed in E.coli, which is also the premise and basis for industrial application.
Figure 3. Electrophoresis identification of recombinant plasmid by EcoR I and Xhol I. M, DL45, 00 DNA Marker; Lane 1, Recombinant plasmid digestion by EcoR I and Xhol I; Lane 2, Recombinant plasmid. The target band is located on the upper side of 1000 bp.
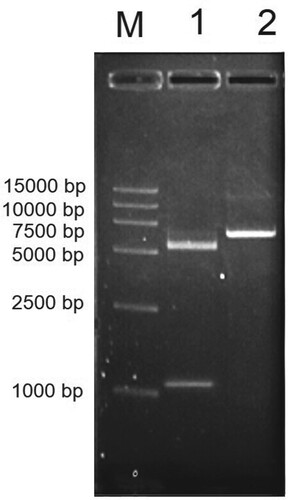
Figure 4. SDS-PAGE analysis of recombinant EST1051. M, Protein MW Marker; Lane 1, unpurified recombinant EST1051; Lane 2, purified recombinant EST1051. The approximate target band (38.4 kDa) including theoretical calculation of the cellulose enzyme molecular weight (35.07 kDa) and protein molecular tags weight (3.33 kDa).
Characterization of recombinant EST1051
The relative enzyme activity was the highest when ρNP-C2 was used as the substrate (Figure ). Therefore, ρNP-C2 was selected as the optimal substrate.
Figure 5. The substrate specificity of acetylxylan esterase EST1051. C2, C4, C6, C8, C10, C12, C14, C16 represent ρNP-C2, ρNP-C4, ρNP-C6, ρNP-C8, ρNP-C10, ρNP-C12, ρNP-C14, ρNP-C16 respectively; the maximum activity was set as 100%.
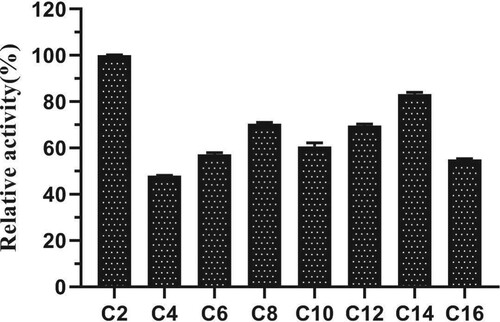
EST1051 displayed an optimal pH of 7.0 toward ρNP-C2. The enzyme activity remained above 60% after 24 h of storage in the buffer solutions at pH 5-8, indicating EST1051 is stale in acidic and neutral conditions (Figure A). The optimal reaction temperature of EST1051 at the optimal pH was measured with ρNP-C2 as substrate. The maximum activity was found at 40 °C. More than 60% of maximum activity was retained at 4 –60°C after 24 h (Figure B), indicating that AXE EST1051 has high adaptability at a wide temperature range.
Figure 6. Effects of (A) pH and (B) temperatures on activity and stability of EST1051. Data points are the average values of triplicate measurements, and error bars represent standard deviations.
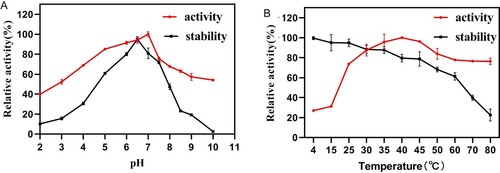
AXE activity was strongly enhanced in the presence of Mg2+ (Figure ). The influence of Mg2+ concentration on EST1051 was further studied. Results show 10 mM Mg2+ promoted enzyme activity to 121.18% and suggest EST1051 is resistant against most metal ions. Enzyme activity can be improved by adding appropriate metal ions in industrial production. The relative activity was maintained at above 80% in all tested organic solvents (Figure ). These results suggest that adding appropriate organic solvents in industrial production may enhance the enzyme activity. NaCl solutions (0 M to 3 M) were used to study the salt tolerance of the recombinant enzyme under the optimal temperature and pH. The enzyme activity was remained above 60% when the salt concentration was 3 M (Figure ), indicating that EST1051 has high tolerance against salt solutions. The kinetic constants Km and Vmax of AXE using ρNP-C2 as the substrate were determined to be 0.923 and 0.061 Mm/min, respectively (Figure ).
Figure 7. Effects of metal ions on enzyme activity of EST1051 using ρNP-C2 as the substrate. Enzyme activity was measured after 180 µL of 40 mM Britton-Robinson buffer (pH 7.0), 10 µL of 1 mM ρNP-C2 and 10 µL of the pure enzyme solution reacted at 40 °C for 20 min. At each time, the Britton-Robinson buffer contained a specific metal ion at the concentration of 1 or 10 mM. Enzymatic activity without metal ions was defined as 100%.
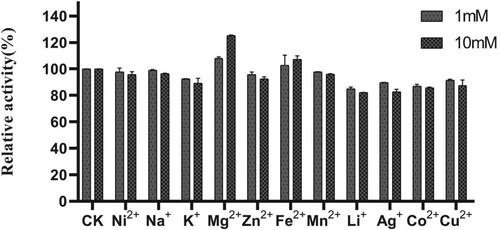
Figure 8. Effects of organic solvents on enzyme activity of EST1051 using ρNP-C2 as the substrate. Enzyme activity was measured after 180 µL of 40 mM Britton-Robinson buffer (pH 7.0), 10 µL of 1 mM ρNP-C2 and 10 µL of a pure enzyme solution reacted at 40 °C for 20 min. At each time, the Britton-Robinson buffer contained different a certain organic solvent with different concentrations. Enzymatic activity without organic solvents was defined as 100%.
Figure 9. Effects of NaCl on enzyme activity of EST1051 using ρNP-C2 as the substrate. Enzyme activity was measured after 180 µL of 40 mM Britton-Robinson buffer (pH 7.0), 10 µL of 1 mM ρNP-C2 and 10 µL of a pure enzyme solution racted at 40 °C for 20 min. At each time, the Britton-Robinson buffer contained NaCl with a specific concentration. Enzymatic activity without NaCl was defined as 100%.
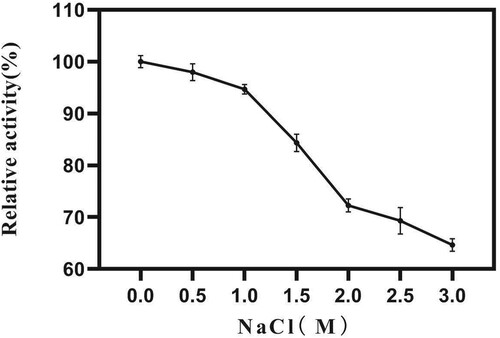
Figure 10. Kinetic parameters of AXE EST1051 using ρNP-C2 as the substrate. The linear regression equation is Y=15.25*X+16.52, R2=0.9946. The intersection points of the line with X and Y are – 1/Km and 1/Vmax, respectively.
Optimization of EST1051 fermentation conditions
The results of Plackett-Burman design display that the most significant effects on EST1051 fermentation are yeast powder, tributyrin, and inoculation quantity. The results of Plackett-Burman experiment and analysis of variance are shown in Tables and respectively. Box–Behnken design was used to optimize the significant factors. The results are shown in Table . A second-order polynomial regression equation was used to calculate the response value (R) of EST1051 yield (R = + 98.50 + 0.71 A + 0.33 B – 2.43 C + 1.28 AB – 1.28 AC + 6.16 BC – 2.28 A2 – 12.29 B2 – 14.78 C2). The analysis of variance for the response surface quadratic model was displayed in Table . The 3D response surface plots (Figure ) generated by the Box–Behnken design graphically represent regression equations. Results show the values of yeast powder, tributyrin and inoculation quantity are 15.90 g/L, 10.00 g/L and 2.91% respectively. Under the optimal conditions, the average AXE yield of three parallel experiments is 1909.32 U/L, which is close to the predicted maximum enzyme yield of 1931.06 U/L, indicating that the model is reliable.
Figure 11. Response surface and contour maps of the influence from (A) yeast powder and tributyrin, (B) yeast powder and inoculation quantity, and (C) yeast powder and inoculation quantity on the fermentation enzyme production.
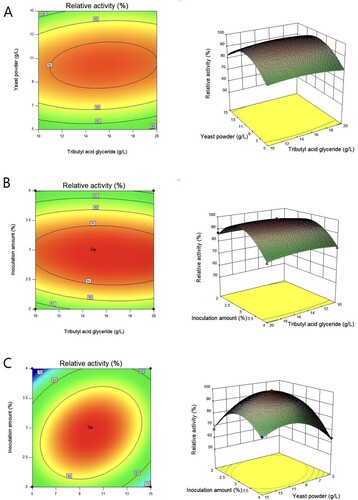
Table 4. Plackett-Burmen design and test results.
Table 5. Analysis of variance for Plackett-Burmen experiment.
Table 6. Box–Behnken design for EST1051 production.
Table 7. Analysis of variance for Box–Behnken experiment.
Enzymatic hydrolysis of WSB
WSB is rich in cellulose, lignin and hemicellulose, which are hydrolyzed into cellobiose or glucose by enzymes such as cellulase and xylan esterase. Cellulase and AXE can degrade cellulose, lignin and hemicellulose to produce cellulosic ethanol, and then produce bio-fuel by microbial fermentation, which can optimize the use of WSB as a resource (Aogaichi and Plaut Citation1977; Janssen et al. Citation1994). The optimal conditions of enzymatic hydrolysis to WSB were studied by using spectrophotometry. Figure shows the optimal hydrolysis conditions. In the experimental group, The saccharification rate reaches 72.58% at pH 6 (Figure A). As pH rises, the saccharification rate is decelerated. At pH 7.5, the saccharification rate is only 42.08%. At 65°C, the saccharification rate minimizes to 29.96%. The saccharification rate increases with the temperature drop and maximizes to 75.04% at 40°C (Figure B). The maximum saccharification rate is 72.13% with 5 mL of enzyme (EST1051 and cellulase, 1:1, v/v) (Figure C). Figure (D) shows the effect of enzymatic hydrolysis time on the saccharification rate. The saccharification rate of WSB maximizes to 70.33% when the enzymatic hydrolysis time is 10 h. In the positive control group, the saccharification rate of WSB reaches 38.79%, 41.98% and 48.78% at pH 6.5, 50°C and enzyme dosage of 4 mL (commercial AXE and cellulase, 1:1, v/v), respectively. These values are lower than the corresponding values in the experimental group. When the reaction time is 24 h, the saccharification rate still increases, indicating the saccharification rate of WSB can be improved after longer reaction time. This result just proves that the synergistic effect of EST1051 and cellulase has high conversion efficiency for WSB. Table shows the orthogonal test results. The primary influence factor on WSB saccharification rate is pH, followed by the secondary factors (enzymatic hydrolysis time, temperature, addition amount). The optimal conditions to reach a peak WSB saccharification rate are as follows: 40°C, 6 mL of enzyme, pH 5.0, and 8 h of enzymatic hydrolysis. The maximum saccharification rate of WSB is about 76.12%. However, the saccharification rate of WSB is only 34.67% ± 1.27% and 78.46% ± 2.37% with the commercial AXE and cellulase mixture for 8 h and 48 h under the optimal conditions. So far, no natural AXE with such high efficiency for water hyacinth degradation has been reported.
Figure 12. Effects of (A) pH, (B) temperature, (C) enzyme amount and (D) time on enzymatic saccharification rate of WSB. Data points and error bars represent mean and standard deviation respectively.
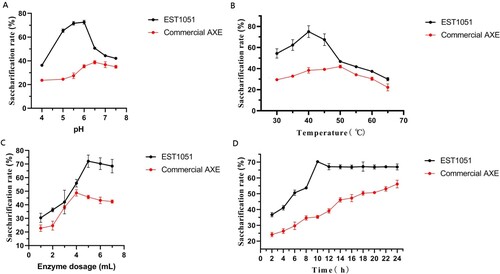
Table 8. Orthogonal test results and analysis.
Discussion
AXEs are among the least known hemicellulose degrading enzymes. The three- dimensional structure of AXEs derived from Trichoderma reesei and Penicillium purpurogenum (Colombres et al. Citation2008) has been determined by X-ray diffraction. The catalytic active site is composed of a catalytic triad (Ser-His-Asp), which is the same as the classic structure of the serine esterase family and corresponds to EST1051 (Ser22-His145-Asp218). All cysteine residues form disulfide bonds, which can maintain the structural rigidity. The binding site of AXE-substrate is very small and only contains one acetyl group, thus maintaining the enzymic specificity (Hakulinen et al. Citation2000, Citation1998).
We constructed the prokaryotic expression vector for the new AXE gene based on the results of enzyme digestion verification, DNA sequencing confirmation and SDS-PAGE. These results indicate that EST1051 is successfully expressed in the prokaryotic system, which lays a foundation for further study.
AXE has a wide range of substrate adaptability. The acetylation sites of AXEs differ depending on the sources. AXE from A. awamori has low activity for α-naphthylacetic-propyl-acetate, but high activity for α-naphthylacetic-methyl-acetate (Hakulinen et al. Citation2000). Some AXEs have the activities of AXE and ferulic acid esterase which hydrolyzes ferulic acid ester to release ferulic acid (Hakulinen et al. Citation1998; Koseki et al. Citation2005). AXE EST1051 has good adaptability at a wide temperature range and excellent thermal stability. Several reports on thermostable AXE show similar catalytic activity at high temperature. Maximum activity of EST1051 occurs at 40°C, which is slightly lower than AXE + CBM1 from Talaromyces leycettanus (Vafiadi et al. Citation2005) and Raxe from Ochrovirga pacifca (Williamson et al. Citation1998). However, the enzyme activity of EST1051 at 80°C is much higher than AXE + CBM1, which is nearly inactive at 80°C. The residual activity at 60°C is 62.41% after incubation for 24 h, which is better than Pc Axe2 (Zhang et al. Citation2019). To determine the usefulness of EST1051 in industrial applications, it is necessary to study the effects of metal ions, organic solvents and high salt on its activity and stability. EST1051 is stable in some common metal ions and organic solvents and maintains above 60% when the salt concentration is 3 M. Determining Km, the most important characteristic constant of enzymes, is a critical method to study enzyme kinetics, which can reflect the magnitude of affinity between an enzyme and the substrate. The Km and Vmax of EST1051 are 0.923 and 0.061 mM/min, respectively. These properties can reduce the application cost of EST1051 and improve its environmental adaptability, which lays a solid foundation for the industrial application. The fermentation conditions of EST1051 were optimized by response surface methodology. The EST1051 yield is 1909.32 U/L, which is about 1.83-fold that before optimization. The optimization experiment increases the possibility of industrial application of EST1051.
Despite some studies on enzymatic straw degradation, the AXE with efficient transformation of wheat straw is rarely reported. In this work, the saccharification rate is up to 76.12% after EST1051 and cellulase react with alkali-pretreated wheat straw for 8 h. Under the optimum conditions, the WSB saccharification rate by the mixture of commercial AXE and cellulase Cel1029 is only 78.46% ± 2.37% after 48 h. The maximum sugar yield reaches 23.48 mg/mL when crude enzyme complex of T. reesei Rut-C30 and AXE transformant T2 react with corn stover plus paddy straw mix (1:1 ratio) after 96 h (Hettiarachchi et al. Citation2019). The maximum sugar yield reaches 1.6 umol/mL when an immobilized enzyme, PersiXyn2bis-AERKIT-6, reacts with rice straw after 50 h (Huy et al. Citation2013). Small-scale degradation experiments prove that EST1051 has the ability to transform wheat straw.
Conclusion
A novel AXE gene Est1051 was cloned and successfully expressed in the prokaryotic system to explore its enzymatic properties and the synergism with cellulase on straw degradation. It showed high activity in a wide temperature range, high thermostability and excellent tolerance against metal ions, organic solvents and salts. The fermentation conditions of the recombinant protein were optimized. The esterase yield reached 1909.32 U/L, which was about 1.83-fold that before optimization. Then the novel AXE and Cel1029 were used to degrade and transform wheat straw and directly damage plant cell walls. AXE and Cel11029 directly converted wheat straw into glucose during a short test period. This study identifies a new AXE, enriches the source of AXE and makes full use of AXE. It also provides a research basis for improving the utilization rate of cellulose-rich plants.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in 4TU. Datacentrum [https://data.4tu.nl/portal] at https://doi.org/10.4121/13414949. The nucleotide sequence was declared to GenBank [https://www.ncbi.nlm.nih.gov/genbank/] with the accession number MK111431.
Additional information
Funding
References
- Adesioye FA, Makhalanyane TP, Vikram S, et al. 2018. Structural characterization and directed evolution of a novel acetyl xylan esterase reveals thermostability determinants of the carbohydrate esterase 7 family [J]. Appl Environ Microbiol. 84(8):e02695–17.
- Agrawal R, Gaur R, Mathur A, et al. 2015a. Improved saccharification of pilot-scale acid pretreated wheat straw by exploiting the synergistic behavior of lignocellulose degrading enzymes[J]. RSC Adv. 5(87):71462–71471.
- Agrawal R, Satlewal A, Gaur R, et al. 2015b. Pilot scale pretreatment of wheat straw and comparative evaluation of commercial enzyme preparations for biomass saccharification and fermentation. Biochem Eng J. 102:54–61.
- Agrawal R, Semwal S, Kumar R, et al. 2018. Synergistic enzyme cocktail to enhance hydrolysis of steam exploded wheat straw at pilot scale[J]. Front Energy Res. 6:122.
- Aiguo Y, Hao L. 2010. Research on issues of resources and environment [M]. International Conference on E-Education. 586–588. doi:10.1109/IC4E.2010.54.
- Aogaichi T, Plaut GWE. 1977. Assay of the esterase activity of thrombin, plasmin and trypsin with a chromogenic substrate p-nitrobenzyl p-toluenesulfonyl-l-arginine. Thromb Haemost. 37(1):253–261.
- Aysenur E, Serdar U, Cloning CS. 2015. Purification and characterization of acetyl xylane esterase from anoxybacillus flavithermus DSM 2641 T with activity on Low molecular-weight acetates [J]. Protein J. 34:237–242.
- Bahiru T, Chandrajit B, Partha R. 2020. Organosolv pretreatments of rice straw followed by microbial hydrolysis for efficient biofuel production. Renewable Energy. 148:923–934.
- Biely P, Cote GL, Kremnický L, et al. 1997. Action of acetylxylan esterase from Trichoderma reesei on acetylated methyl glycosides[J]. FEBS Lett. 420(2):121–124.
- Biely P, MacKenzie CR, Schneider H. 1988a. Production of acetyl xylan esterase by Trichoderma reesei and schizophyl- lure commune[J]. Can J Microbiol. 34:767–772.
- Biely P, MacKenzie CR, Schneider H. 1988b. Acetylxylan esterase of Schizophyllum commune[J]. Methods Enzymol. 160:700–707.
- Biely P, MacKenzie CR, Schneider H, et al. 1986. Cooperativity of esterase and xylanases in the enzymatic degration of acetyl xylan [J]. BioTechnology. 4:731–733.
- Biely P, Puls J, Schneider H. 1985. Acetyl xylan esterases in fungal cellulolytic systems[J]. FEBS Lett. 186(1):80–84.
- Cheeseman MT, Bates PA, Crampton JM. 2001. Preliminary characterisation of esterase and platelet-activating factor (PAF)-acetylhydrolase activities from cat flea (ctenocephalides felis) salivary glands. Insect Biochem Mol Biol. 31:157–164.
- Colombres M, Garate JA, Lagos CF, et al. 2008. An eleven amino acid residue deletion expands the substrate specificity of acetyl xylan esterase II (AXE II) from Penicillium purpurogenum. J Comput Aid Mol Des. 22(1):19–28.
- Coniglio RO, Gabriela V, Díaz MI, Fonseca ML. 2020. Enzymatic hydrolysis of barley straw for biofuel industry using a novel strain of trametes villosa from paranaense rainforest. Prep Biochem Biotechnol. 2:1–10.
- Cybinski DH, Layton I, Lowry JB, et al. 1999. An acetylxylan esterase and a xylanase expressed from genes cloned from the ruminal fungus Neocallimastix patriciarum act synergistically to degrade acetylated xylans [J]. Appl Microbiol Biotechnol. 52(2):221–5.
- Ding SJ, Cao J, Zhou R, et al. 2007. Molecular cloning and characterization of a modular acetyl xylan esterase from the edible straw mushroom Volvariella volvacea. Fems Microbiol Lett. 274(2):304–310.
- Dupont C, Daigneault N, Shareck F, et al. 1996. Purification and characterization of an acetyl xylan esterase produced by Streptomyces lividans[J]. Biochem J. 319:881–886.
- Fujimoto Z, Kuno A, Kaneko S, et al. 2000. Crystal structure of streptomyces olivaceoviridis E-86 beta-xylanase containing xylan-binding domain[J]. J Mol Biol. 300(3):575–585.
- Ghysel F, Godin B, Flamin C, et al. 2010. Forages as sources of energy: comparison of three methods of production, with their consequences and opportunities. Fourrages. 203:163–173.
- Hakulinen N, Tenkanen M, Rouvinen J. 1998. Crystallization and preliminary X-ray diffraction studies of the catalytic core of acetyl xylan esterase from Trichoderma reesei. Acta Crystallogr D. 54(3):430–432.
- Hakulinen N, Tenkanen M, Rouvinen J. 2000. Three-dimensional structure of the catalytic core of acetylxylan esterase from Trichoderma reesei: insights into the deacetylation mechanism. J Struct Biol. 132(3):180–190.
- Halgasová N, Kutejová E, Timko J. 1994. Purification and some characteristics of the acetylxylan esterase from Schizophyllum commune[J]. Biochem J. 298 Pt 3(3):751–755.
- Hea JC, Seung MP, Hak RK, et al. 2002. Cloning the gene encoding acetyl xylan esterase from Aspergillus ficuum and its expression in Pichia pastoris. Enzyme Microb Tech. 31(4):384–391.
- Hettiarachchi SA, Kwon YK, Lee Y, et al. 2019. Characterization of an acetyl xylan esterase from the marine bacterium Ochrovirga pacifica and its synergism with xylanase on beechwood xylan. Microb Cell Fact. 18(1):122.
- Huang C, Han L, Liu X, Ma L. 2010. The rapid estimation of cellulose, hemicellulose, and lignin contents in rice straw by near infrared spectroscopy. Energy Sources Part A Recovery Utilization and Environmental Effects. 33(2):114–120.
- Huy ND, Thiyagarajan S, Kim DH, et al. 2013. Cloning and characterization of a novel bifunctional acetyl xylan esterase with carbohydrate binding module from phanerochaete chrysosporium. J Biosci Bioeng. 115(5):507–513.
- Janssen PH, Monk CR, Morgan HW. 1994. A thermophilic, lipolytic Bacillus sp. and continuous assay of its p-nitrophenyl-palmitate esterase activity. FEMS Microbiol Lett. 1-2:195–200.
- Kirtay E. 2009. The role of renewable energy sources in meeting Turkey’s electrical energy demand [J]. Energy Education Science and Technology Part A: Energy Science and Research. 23:15–30.
- Koseki T, Miwa Y, Akao T, et al. 2006. An Aspergillus oryzae acetyl xylan esterase: molecular cloning and characteristics of recombinant enzyme expressed in Pichia pastoris. J Biotechnol. 121(3):381–389.
- Koseki T, Miwa Y, Fushinobu S, et al. 2005. Biochemical characterization of recombinant acetyl xylan esterase from Aspergillus awamori expressed in Pichia pastoris: mutational analys is of catalytic residues. BBA-Proteins Proteom. 1749(1):7–13.
- Krastanova I, Guarnaccia C, Zahariev S, et al. 2005. Heterologous expression, purification, crystallization, X-ray analysis and phasing of the acetyl xylan esterase from Bacillus pumilus[J]. BBA-Proteins Proteom. 1748(2):222–230.
- Kuno A, Kaneko S, Ohtsuki H, et al. 2000. Novel sugar-binding specificity of the type XIII xylan-binding domain of a family F/10 xylanase from Streptomyces olivaceoviridis E-86[J]. FEBS Lett. 482(3):231–236.
- Miao X, Lin J, Bian F. 2020. Utilization of discarded crop straw to produce cellulose nanofibrils and their assemblies. J Bioresources Bioproducts. 5(1):26–36.
- Prade RA. 1996. Xylanases: from biology to biotechnology. Biotechnol Genet Eng Rev. 13(1):101–132.
- Prashant MAP, Marta DM, Sunli C, et al. 2016. Expression of fungal acetyl xylan esterase in arabidopsis thaliana improves saccharification of stem lignocellulose[J]. Plant Biotechnol J. 14:387–397.
- Shohreh A, Farzaneh J, Parvin H, et al. 2020. An efficient nano-biocatalyst for lignocellulosic biomass hydrolysis: xylanase immobilization on organically modified biogenic mesoporous silica nanoparticles. Int J Biol Macromol. 164:3462–3473.
- Tamilvendan M, Rui L, Zhihua Z, et al. 2017. Optimization of acetyl xylan esterase gene expression in Trichoderma reesei and its application to improve the saccharification efficiency on different biomasses. Process Biochem. 58:160–166.
- Tsujiyama S, Nakano N. 1996. Detection and production condition of acetylxylan esterase from a wood-rotting fungus, Coriolus versicolor[J]. Mycoscience. 37(1):103–104.
- Vafiadi C, Topakas E, Wong KKY, et al. 2005. Mapping the hydrolytic and synthetic selectivity of a type C feruloyl esterase (St Fae C) from sporotrichum thermophile using alkyl ferulates. Tetrahedron-asymmetr. 16(2):373–379.
- Wang Y, Spratling BM, ZoBell DR, et al. 2004. Effect of alkali pretreatment of wheat straw on the efficacy of exogenous fibrolytic enzymes. J Anim Sci. 82(1):198–208.
- Watanabe M, Ishikawa K. 2014. Crystallization and preliminary X-ray crystallographic analysis of a putative acetylxylan esterase from Talaromyces cellulolyticus [J]. Acta Crystallogr F Struct Biol Commun. 70(Pt 12):1664–1667.
- Weina L, Di L, Wang X, et al. 2015. Cloning, expression and characterization of acetyl xylan esterase from Streptomyces griseus[J]. Biotechnology Bulletin. 31(02):153–159.
- Wenjing W, Huijun W, Xiaoyu W, et al. 2012. Cloning and bioinformatic analysis of acetyl xylan esterase gene from Streptomyces albus[J]. Biotechnology Bulletin. 06:122–128.
- Williamson G, Kroon PA, Faulds CB. 1998. Hairy plant polysaccharides: a close shave with microbial esterases. Microbiology. 144(8):2011–2023.
- Zhang YQ, Yang H, Yu XR, et al. 2019. Synergistic effect of acetyl xylan esterase from Talaromyces leycettanus JCM12802 and xylanase from Neocallimastix patriciarum achieved by introducing carbohydrate-binding module-1. AMB Expression. 9(1):13.