
Abstract
Single-cell transcriptome sequencing (scRNA-seq) is an unparalleled technology in the field of transcriptomics, which can study cells in a comprehensive and unbiased manner, thus characterizing complex biological processes at the cellular level. At the same time, scRNA-seq can produce a large amount of experimental data; therefore, the extraction of important information is required. In this review, we summarize the common applications of scRNA-seq analysis, such as of cellular communication, pseudo-timing, transcription factors, and functional enrichment, to mine cellular information and reveal the regulatory relationships between genes and trajectories of different cell lineages during development. We also describe the latest technological progress of scRNA-seq use in organoid, pathogenic gene variants, tumor tissues, stem cells, viruses, antibody preparation, and so on, in order to improve its applicability in future research in the life science field.
Introduction
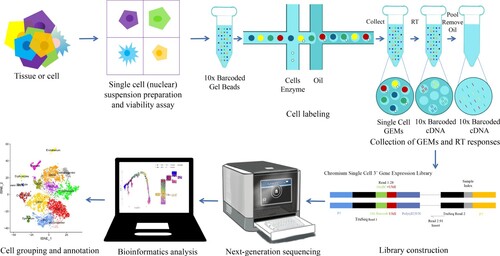
Single-cell RNA sequencing (scRNA-seq) is an emerging technique for sequencing the transcriptome at the single-cell level, allowing for the study of gene expression within a single cell. At the same time, it solves the problem of cell heterogeneity that cannot be solved by tissue sample sequencing, making it possible to analyze the behavior and mechanism of a single cell and its relationship with the body, thereby providing insights to enable related biological problems to be solved (Stuart and Satija Citation2019; Paik et al. Citation2020). The scRNA-seq workflow includes cell suspension preparation, cell viability detection, 10X Genomics formation of GEMs, collection of GEMs and RT reactions, separation of cDNA from oil droplets, library construction, on-board sequencing, and data analysis (Figure ). scRNA-seq analysis of the traditional transcriptome, bulk genome and transcriptome analysis provided a wealth of insights into tissue growth and evolution. However, the signals displayed by specific cell populations or states are masked during bulk sequencing, and these specific cell populations or states is sometimes very critical to understanding cell function. Therefore, examination of individual cells by scRNA-seq can overcome the limitations of analyzing the traditional bulk level and enable more detailed analyzes at the cellular and molecular levels (Gohil et al. Citation2021).
Figure 1. Single cell RNA sequencing (scRNA-seq).
In addition to the construction of conventional cell maps, scRNA-seq data analysis often requires deeper investigations into the biological significance of gene expression data. It can be seen that advanced analysis is particularly important for mining important biological information (Figure ). Currently, the most widely used advanced single-cell analyzes are of pseudo-time, cellular communication, transcription factors and single-cell enrichment. Pseudo-time analysis can simulate cell dynamics by ranking single cells along trajectories based on the similarity in expression patterns between sequenced cells (Meistermann et al. Citation2021). Cell communication analysis is used to infer the interactions between different cells by determining the expression and pairing of receptors and ligands in different cell types (Efremova et al. Citation2020; Jin et al. Citation2021). Transcription factor analysis is the efficient identification of co-expression modules between transcription factors (TFs) and potential target genes using auxiliary software (Van de Sande et al. Citation2020). Single-cell enrichment analysis refers to statistical analysis using various databases and analysis tools to mine gene function categories that are significantly related to the biological problem studied in the database. At present, the amount of data generated by scRNA-seq has exploded, and a large number of analytical methods and tools have been developed (Luecken and Theis Citation2019), especially for advanced analysis and applied research methods, making it difficult to determine the best analysis method. In this review, we explored the most commonly used advanced analytical tools and related application strategies for scRNA-seq. The application of scRNA-seq in assisting the analysis of organoid models, cell specificity of pathogenic gene variants, cell specificity of tumor tissues, and stem cell development and differentiation. We aim to provide directions for improved application of scRNA-seq in single-cell research and to discuss possible future development directions.
Figure 2. Common advanced analysis tools in scRNA-seq.
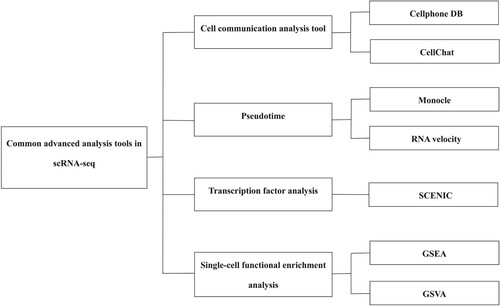
Common advanced analysis tools in scRNA-seq
Cell communication analysis tool
Cellphone DB
CellPhoneDB is currently the most widely used software for cellular communication analysis and includes a database consisting of ligands, receptors, and their interactions. It can conduct a comprehensive and systematic analysis of molecules involved in the communication between cells and study mutual communication and communication networks between different cell types. CellPhoneDB's ligand–receptor database stores a total of 978 proteins, 501 secreted proteins and 585 membrane proteins, which are involved in 1,396 interactions. The CellPhoneDB database also takes into account the subunit structures of ligands and receptors and accurately represents heteromeric complexes, 466 of which are heteromeric, which are critical for the accurate study of cellular communication mediated by multi-subunit complexes. In addition, there are 474 secreted proteins, 490 membrane proteins, and 250 integrin proteins involved in the interactions that can be analyzed by CellPhoneDB (Efremova et al. Citation2020). The CellPhoneDB is widely used to improve the understanding of the relationships between cell subgroups. For example, in the immune microenvironment of bladder cancer, CellPhoneDB analysis focused on two cell subgroups, inflammatory cancer-associated fibroblasts (iCAFs) and myo-cancer-associated fibroblasts (mCAFs), and examined the communication between these two cell subgroups and all other cell subgroups, and constructed a mechanism diagram based on the cell communication relationship (Chen Z et al. Citation2020). In the microenvironment of esophageal squamous cell carcinoma, CellPhoneDB analysis identified an interaction between macrophages ligands and receptors on other cell types in the tumor or adjacent tissues (Chen Z et al. Citation2021). In hematopoietic stem cells and multipotent progenitor cells (HSC/MPP), CellPhoneDB was used to construct an regulatory interaction network between microenvironment cells and HSC/MPPs, to identify potential pro-amplification molecules. For example, understanding the roles of molecules such as the growth factors MDK and PTN contributes to our understanding of cellular interactions (Gao et al. Citation2022). In mouse hair follicles, developmental maps were mapped using scRNA-seq data and receptor–ligand pairs enriched in the same cell subset were found to show strong autocrine signals, using CellPhoneDB, indicating that robust intercellular communication is involved in early hair follicle development (Ge et al. Citation2020).
Cellchat
CellChat can accurately identify and display cell-to-cell communication signals and systematically analyze them with the aim of discovering new cell-to-cell communication networks and building cell–cell communication maps in different tissues (Shao et al. Citation2020; Fang et al. Citation2022). Based on the law of mass action, CellChat combines single-cell expression profiles with known ligands, receptors, and their cofactors (also known as a heterogeneous molecular complex, which avoids the problem of using only one ligand or one receptor gene pair while ignoring many receptors that function as multi-subunit complexes) to calculate the probability of cell–cell communication (which can also be understood as interaction intensity). Furthermore, ligand–receptor pairs with significant interactions can be identified using ligand–receptor interaction probability and disturbance tests. Due to its ability to sum the number or strength of ligand–receptor relationship pairs that interact significantly between cell types to calculate an integrated cell–cell communication network, CellChat's predictive power goes beyond classifying cell populations and establishing their lineage relationships. Ultimately, accurate inference, systematic quantification, comparative analysis, and intuitive visualization of complex cellular communication networks can be achieved using CellChat (Jin et al. Citation2021). Researchers have successfully applied the CellChat tool to analyze single-cell transcriptome datasets of skin development, wound healing, and skin diseases in mice and humans. It has not only revealed many signaling pathways that have been verified by biological experiments but also the regulatory mechanisms of cell communication that have not been reported previously in literature. Computational analysis by CellChat predicted that two signaling pathways, Edn3-Ednrb and Pros1-Axl, regulate the formation of dermal coagulation in the early development of neonatal mouse skin hair follicles. Furthermore, RNAscope in situ hybridization was used to verify the co-expression of related genes in these two signaling pathways in the cells of E14.5-day mouse skin, revealing the potential signaling regulatory mechanism of mouse hair growth (Jin et al. Citation2021).
Pseudotime
Monocle
Monocle tracks changes in gene expression along a trajectory, determined ‘pseudo-time’. Pseudo-time analysis using the tool Monocle involves the arrangement of each cell on the corresponding track according to the time sequence of gene expression in each cell (Qiu et al. Citation2017). At the same time, according to the gene expression status, samples are divided into several cell groups in the state of differentiation, and an intuitive tree map of pedigree development is generated, which can predict the differentiation and development trajectory of cells. However, the results of pseudo-time analysis need to include the starting and end points of differentiation according to the trajectory distribution of cell types and the expression changes of characteristic genes. In the study of differentiation or development, we can analyze the key differentiation nodes by pseudo-time analysis, and key cell subsets or genes can be identified.
In scRNA-seq of early birth testicular tissue, germ cell subsets were divided into four developmental stages based on the expression of genes related to germ cell development: undifferentiated spermatogonia (UTF1 + MKI67-), differentiated spermatogonia (KIT + MKI67+), primary spermatocytes (STRA8 + GPR85+), and sperm cells (PRM3+). This developmental trajectory was further verified using Monocle quasi-sequential analysis (Guo J et al. Citation2020). Researchers have used scRNA-seq to delve deeper into the process of somatic cell reprograming induced by small molecule compounds. Monocle pseudo-time analysis of data at 12 time points in the process of somatic cell reprograming induced by small molecules revealed that the reprograming process eventually differentiated into two branches: one branch successfully differentiated into pluripotent stem cells, and the other branch failed to differentiate. Molecular induction of the key cell subset Ci2C-like at the branch point effectively promoted the activation of Ci2C-like-related genes, shortening the induction period of chemical reprograming from 40 to 16 days (Zhao et al. Citation2018). The authors performed scRNA-seq on lung squamous cell carcinoma and found that basal cells, club cells, and rod cells can develop into lung squamous carcinoma (LUSC) tumor cells. Through Monocle analysis, basal cells appeared to be in an intermediate state in the transition from rod cells to tumor cells (Wu F et al. Citation2021). In conclusion, Monocle is a classic tool for pseudo-timing analysis of scRNA-seq data that uses algorithms to learn the sequence of gene expression changes that each cell must undergo during state transitions. Once the overall ‘trajectory’ of gene expression changes is known, Monocle can place each cell in the appropriate position in the trajectory.
RNA velocity
Single-cell sequencing techniques can sensitively and accurately reveal RNA abundance, which is a powerful indicator of the state of a single cell. However, this can only capture static snapshots at a certain point in time, making it challenging to analyze phenomena that involve dynamic changes over time (such as embryogenesis or tissue regeneration). La Manno et al. (La Manno et al. Citation2018) proposed the vector of RNA velocity in response to this problem. RNA velocity uses a dynamic model to evaluate RNA abundances in the instantaneous state of the cell. It is an indicator of dynamic changes in transcripts and can predict the future states of cells. The difference with RNA velocity lies in the use of RNA splicing, which is not commonly used in scRNA-seq, to predict the upregulation and downregulation of mRNA expression using the ratio of spliced mRNA to unfinished mRNA in cells. It can be used to study cell differentiation, pedigree development, and dynamic changes of cell composition in tumor microenvironment and so on. RNA velocity analysis does not need to provide the starting and ending points of cell trajectory analysis and can analyze cells with unknown differentiation relationships. At the same time, it also has the advantages of strong data compatibility and providing a perspective of single-cell level analysis (Romanov et al. Citation2020).
The migration of macrophages in the tumor microenvironment and the origin of newly discovered dendritic cell subsets were revealed by RNA velocity analysis of scRNA-seq data from hepatocellular carcinoma cells (Zhang Q et al. Citation2019). The scRNA-seq data of mouse chromaffin cells were evaluated by RNA velocity analysis, which accurately reproduced the transcriptional kinetics of the data, including gene activation and inhibition and the direction of cell differentiation. This proved the ability of the RNA velocity model to determine the rate and direction of transcriptome changes in the dynamic process of single-cell sequencing. From the above findings, it can be seen that RNA velocity is a remarkable analysis model that provides a unique perspective, which illuminates the subject of cell differentiation, and be applied and optimized in future research (La Manno et al. Citation2018).
Transcription factor analysis
SCENIC
The identity of a single cell is largely defined by its potential gene regulatory network in which transcription factors (TF) drive the expression of their target gene, in combination with the expression of other specific genes, thereby establishing a gene expression profile. Single-cell regulatory network inference and clustering (SCENIC) is a method for reconstructing gene regulatory networks and identifying cell status by evaluating scRNA-seq data, based on co-expression and motif analyzes, reconstruction and evaluation of the activity of regulatory factors (TFs and their target genes) in individual cells, and use of cellular activity to identify meaningful clusters of cells. Since such methods aim to predict combinations of upstream regulators for a specific cell type, they facilitate subsequent validation and manipulation of cell types (Aibar et al. Citation2017; Van de Sande et al. Citation2020).
Aibar et al. (Aibar et al. Citation2017) used the SCENIC algorithm to reconstruct gene regulatory networks and identify cell statuses in tumors and mouse brains following scRNA-seq. They proposed that cis-regulatory network analysis can be used to guide the identification of transcription factors and cell status. Rambow et al. (Rambow et al. Citation2018) used SCENIC to analyze scRNA-seq data of minimal residual disease (MRD) cells in melanomas and predicted that the nuclear receptor RXRG is a key driver of neural crest stem cells (NCSCs). They found RXR antagonists can reduce NCSC accumulation in MRD and delay the occurrence of drug resistance. Xie et al. (Xie et al. Citation2021) used SCENIC software to calculate the regulator activity score of scRNA-seq data from 32 kinds of blood cells, analyzed the TF regulatory network involved in the process of hematopoietic differentiation, and discovered new potential hematopoietic differentiation regulatory factors (such as CREM, RXRA, and FOXO3). This suggests that the lymphoid transcriptional regulatory network is cell specific, although neutrophils and monocytes can be regulated by common transcription factors. In scRNA-seq of bladder cancer, SCENIC analysis identified that the specific activation of MEF2D and MEF2C in myo-cancer-associated fibroblasts (mCAFs) plays an important role in the transcriptional regulation of muscle cell lineage. Additionally, TCF21 and TWIST2 motifs are specifically activated in inflammatory cancer-associated fibroblasts (iCAFs). In previous studies, TCF21 was associated with coronary heart disease, whereas TWIST2 was a driver of epithelial–mesenchymal transition (EMT). SCENIC analysis revealed that the activities of three genes, BACH1, MAFG, and NFE2, were inhibited along the pseudo-time trajectory, while MAF, STAT1, and STAT2 were activated, which may be the cause of M2 polarization in monocytes (Chen Z et al. Citation2020). Thus, SCENIC can resolve TF activity in single cells. Studying differences in transcription factor activity using scRNA-seq can not only improve our understanding of cellular heterogeneity but is also an efficient approach to analyze the key regulatory mechanisms of specific TFs.
Single-cell functional enrichment analysis
GSEA
Gene set enrichment analysis (GSEA) analyzes all genes instead of only differential genes, making it easier to discover the impact of subtle changes on biological pathways. GSEA requires comparative analysis between samples in advance and is typically used in case–control studies to obtain the trends in upregulated or downregulated expression of a certain pathway gene set in the experimental group and the control group. At the same time, GSEA can also compare the same cell type between different experimental groups or different cell types. Zeng et al. (Zeng, Liu, et al. Citation2019b) used GSEA to perform enrichment analysis in thymic epithelial cell (TEC) subtypes TEC2 and TEC3, and found that the BMP and WNT signaling pathways were significantly enriched in TEC2 and TEC3, respectively. Results showed significant upregulation of genes involved in metabolism and antigen presentation in TEC3, suggesting that TEC3 is more developed than TEC2 and revealing the early development and maturation features of thymic epithelial cells and the molecular features underlying these processes. In tumor research, malignant tumor cells can be distinguished from non-malignant cells by GSEA. Zhang et al. (Zhang M et al. Citation2021a) used scRNA-seq to study the origin of gastric metaplastic cells and gastric cancer heterogeneity via GSEA of malignant and non-malignant cells. Compared to non-malignant epithelial cells, malignant epithelial cells were found to be enriched in tumor necrosis factor, transcriptional activation signaling pathway factors, and other genes that are critical for the development and progression of cancer, revealing the underlying molecular mechanisms of gastric cancer development. In scRNA-seq of bladder cancer cells, GSEA determined that iCAFs were significantly enriched in genes associated with extracellular matrix degradation and cytokine-cytokine receptor interaction pathways, while mCAFs were enriched in genes associated with muscle contraction and PGC1A pathways (Chen Z et al. Citation2020). In conclusion, the advantage of GSEA is that there is no need to specify a threshold (p-value or FDR) to screen for differential genes, and we can analyze the gene set of interest without prior knowledge, which may not necessarily include significantly differentially expressed genes. GSEA analysis can include genes that are easily missed by Gene Ontology (GO) / Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis and have insignificant differential expression but important biological significance.
GSVA
Gene set variation analysis (GSVA) does not require pre-grouping of sample; the enrichment scores for a specific gene set in each sample or cell type are calculated, and then compared across different samples or cell types. Although GSVA is a powerful enrichment analysis tool, its application is usually limited to case–control studies. For large-sample studies with complex phenotypes, such as projects like TCGA and single-cell atlas, analysis is more difficult. Eight years after the release of GSVA, the Broad Institute developed the GSVA algorithm to expand the application of gene set analysis. GSVA does not require prior differential analysis between samples; it can be used to calculate the variation score of a specific gene set in each sample based on the expression matrix (Hanzelmann et al. Citation2013). Zhang et al. (Zhang M et al. Citation2020) used scRNA-seq to study intrahepatic cholangiocarcinoma tumor cell heterogeneity and conducted enrichment analysis of malignant cells (subgroups 0-3) using GSVA. Subgroup 0 was enriched for epithelial–mesenchymal transition signals, subgroup 1 for cell cycle and hypoxia-dominant signals, and subgroup 2 for interferon response signals. The results of these analyzes revealed a high degree of heterogeneity among tumor cells. Luo et al. (Luo et al. Citation2021) used GSVA to assess the unique biological characteristics of healthy and end-stage renal disease (ESRD)-derived monocytes. Cui et al. (Cui et al. Citation2019) used GSVA to study the enrichment of endothelial cells and cardiac fibroblasts in the BMP signaling pathway. Hoang et al. (Hoang et al. Citation2019) used GSVA to study differences in pathway expression in different stages of liver fibrosis and different non-alcoholic fatty liver disease (NAFLD) activity scores. In conclusion, GSVA is a non-parametric and unsupervised learning method, which is mainly used to evaluate the gene set enrichment results of microarray nuclear transcriptomes, and to quantify the gene enrichment results simultaneously, which can make subsequent statistical analysis more convenient.
GO analysis
Gene ontology (GO) divides the functions of genes into three parts: cellular component (CC), molecular function (MF), and biological process (BP). Using the GO database, the expression of the target gene at the CC, MF, and BP levels can be obtained. Each category explains the biological function of genes at different levels and can focus on GO terms in combination with specific biological problems. At the same time, there is an inclusive relationship between GO terms, and a complex structural network can be constructed between them. The lower the level of the GO term, the more specific the functional description is; i.e. the lower the level, the better gene can explain biological problems. Therefore, we should pay attention to significantly enriched low-level GO terms that may explain biological problems in detail. In addition, the statistical hypothesis of GO enrichment analysis cannot fully represent the importance of gene function, so it is necessary to combine biological problems and gene functional annotation to judge whether the gene changes have biological significance (Denny et al. Citation2018). Shen et al. (Shen et al. Citation2021) confirmed that 10 genes enriched from liver tumors were associated with the specific activity of autophagy by analyzing the scRNA-seq data using GO. Darmanis et al. (Darmanis et al. Citation2017) performed scRNA-seq on glioma and paracancerous cells and found that the function of invasive cell enrichment was highly correlated with cancer cell metastasis through GO database analysis. At the same time, it was deduced that the mechanism of glioma proliferation involves cancer cell metastasis from the inside of the tumor to the pericancerous tissue, where cells settle and become invasive cells. In conclusion, GO enrichment analysis is a relatively simple, widely used technique for scRNA-seq, which is helpful for understanding genes and gene products.
KEGG analysis
KEGG database systematically analyzes the metabolic pathways of gene products in cells and the functions of these gene products. KEGG integrates the databases of genomes, chemical molecules, and biochemical systems, including metabolic pathways, drugs, gene sequences, genomes, and diseases. The KEGG pathway database is the most widely used public database of metabolic pathways. The main feature of the KEGG database is that the genes and metabolites (chemical information) are presented in detail in the form of network diagrams, which provide a very intuitive and comprehensive overview of the signal transmission processes of genes and metabolites in the body. Hu et al.(Hu et al. Citation2020) performed KEGG analysis of differential genes from scRNA-seq data of clear cell renal cell carcinoma (ccRCC) and revealed that the hypoxic response, lipid biosynthesis, and localization pathways were enriched in cancer cells, whilst lipid catabolism was inhibited. Thus, KEGG-assisted analysis provided further insight into the immune properties of the tumor microenvironment (TME) of ccRCC. Zhang et al. (Zhang Y et al. Citation2021b) identified seven chondrocyte subsets using scRNA-seq implicated in intervertebral disc degeneration (IVDD): fibrochondrocyte progenitor cells (FCPs), chondrocyte progenitor cells (CPCs), homeostatic chondrocytes (HomCs), and C1, C2, C3, and C4. KEGG analysis showed that ferroptosis-related (Jiang et al. Citation2021) genes were significantly enriched in the mild IVDD group (FTL, HMOX, and CP) compared to the control (no IVDD) or severe IVDD group. Compared with controls, ferroptosis signaling was upregulated in C2, C4, CPCs, and HomCs in the IVDD group, suggesting that ferroptosis is involved in the pathogenesis of intervertebral disc degeneration. One of the remarkable features of KEGG, which is different from other databases, is that it has powerful graphical functions. It uses intuitive graphics to display the enrichment pathways of differential genes from scRNA-seq data, which helps to deepen the understanding of differential genes and the mining of relevant information for research.
Applications of single-cell transcriptome sequencing
Establishment of organoid models
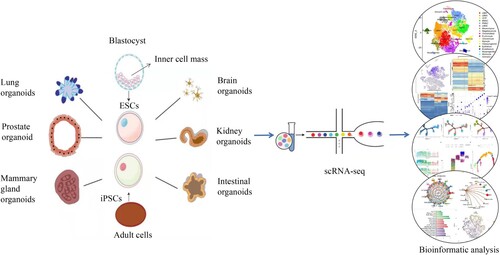
Organoids are composed of primary tissues (single cells or tissue subunits), embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs), established cell lines, and intact or segmented organs (such as explants consisting of multiple tissue types). This technology is defined as a cell-derived in vitro 3D model capable of self-renewal and self-organization, and exhibits organ functions similar to the origin tissues. Researchers have succeeded in obtaining organoids from human patients and animal models, which mimic different tissues, organs, and systems that reflect the natural, in vivo physiology (Fatehullah et al. Citation2016; Rossi et al. Citation2018). At present, scientists have established organoids such as intestinal organoids (Sato et al. Citation2009), retinal organoids (Shirai et al. Citation2016), brain organoids (Lancaster et al. Citation2013; Qian et al. Citation2016), stomach organoids (McCracken et al. Citation2014), liver organoids (Takebe et al. Citation2013), pancreas organoids (Boj et al. Citation2015), prostate organoids (Karthaus et al. Citation2014), breast organoids (Sachs et al. Citation2018), and embryonic organoids (van den Brink et al. Citation2014; Warmflash et al. Citation2014). Organoids can simulate specific aspects of three-dimensional structures, cell type composition, and organ function, while maintaining the advantages of simplified and easily accessible cell culture models. However, it is unclear how precisely these organoid systems reproduce the expression of cell state-specific genes in the tissues they model. Single-cell sequencing has provided new analytical methods to characterize these organoid models with greater resolution and less bias than previous immunohistological or RNA-seq analyzes of organoid development (Picelli Citation2017). scRNA-seq is particularly useful for studying cellular diversity and complex organoids, helping to understand the variability of organoid development (Brazovskaja et al. Citation2019)(Figure ).
Figure 3. Single-cell transcriptome sequencing of organoids.
Human dermal fibroblasts (HDFs) were reprogramed to generate in vitro three-dimensional models of human blastocysts, termed induced blastocysts (iBlastoids). iBlastoids can mimic the overall structure of blastocysts, with an inner cell mass of ectoderm and primitive endoderm-like cells, a blastocyst-like cavity, and an outer layer of trophectoderm-like cells. The existence of ectoderm, primitive endoderm, and trophectoderm-like cells was further confirmed by scRNA-seq, and the gene expression of each germ layer was also determined. iBlastoids provide an opportunity to study the development of human embryos, preimplantation blastocysts, and post-implantation embryos in vitro (Liu X et al. Citation2021). In addition, Li et al. (Li et al. Citation2019) cultured mouse EPS cells into blastocysts in vitro and scRNA-seq analysis showed that blastocysts and fertilized blastocysts had similar gene expression characteristics. Further experiments demonstrated that blastocysts can develop in vitro into structures similar to those of early postimplantation embryos. Researchers believe that by further optimizing the system and including gene editing technology, blastocysts with more complete functions can be obtained, which could develop to the stage of forming different organ primordia, thus becoming the seeds of organoids that could be used for organ transplantation. Among intestinal organoids, a novel intestinal organoid culture system consisting of eight chemical components was established in vitro with injury-regenerative characteristics, called hyperplastic intestinal organoids (hyper organoids). Researchers analyzed hyper organoids using scRNA-seq to identify distinct subpopulations of regenerative stem cells in organoids and found that they had the same molecular characteristics as in vivo injury model stem cells. Further analysis of the key regulatory small molecules involved in genome reprograming and regeneration of hyper organoids led to the establishment of a novel organoid model that could perform injury regeneration. The use of organoid technology provides broader application prospects for future research on organ regeneration, establishment of disease injury models, and drug screening (Qu et al. Citation2021). Camp et al. (Camp et al. Citation2015) used scRNA-seq for the first time to directly compare cerebral organoids with the early fetal neocortex and found that the cellular composition, lineage relationships, and gene expression programs of cerebral organoids largely recapitulated regions of the organoid cortex. In addition, Cowan et al. (Cowan et al. Citation2020) compared retinal organoids and human retinas by scRNA-seq and found that the two cell types developed similarly, and the organoid cell-type transcriptomes converged to those of adult peripheral retinal cell types. These studies further underscore the importance of high-resolution characterization of single-cell states emerging in organoid culture systems. Finally, the combination of organoid construction and single-cell sequencing could improve the accuracy, precision, and efficiency of organoid model building.
Disease-causing genes are cell-specific
scRNA-seq results can show that specific cell types cause specific diseases, providing insights for target-directed therapy. At the same time, scRNA-seq can also help the understanding of the pathogenic loci found in genome-wide association analysis (GWAS), identify cell-type-specific pathogenic gene variants, and accelerate the development of novel drug targets. In conclusion, scRNA-seq is a powerful tool for studying the mechanisms of gene regulation in different cell types and the pathogenesis of complex human diseases. The expression of disease-related genes in the adult retina is cell-type-specific. In retinal scRNA-seq, genes for color blindness were found and validated mainly in cone cells, whilst genes for night blindness were mainly expressed in rod cells (Cowan et al. Citation2020). Lenis et al. (Lenis et al. Citation2018) also found that Stargardt disease may arise from foveal-specific dysfunction of pigment epithelial cells. Kidney-related pathogenic genes are expressed in specific cell types. scRNA-seq of mouse kidneys found 21 genes associated with proteinuria, and these genes were exclusively expressed in the visceral epithelial cells of the renal capsule. The causative gene associated with renal tubular acidosis was expressed only in the intercalated cells. In addition, the causative genes of blood pressure, chronic kidney disease, serum metabolite levels, kidney stones, and renal tubular acidosis are also expressed in specific cell types (Park et al. Citation2018). Integrating scRNA-seq, snATAC-seq and GWAS information, Sheng et al. (Sheng et al. Citation2021) found that in the kidney, immune cells were significantly associated with multiple sclerosis, whereas renal endothelial cells and distal tubular cells were closely associated with hypertension. In addition, proximal tubular cells are significantly associated with glomerular filtration rate (eGFR) and uric acid metabolism. At the same time, the study also identified the AGT gene as the causative gene of hypertension through the colocalization analysis of GWAS information and cell type interaction eQTL (eQTL(ci)) affected by specific cells.
Disease-related genes are also expressed in specific placental cell types. In placental scRNA-seq, differentially expressed genes (DEGs) in early onset preeclampsia (EOPE) were found to be cell-specific, and EOPE-upregulated DEGs were found to be highly enriched in placental extravillous trophoblast cells (EVT). The importance of EVT in EOPE pathogenesis has been previously confirmed (Guo F et al. Citation2021). In addition, chimeric antigen receptor T-cell immunotherapy (CAR-T) has been developed. Targeting CD19 with CAR-T has shown significant clinical efficacy in the treatment of B-cell leukemia and lymphoma, however, CAR-T therapy has serious side effects. Parker et al. (Parker et al. Citation2020) analyzed published human brain scRNA-seq data and found that vascular wall cells also express the CD19 gene, and that human brain wall cells are critical for the integrity of the blood–brain barrier. Through simple functional experiments, it was proven that CAR-T cells attack the blood vessel wall cells in brain tissue, resulting in leakage of the blood–brain barrier. This finding provides a theoretical foundation for improving the clinical safety of CAR-T therapy and insights into the efficacy of targeted therapy. It can be seen from the above that pathogenic loci can be found by GWAS, as well as cell-type-specific pathogenic genes, which can subsequently accelerate the development of new drug targets. In addition, the combined application of scRNA-seq and data analysis methods to study gene regulatory mechanisms in different cell types provides a powerful tool for studying the pathogenesis of complex human diseases.
Cell specificity of tumor tissue
The tumor microenvironment is a complex integrated system formed by the interaction of tumor cells with the surrounding tissues and immune cells. Its existence enhances tumor cell proliferation, migration and immune evasion abilities, thereby promoting the occurrence and development of tumors. scRNA-seq analysis provides a way to comprehensively study immune cells in the tumor microenvironment. In particular, there are many studies on cancer-associated fibroblasts (CAFs), macrophages, and T cells, which are of great significance for the occurrence and development of tumors (Figure ).
Figure 4. Single-cell transcriptome sequencing of the tumor microenvironment.
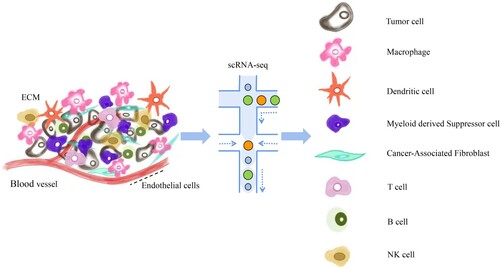
Cancer-associated fibroblast
CAFs are fibroblasts that are activated by cancer cells in tumor tissues and resemble myofibroblasts. Malignant transformation of the CAF phenotype is closely related to tumor progression, and CAFs are the main tissue component in the tumor microenvironment. Generally, the effects of CAF secretion and intercellular communication on tumor cell proliferation, migration, and apoptosis is of great concern (Arneth Citation2019). scRNA-seq was performed on primary tissues from triple-negative breast cancer (TNBC) tumors. The results mainly focused on myofibroblasts (myCAFs) and inflammatory fibroblasts (iCAFs) and identified that communication between myCAFs and myeloid cells is achieved through TGFB1-TGFBR1 interactions. Additionally, the communication between iCAFs and T cells is achieved through CXCL12-CXCR4 interactions, whilst communication between iCAFs and B cells occurs via CXCL13-CXCR5 interactions. This indicated that different stromal cell subsets expressed different immunoregulatory genes, suggesting that different stromal cell subsets of TNBCs may be involved in the process of immune escape. In addition, iCAF signature genes were significantly associated with T cell dysfunction. Among them, patients with a lower iCAF signal had higher levels of cytotoxic T lymphocytes (CTL), which can significantly improve survival rate. However, in patients with a higher iCAF signal, CTL levels were not associated with prognosis and immune escape events occurred. In conclusion, this study provides guidance for future developments in targeted therapy (Wu SZ et al. Citation2020).
Tumor-associated macrophages
Tumor-associated macrophages (TAMs) are the most abundant immune cells in the tumor microenvironment, which infiltrate the tumor tissues. Studies have shown that M1-type macrophages play key roles in inhibiting tumor progression, and promoting inflammation and immune activity, while M2-type macrophages play roles in tissue repair, immune escape, and promotion of tumorigenesis and development. The proportion of macrophages in the tumor microenvironment is highly correlated with tumor development, severity, and prognosis (Vitale et al. Citation2019). scRNA-seq of pleural effusion and peripheral blood samples from patients with malignant pleural effusion (MPE) due to non-small cell lung cancer showed greater depletion of cytotoxic T cells and increased Treg cells in MPE than in the blood, similar to previous findings in studies of non-small cell lung cancer. Patients with high expression of both cytotoxic T cells and T cell-depleted signature genes showed significantly reduced survival. Regulatory B cells (Breg) were enriched in MPE samples, whereas plasma cells were enriched in the blood samples. Macrophages enriched in MPE patient samples are dominated by the M2 type, which plays a role in promoting tumorigenesis. Taken together, these findings indicate that immune cells in MPE enhance immune evasion and tumorigenesis (Huang et al. Citation2021).
Tumor-associated T cells
Tumor-associated T cells are the main immune cell population and play an important role in immune-mediated killing of tumor cells. In the tumor microenvironment, the T cell subtype CD8 + T cells can kill tumor cells, and are mainly regulated by regulatory T cells (Tregs) in CD4 + T cells. In studies of the tumor microenvironment, researchers used the ratio of CD4 + FOXP3 + T cells in various tumors as a benchmark for the immune activity of T cells in the tumor microenvironment. In addition, tumor cells and the microenvironment together determine the progression of the disease and response to treatment. In particular, understanding the ecological environment of tumor cells is helpful in guiding individualized therapy and providing new insights into the mechanisms of drug resistance and potential therapeutic targets (Hinshaw and Shevde Citation2019; Vishnubalaji and Alajez Citation2021). scRNA-seq in lung adenocarcinomas showed that CD8+ T cells and Tregs were enriched in tumors, but natural killer cells were depleted during tumor progression. The analysis revealed a shift in lymphocyte composition and gene expression towards immunosuppression during lung adenocarcinoma progression. Concurrently, stromal cells underwent a phenotypic shift towards tissue remodeling and angiogenesis during lung adenocarcinoma progression (Wang Z et al. Citation2021b). scRNA-seq of esophageal squamous cell carcinoma (ESCC) tumors and paired adjacent esophageal tissue revealed that T cells, NK cells, monocytes/macrophages, dendritic cells (DCs), B cells, plasma cells, and mast cells were predominantly present in tumors compared with paired adjacent tissues. The presence of these cells suggests the presence of an inflammatory and immunosuppressive tumor microenvironment (TME) in ESCC. Simultaneously, the interactions between macrophages and Tregs through ligand receptors may lead to an immunosuppressive state and disease progression (Zheng et al. Citation2020).
It can be seen from the above findings that immune cells are an important part of the tumor microenvironment, and each cell type plays a crucial role. With the increase in tumor microenvironment research, more efficient tumor treatment methods and drugs may emerge in the near future (Pan et al. Citation2019).
Stem cell development and differentiation
Stem cells have proliferation, differentiation, self-renewal, and replication capabilities, and can produce highly differentiated functional cells. Stem cells have important applications in disease research, gene therapy, tissue engineering, developmental biology models, systems biology research, and other fields (Yamanaka Citation2020). With the help of scRNA-seq, the transcriptome characteristics of individual stem cells can be analyzed, and further computational methods can be used to determine cell states and reconstruct lineage differentiation characteristics based on transcriptome characteristics. Consequently, scRNA-seq has played a powerful role in stem cell and tissue development research, accelerating our understanding of stem cell differentiation and tissue development-related processes and regulatory pathways. scRNA-seq analysis can be used to elucidate the origin and heterogeneity of stem cells, as well as for the continuous observation of the differentiation process of stem cells, which is very important for their application. The main stem cell types and their characteristics are summarized in Table . Tan et al. (Tan et al. Citation2021) studied the chimeric ability of human expanded pluripotent stem cells (hEPS) in cynomolgus monkeys using a monkey embryo in vitro culture system. They further combined it with scRNA-seq and other technologies to analyze the developmental trajectories of human and monkey cells. The researchers were surprised to find that hEPS gradually became similar in expression characteristics to recipient cynomolgus monkey embryo cells as cell development progressed and found that hEPS exhibited chimeric properties in cynomolgus monkey embryos. In embryonic development, hEPS progress into the gastrulation stage and express mesodermal and endodermal marker genes. Surprisingly, hEPS are difficult to differentiate into trophoblast cells in cynomolgus monkey embryos but are easier to differentiate into epiblast (EPI) cells. On the other hand, the regulation and differentiation of skeletal stem cells (SSCs), particularly human skeletal stem cells (hSSCs), are still largely not understood. SSCs are self-renewing and can generate osteoblasts, chondrocytes, and reticular bone marrow stromal cells, but not adipocytes (Bianco and Robey Citation2015). Through scRNA-seq quantitative analysis, differences in the gene expression of fetal and adult hSSCs during differentiation were found, which may partially explain the different proportions of ossicles, cartilage, and bone formed by hSSCs from different sources. In acute skeletal injury, hSSCs undergo significant local expansion at the injury site (Chan et al. Citation2018). He et al. (He et al. Citation2021) performed scRNA-seq of limb buds from 5-week human embryos and long bones from 8-week human embryos. A group of embryonic skeletal stem and progenitor cells (eSSPCs) located in the perichondrium, with the potential to self-renew and differentiate into cartilage and osteoblasts, was discovered by combining in vitro and in vivo functional verification. This suggests that human SSCs originate from the embryonic perichondrium. At the same time, this is the first study of the origin of SSCs in early human embryos, which is of great significance to achieve an in-depth understanding of the mechanism of human skeletal development and damage repair. In conclusion, the use of scRNA-seq to assist in the identification of hSSCs, reveal a bone development mechanisms, and responses to injury, will guide future approaches for bone regeneration.
Table 1. Summary of main stem cell types and their key characteristics.
Hematopoietic stem cells (HSCs) are derived from early embryonic precursor cells such as hematopoietic endothelial cells and pre-hematopoietic stem cells (pre-HSCs), and their molecular identities remain unclear. Zhou et al. (Zhou et al. Citation2016) used scRNA-seq to analyze HSCs in endothelial cells, aorta-gonad-mesonephros, and embryonic liver cells. Pre-HSCs were found to have a unique transcriptional mechanism, arterial characteristics, metabolic status, signaling pathways, and transcription factor networks. Notably, activation of the mechanistic target of rapamycin (mTOR) is essential for the emergence of HSCs but not for hematopoietic progenitor cells. This study paves the way for dissecting the complex molecular mechanisms that regulate progressive generation of HSCs in vivo. Zeng et al. (Zeng, He, et al. Citation2019a) performed scRNA-seq on Carnegie stage 12–14 (CS12-14) human embryonic cells. The results showed that blood-derived endothelial cells (HECs) displayed distinct characteristics of arterial endothelial cells (ECs), with upregulated expression of RUNX1, MYB, and ANGPT1. We further mapped the developmental path of HECs from arterial ECs to hematopoietic stem progenitor cells, which provides important theoretical support and guidance for hematopoietic stem cell regeneration and determining the hematopoietic fate of arterial ECs. In addition, Gao et al.(Gao et al. Citation2022) performed scRNA-seq on mouse embryonic livers from E11.5-E14.5. Differential gene screening revealed that the cell surface marker gene CD93 was enriched in hematopoietic stem/progenitor cells (HSC/MPP1), and with additional cell biological function tests, it was found that CD93-positive cells have stronger clone formation, recombination, and self-renewal capabilities. This indicates that CD93 could be used to generate HSCs/MPPs with strong stemness capabilities.
In a study of adipose stem cells using scRNA-seq, Ferrero et al. (Ferrero et al. Citation2020) found that adipose stem and progenitor cells (ASPC) are heterogeneous. Three subsets were included, namely adipose stem cells (ASCs), preadipocytes (PreAs), and adipogenesis regulators (Aregs). Both PreAs (ICAM1+ and VAP-1+) and Aregs (CD142+) originate from ASCs (DPP4+ and CD55+), whereas in adult mice, Aregs (CD142+) inhibited adipogenesis through a paracrine mechanism. In the study of musculoskeletal stem cells (MSSC), Yin et al. (Yin et al. Citation2020) constructed the developmental differentiation trajectory and cellular composition of mouse MSSCs by performing scRNA-seq on mouse limbs at different developmental stages. Furthermore, single-cell analysis of limb tissue in Scx-knockout mice showed that Scx regulates the self-renewal and proliferative potential of MSSCs. Multi-tissue RNA sequencing demonstrated that tissues of the musculoskeletal system, including muscle, bone, meniscus, and cartilage, were abnormally developed in Scx-knockout mice. It is also worth mentioning that scRNA-seq can be used to understand the cellular structure of cancer stem cells (Tirosh et al. Citation2016), the mobilization of regenerative stem cells in the regenerating small intestine (Ayyaz et al. Citation2019), and the cell differentiation potential and branching in the olfactory stem cell lineage trajectory (Fletcher et al. Citation2017). Thus, scRNA-seq helps to explore the structure and differentiation of stem cells with remarkably high resolution.
Mutual transformation between cells
In a variety of biological systems, cells show a series of different states, and transition into these states in a certain order of time. The most typical process is cell differentiation, where cells gradually differentiate from immature cells into mature cells. After the corresponding samples are sequenced using scRNA-seq technology, the transformation process between different cell states can be studied based on the sequencing data. The transformation process involves arranging different cells according to the quasi-time series from the start state, intermediate state, and end state according to the expression of genes in the cells to facilitate the interpretation of the underlying mechanism of cell state transformation. scRNA-seq of kidneys, found that the main cells and intercalated cells of the kidney could be transformed into each other by pseudo-chronological analysis, and it was confirmed that the activation of the Notch signaling pathway led to the transformation of leap cells to master cells, thus altering the ratio of the two types of cells, which may be the cause of metabolic acidosis (Park et al. Citation2018). Tosti et al. (Tosti et al. Citation2021) found that acinar cells could be divided into four subtypes: acinar-β, acinar-REG+, idling acinar cells (acinar-I), and secretory acinar cells (acinar-S). The latter two subtypes were identified for the first time in this study. Due to active protein synthesis in secretory acinar cells, the pressure of the endoplasmic reticulum increases, which enhances apoptosis of secretory acinar cells. It was inferred that resting acinar cells can be transformed into secretory acinar cells under stimulation. In this study, a single-cell map of all cell types in the pancreatic tissue was constructed for the first time.
In the study of meniscus, Sun et al. (Sun et al. Citation2020) performed scRNA-seq on samples of healthy and degenerated meniscus, followed by trace analysis by Monocle. The results showed that fibrochondral progenitor cells (FCP) and proliferating fibrochondrocytes (ProFC) were found in healthy meniscus, and then differentiated into fibrochondrocytes (FC) and prehypertrophic chondrocytes (PreHTC), and finally differentiated into regulatory chondrocytes (RegC) and PreHTCs. FCP and ProFCs are found in the degenerative meniscus, which first differentiate into degenerative meniscal progenitor cells (DegP) and chondrogenic progenitor cells (CPC), suggesting that abnormal cellular states occur during meniscal degeneration. Finally, the cell trajectories showed that DegP was a key factor in meniscal degeneration, and that TGFβ1 might delay the degeneration of the meniscus. Testicular tissues collected at early birth were subjected to scRNA-seq. Based on the expression of genes related to germ cell development, germ cell subsets were divided into four developmental stages: undifferentiated spermatogonia (UTF1+ MKI67−), differentiated spermatogonia (KIT+ MKI67+), primary spermatocytes (STRA8+ GPR85+), and spermatids (PRM3+). The developmental trajectory of germ cells was further verified using Monocle analysis (Guo J et al. Citation2020). In addition, Wu et al. (Wu F et al. Citation2021) performed scRNA-seq on advanced non-small cell lung cancer (NSCLC) and identified T helper 17 (Th17) cells for the first time. This revealed a potential transitional relationship between Th17 and Tregs. Through malignant pleural effusion and peripheral blood scRNA-seq analyzes, Huang et al. (Huang et al. Citation2021) found that naïve CD4+ T cells differentiate into follicular helper T (Tfh) cells through Th1/17 cells, while naïve CD8+ T cells eventually become depleted by fine cytotoxic T cells. Through scRNA-seq, we can understand the transformation between cells, as well as perform in-depth analysis of the molecular mechanism of cell transformation, which is of great significance in medicine, organism development, and other fields.
Intercellular interaction
In an organism, cell-to-cell interactions (CCI) in different cell types and tissues can coordinate the development of organisms. With developments in cell research, particularly the technology used, CCI and communication between cells (CCC) can now be inferred from gene expression. The simultaneous use of multiple research methods can reveal the fundamental roles of cells in their environment and how cellular functions are shaped. This has great potential for future applications, particularly in biomedicine and biotherapy. Particularly, with the increasing popularity of scRNA-seq, the study of CCI has greatly accelerated, especially the analysis of interactions between cell signaling molecules displayed by RNA expression. CCIs are closely associated with many biological processes, such as embryonic development, organogenesis, carcinogenesis, inflammation, drug effects, and drug resistance.
scRNA-seq of the brain tissues of young and old mice revealed that the number of T cells increased in the neural niches of old mice. Immunofluorescence staining and FACS analysis proved that T cells inhibit the proliferation of neural stem cells by secreting interferon-gamma, which provides an explanation for the functional decline in neural stem cells during aging (Dulken et al. Citation2019). scRNA-seq was also performed on the tail hematopoietic tissues of zebrafish. It was found that the endothelial cell-specific factor Gpr182 was necessary for the expansion of hematopoietic stem cells (HSPCs) in tail hematopoietic tissue and provided a stable research basis for the establishment of support for hematopoietic stem cell expansion in vitro(Xia et al. Citation2021). Wang et al. (Wang X et al. Citation2021a) performed scRNA-seq on cartilage tissues from healthy humans, Kashin-Beck disease (KBD), and osteoarthritis (OA). This study found that a new cell group, mitochondrial chondrocytes (MTCs), was significantly enlarged in KBD, which may represent an important mechanism of chondrocyte degeneration in KBD. Ge et al. (Ge et al. Citation2020) performed scRNA-seq on the skin of E13.5 (induction), E16.5 (organogenesis) fetal mice and neonatal mice (cell differentiation stage, P0). By predicting the interaction between ligands and receptors, it was found that there was strong cellular communication among stromal cell progenitor cells, interfollicular epidermis cells (IFE), and dermal condensate cells (DC) in the E16.5; thus there is strong autocrine regulation of DCs. Strong intercellular communication was also observed in the dermis and epidermis of E13.5 tissues.
Ramachandran et al. (Ramachandran et al. Citation2019) used scRNA-seq to identify pathogenic cell types that may lead to liver cirrhosis. By comparing the levels of macrophages, endothelial cells, and mesenchymal cells of normal liver and liver cirrhosis tissues, they identified subtypes of these three cell types, and the number of cells was upregulated in patient samples, and all cells had increased expression of fibrosis-related genes. Among them, scar-associated macrophages appeared to promote the expression of fibrocollagen genes in stellate cells and regulate the proliferation of scar-associated mesenchymal cells through the TNFRSF12A pathway. In addition, scar-associated endothelial cells increase the fibrillar collagen content in mesenchymal cells through the PDGFRA and Notch signaling pathways. Therefore, scRNA-seq can identify the subdivision of known cell types, thereby providing a method to solve biological problems.
Other
scRNA-seq can also be used in eukaryotes, viruses, antibody preparation, and other auxiliary research, such as to produce transcriptome maps of unicellular eukaryotes to reveal the individual differences and gene expression at different life cycle stages. For example, Plasmodium is a single-cell eukaryote and its complex life cycle shows surprising cellular plasticity. However, approximately 40% of the gene function is still unknown, which seriously hinders the research and development of antimalarial drugs. Howick et al. (Howick et al. Citation2019) studied 1787 P. berghei cells using scRNA-seq and divided them into 20 subgroups. Genes encoding the invasion-related proteins CelTos and CSP were expressed in cluster16. A total of 79 cluster genes were highly expressed in zygotes and sporozoites during invasion. Of these 79 genes, the functions of 34 genes are unknown. As the expression of these 34 genes is highly related to the expression of known invasion-related genes, it is worthy of further study.
Exploration of scRNA-seq in viruses. Using the results of viral scRNA-seq, Bost et al. (Bost et al. Citation2020) used the Viral-Track data analysis tool to identify virus-containing transcripts in cells to study the interaction between the virus and host cells. First, hepatitis B virus (HBV) was successfully identified in the liver tissue of hepatitis B patients, and HBV transcripts were highly enriched in hepatocytes and apoptotic hepatocytes. HBV transcripts were also detected in two macrophages types (CD163+ and TREM2+), one dendritic cell type (cDC1, XCR1+), one vascular endothelial cell type (OIT3+), and one epithelial cell type (KRT7+). These findings are worthy of further research. Notably, according to whether the virus transcript could be detected, cells of the same type were divided into two groups, and the differentially expressed genes were compared to confirm that after the virus infected the host cell, the expression of those genes in the host cell changed. Collectively, this study demonstrated the advantages of scRNA-seq of viruses to confirm, at the single-cell level, the cell types that the virus infects in vivo.
scRNA-seq in auxiliary antibody preparation can also be beneficial. Setliff et al. (Setliff et al. Citation2019) developed a new technique to link B-cell receptors to antigen specificity through sequencing (LIBRA-seq). Linking a specific DNA tag to each antigen converts the binding information of the BCR and antigen into events that can be detected by sequencing, thereby establishing the corresponding relationship between the BCR and antigen. Through scRNA-seq, the sequence of the BCR in each B cell and the type of antigen recognized by its BCR was determined. As the variable region sequence of the BCR is identical to that of the antibody, once the BCR sequence was obtained, the antibody was prepared at a large scale in vitro. Researchers found B cells in HIV patients that could bind to HIV antigens, prepared antibodies from the obtained BCR sequences, and found that they could significantly neutralize HIV antigens. In summary, scRNA-seq has broad application prospects in areas of research such as viruses, antibody preparation, unicellular eukaryotes.
Conclusion and future perspectives
It can be predicted that analyzing the changes and relationships between cells and analyzing the cellular and molecular mechanisms of trait formation will have a wide application prospect in analyzing the genetic mechanism of complex traits in humans and animals as well as in the prevention and treatment of diseases. There are numerous exciting medical applications that can utilize this technology. For example, a portable, low-cost, high-throughput Seq-Well technology can make scRNA-seq more widely used and accelerate scientific and clinical discoveries (Gierahn et al. Citation2017). Simultaneously, the system can also measure the protein secretion of each cell type, such as cytokines. There are cytokines secretion can be significantly different depending on the spatial and temporal properties of the cell type, state, lineage, and microenvironment. Monolithic hydrogel nanowells-in-microwells enable simultaneous single-cell secretion and phenotypic analysis (Choi et al. Citation2020). Future work may include the use of micromanipulation or syringes for single-cell sequencing, and other methods to separate high secretion. An in-depth understanding of the relationship between cytokine secretion and cell phenotypes is of great significance to further understand many biological processes and inform drug development in precision medicine. Moreover, liquid biopsy combined with the single-cell technique can obtain information on the genome and transcriptome and provide the molecular characteristics and target information associated with tumor metastasis (Keller and Pantel Citation2019). Thus, liquid biopsy combined with single cell sequencing could be a crucial technical path for future developments in precision cancer treatments.
With the development of scRNA-seq technology, this technology has a single limitation and restricts the deepening of research. In particular, cell survival is a complex dynamic process, and the study of a single transcript is not sufficient to fully reflect the changes in this process. Single-cell multigroup technology can capture various genomic information in the same cell, including single-cell transcriptome, proteome, genome, and epigenome information, which can more comprehensively reflect cell characteristics. For example, the combined analysis of single-cell transcripts and proteomes can provide a more detailed classification of cell subsets, in which RNA expression and protein sequencing (REAP-seq) (Peterson et al. Citation2017), and cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) (Stoeckius et al. Citation2017) can simultaneously measure proteins and mRNA in a single cell and can be annotated to identify rare cell types and states. In addition, simultaneous genomic and transcriptome sequencing of a single cell is the ideal combination to determine genetic variation and gene expression, in which genome and transcriptome sequencing (G&T-seq) (Macaulay et al. Citation2015) and TARGET-seq (Rodriguez-Meira et al. Citation2020) techniques can simultaneously sequence genomic DNA and full-length mRNA in a single cell, which can reveal the effect of genetic variation at the transcriptional level. Moreover, single-cell chromatin accessibility and transcriptome sequencing (scCAT-seq) (Liu L et al. Citation2019), single-nucleus chromatin accessibility and mRNA expression sequencing (SNARE-seq) (Chen S et al. Citation2019), and single-cell nucleosome, methylation, and transcription sequencing (scNMT-seq) (Clark et al. Citation2018) techniques can simultaneously detect chromatin accessibility and transcriptomes in a single cell, which is helpful in exploring the effects of gene expression variation on cell plasticity and diversity. Therefore, single-cell multi-group technology may be a powerful tool for the subsequent analysis of life processes and more comprehensive analysis of biological mechanisms.
In summary, with the continuous improvement of technology and methods, scRNA-seq is becoming an indispensable tool in many biomedical fields. It is predicted that single-cell multiplex technology will play a more powerful role in single-cell research of complex organs and tissues in the future. It is expected that the demand and application of scRNA-seq technology will increase greatly in the future, and the technology will become more refined, high-throughput, affordable, and easier to use in scientific research laboratories and clinical laboratories. Especially in the new era of precision medicine, the study of the characteristics of high intercellular heterogeneity and clonal evolution in the occurrence, development, and treatment of diseases brings hope for the accurate diagnosis and treatment of diseases. In particular, it can be used to monitor the progress, efficacy, and prognosis of hematological tumors, and is likely to find potential therapeutic targets, providing a basis for accurate diagnosis, dynamic monitoring, and individualized treatment of the disease. More importantly, innovative single-cell technology is expected to greatly promote the effective control of diseases in IVF and early pregnancy screening and diagnosis of chromosomal and genetic diseases by improving the efficiency and detection quality. Thus, scRNA-seq is of great significance to improve human genetic health.
Acknowledgements
RuoHan Zhao and YiCheng Bai contributed to the conception and design of the study, wrote the initial draft of the paper, and revised the paper. JingYing Zhao, Mei Hu, XinYan Zhang, and Min Yang revised the manuscript for important intellectual content. TengFei Dou and JunJing Jia have approved the final manuscript. All authors agree to be accountable for all aspects of this study.
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, et al. 2017. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 14(11):1083–1086.
- Arneth B. 2019. Tumor microenvironment. Medicina (Kaunas). 56(1):15.
- Ayyaz A, Kumar S, Sangiorgi B, Ghoshal B, Gosio J, Ouladan S, Fink M, Barutcu S, Trcka D, Shen J, et al. 2019. Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature. 569(7754):121–125.
- Bianco P, Robey PG. 2015. Skeletal stem cells. Development. 142(6):1023–1027.
- Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, et al. 2015. Organoid models of human and mouse ductal pancreatic cancer. Cell. 160(1-2):324–338.
- Bost P, Giladi A, Liu Y, Bendjelal Y, Xu G, David E, Blecher-Gonen R, Cohen M, Medaglia C, Li H, et al. 2020. Host-Viral infection maps reveal signatures of severe COVID-19 patients. Cell. 181(7):1475–1488 e1412.
- Brazovskaja A, Treutlein B, Camp JG. 2019. High-throughput single-cell transcriptomics on organoids. Curr Opin Biotechnol. 55:167–171.
- Camp JG, Badsha F, Florio M, Kanton S, Gerber T, Wilsch-Brauninger M, Lewitus E, Sykes A, Hevers W, Lancaster M, et al. 2015. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci U S A. 112(51):15672–15677.
- Chan CKF, Gulati GS, Sinha R, Tompkins JV, Lopez M, Carter AC, Ransom RC, Reinisch A, Wearda T, Murphy M, et al. 2018. Identification of the human skeletal stem cell. Cell. 175(1):43–56.e21. e21.
- Chen S, Lake BB, Zhang K. 2019. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat Biotechnol. 37(12):1452–1457. eng.
- Chen Z, Zhao M, Liang J, Hu Z, Huang Y, Li M, Pang Y, Lu T, Sui Q, Zhan C, et al. 2021. Dissecting the single-cell transcriptome network underlying esophagus non-malignant tissues and esophageal squamous cell carcinoma. EBioMedicine. 69:103459.
- Chen Z, Zhou L, Liu L, Hou Y, Xiong M, Yang Y, Hu J, Chen K. 2020. Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nat Commun. 11(1):5077.
- Choi JR, Lee JH, Xu A, Matthews K, Xie S, Duffy SP, Ma H. 2020. Monolithic hydrogel nanowells-in-microwells enabling simultaneous single cell secretion and phenotype analysis. Lab Chip. 20(24):4539–4551. eng.
- Clark SJ, Argelaguet R, Kapourani CA, Stubbs TM, Lee HJ, Alda-Catalinas C, Krueger F, Sanguinetti G, Kelsey G, Marioni JC, et al. 2018. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat Commun. 9(1):781. eng.
- Cowan CS, Renner M, De Gennaro M, Gross-Scherf B, Goldblum D, Hou Y, Munz M, Rodrigues TM, Krol J, Szikra T, et al. 2020. Cell types of the human retina and its organoids at single-cell resolution. Cell. 182(6):1623–1640 e1634.
- Cui Y, Zheng Y, Liu X, Yan L, Fan X, Yong J, Hu Y, Dong J, Li Q, Wu X, et al. 2019. Single-Cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 26(7):1934–1950 e1935.
- Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, Zhang Y, Neff N, Kowarsky M, Caneda C, et al. 2017. Single-Cell RNA-Seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep. 21(5):1399–1410.
- Denny P, Feuermann M, Hill DP, Lovering RC, Plun-Favreau H, Roncaglia P. 2018. Exploring autophagy with gene ontology. Autophagy. 14(3):419–436.
- Dulken BW, Buckley MT, Navarro Negredo P, Saligrama N, Cayrol R, Leeman DS, George BM, Boutet SC, Hebestreit K, Pluvinage JV, et al. 2019. Single-cell analysis reveals T cell infiltration in old neurogenic niches. Nature. 571(7764):205–210.
- Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. 2020. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc. 15(4):1484–1506.
- Fang Z, Tian Y, Sui C, Guo Y, Hu X, Lai Y, Liao Z, Li J, Feng G, Jin L, et al. 2022. Single-Cell transcriptomics of proliferative phase endometrium: systems analysis of cell-cell communication network using CellChat. Front Cell Dev Biol. 10:919731. eng.
- Fatehullah A, Tan SH, Barker N. 2016. Organoids as an in vitro model of human development and disease. Nat Cell Biol. 18(3):246–254.
- Ferrero R, Rainer P, Deplancke B. 2020. Toward a consensus view of mammalian adipocyte stem and progenitor cell heterogeneity. Trends Cell Biol. 30(12):937–950.
- Fletcher RB, Das D, Gadye L, Street KN, Baudhuin A, Wagner A, Cole MB, Flores Q, Choi YG, Yosef N, et al. 2017. Deconstructing olfactory stem cell trajectories at single-cell resolution. Cell Stem Cell. 20(6):817–830 e818.
- Gao S, Shi Q, Zhang Y, Liang G, Kang Z, Huang B, Ma D, Wang L, Jiao J, Fang X, et al. 2022. Identification of HSC/MPP expansion units in fetal liver by single-cell spatiotemporal transcriptomics. Cell Res. 32(1):38–53.
- Ge W, Tan SJ, Wang SH, Li L, Sun XF, Shen W, Wang X. 2020. Single-cell transcriptome profiling reveals dermal and epithelial cell fate decisions during embryonic hair follicle development. Theranostics. 10(17):7581–7598.
- Gierahn TM, Wadsworth MH, Hughes TK, Bryson BD, Butler A, Satija R, Fortune S, Love JC, Shalek AK. 2017. Seq-well: portable, low-cost RNA sequencing of single cells at high throughput. Nat Methods. 14(4):395–398. eng.
- Gohil SH, Iorgulescu JB, Braun DA, Keskin DB, Livak KJ. 2021. Applying high-dimensional single-cell technologies to the analysis of cancer immunotherapy. Nat Rev Clin Oncol. 18(4):244–256.
- Guo F, Zhang B, Yang H, Fu Y, Wang Y, Huang J, Cheng M, Li X, Shen Z, Li L, et al. 2021. Systemic transcriptome comparison between early- and late-onset pre-eclampsia shows distinct pathology and novel biomarkers. Cell Prolif. 54(2):e12968.
- Guo J, Nie X, Giebler M, Mlcochova H, Wang Y, Grow EJ, DonorConnect KR, Tharmalingam M, Matilionyte G, et al. 2020. The dynamic transcriptional cell atlas of testis development during human puberty. Cell Stem Cell. 26(2):262–276.e4. e264.
- Hanzelmann S, Castelo R, Guinney J. 2013. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics. 14:7.
- He J, Yan J, Wang J, Zhao L, Xin Q, Zeng Y, Sun Y, Zhang H, Bai Z, Li Z, et al. 2021. Dissecting human embryonic skeletal stem cell ontogeny by single-cell transcriptomic and functional analyses. Cell Res. 31(7):742–757.
- Hinshaw DC, Shevde LA. 2019. The tumor microenvironment innately modulates cancer progression. Cancer Res. 79(18):4557–4566. eng.
- Hoang SA, Oseini A, Feaver RE, Cole BK, Asgharpour A, Vincent R, Siddiqui M, Lawson MJ, Day NC, Taylor JM, et al. 2019. Gene expression predicts histological severity and reveals distinct molecular profiles of nonalcoholic fatty liver disease. Sci Rep. 9(1):12541.
- Howick VM, Russell AJC, Andrews T, Heaton H, Reid AJ, Natarajan K, Butungi H, Metcalf T, Verzier LH, Rayner JC, et al. 2019. The malaria cell atlas: single parasite transcriptomes across the completeplasmodiumlife cycle. Science. 365:6455.
- Hu J, Chen Z, Bao L, Zhou L, Hou Y, Liu L, Xiong M, Zhang Y, Wang B, Tao Z, et al. 2020. Single-Cell transcriptome analysis reveals intratumoral heterogeneity in ccRCC, which results in different clinical outcomes. Mol Ther. 28(7):1658–1672.
- Huang ZY, Shao MM, Zhang JC, Yi FS, Du J, Zhou Q, Wu FY, Li S, Li W, Huang XZ, et al. 2021. Single-cell analysis of diverse immune phenotypes in malignant pleural effusion. Nat Commun. 12(1):6690. eng.
- Jiang X, Stockwell BR, Conrad M. 2021. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 22(4):266–282.
- Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, Myung P, Plikus MV, Nie Q. 2021. Inference and analysis of cell-cell communication using CellChat. Nat Commun. 12(1):1088. eng.
- Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, Dowling CM, Gao D, Begthel H, Sachs N, et al. 2014. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell. 159(1):163–175.
- Keller L, Pantel K. 2019. Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat Rev Cancer. 19(10):553–567. eng.
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lonnerberg P, Furlan A, et al. 2018. RNA velocity of single cells. Nature. 560(7719):494–498.
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. 2013. Cerebral organoids model human brain development and microcephaly. Nature. 501(7467):373–379.
- Lenis TL, Hu J, Ng SY, Jiang Z, Sarfare S, Lloyd MB, Esposito NJ, Samuel W, Jaworski C, Bok D, et al. 2018. Expression of ABCA4 in the retinal pigment epithelium and its implications for stargardt macular degeneration. Proc Natl Acad Sci U S A. 115(47):E11120–E11127.
- Li R, Zhong C, Yu Y, Liu H, Sakurai M, Yu L, Min Z, Shi L, Wei Y, Takahashi Y, et al. 2019. Generation of blastocyst-like structures from mouse embryonic and adult cell cultures. Cell. 179(3):687–702 e618.
- Liu L, Liu C, Quintero A, Wu L, Yuan Y, Wang M, Cheng M, Leng L, Xu L, Dong G, et al. 2019. Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity. Nat Commun. 10(1):470. eng.
- Liu X, Tan JP, Schroder J, Aberkane A, Ouyang JF, Mohenska M, Lim SM, Sun YBY, Chen J, Sun G, et al. 2021. Modelling human blastocysts by reprogramming fibroblasts into iBlastoids. Nature. 591(7851):627–632.
- Luecken MD, Theis FJ. 2019. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol. 15(6):e8746.
- Luo T, Zheng F, Wang K, Xu Y, Xu H, Shen W, Zhu C, Zhang X, Sui W, Tang D, et al. 2021. A single-cell map for the transcriptomic signatures of peripheral blood mononuclear cells in end-stage renal disease. Nephrol Dial Transplant. 36(4):599–608.
- Macaulay IC, Haerty W, Kumar P, Li YI, Hu TX, Teng MJ, Goolam M, Saurat N, Coupland P, Shirley LM, et al. 2015. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat Methods. 12(6):519–522. eng.
- McCracken KW, Cata EM, Crawford CM, Sinagoga KL, Schumacher M, Rockich BE, Tsai YH, Mayhew CN, Spence JR, Zavros Y, et al. 2014. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature. 516(7531):400–404.
- Meistermann D, Bruneau A, Loubersac S, Reignier A, Firmin J, Francois-Campion V, Kilens S, Lelievre Y, Lammers J, Feyeux M, et al. 2021. Integrated pseudotime analysis of human pre-implantation embryo single-cell transcriptomes reveals the dynamics of lineage specification. Cell Stem Cell. 28(9):1625–1640e1626.
- Paik DT, Cho S, Tian L, Chang HY, Wu JC. 2020. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat Rev Cardiol. 17(8):457–473.
- Pan Y, Lu F, Fei Q, Yu X, Xiong P, Yu X, Dang Y, Hou Z, Lin W, Lin X, et al. 2019. Single-cell RNA sequencing reveals compartmental remodeling of tumor-infiltrating immune cells induced by anti-CD47 targeting in pancreatic cancer. J Hematol Oncol. 12(1):124. eng.
- Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, Li M, Barasch J, Susztak K. 2018. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science. 360(6390):758–763.
- Parker KR, Migliorini D, Perkey E, Yost KE, Bhaduri A, Bagga P, Haris M, Wilson NE, Liu F, Gabunia K, et al. 2020. Single-Cell analyses identify brain mural cells expressing CD19 as potential off-tumor targets for CAR-T immunotherapies. Cell. 183(1):126–142.e17. e117.
- Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC, Moore R, McClanahan TK, Sadekova S, Klappenbach JA. 2017. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol. 35(10):936–939. eng.
- Picelli S. 2017. Single-cell RNA-sequencing: the future of genome biology is now. RNA Biol. 14(5):637–650.
- Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, et al. 2016. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell. 165(5):1238–1254.
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, Trapnell C. 2017. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods. 14(10):979–982.
- Qu M, Xiong L, Lyu Y, Zhang X, Shen J, Guan J, Chai P, Lin Z, Nie B, Li C, et al. 2021. Establishment of intestinal organoid cultures modeling injury-associated epithelial regeneration. Cell Res. 31(3):259–271.
- Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, Portman JR, Matchett KP, Brice M, Marwick JA, et al. 2019. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 575(7783):512–518.
- Rambow F, Rogiers A, Marin-Bejar O, Aibar S, Femel J, Dewaele M, Karras P, Brown D, Chang YH, Debiec-Rychter M, et al. 2018. Toward minimal residual disease-directed therapy in melanoma. Cell. 174(4):843–855 e819.
- Rodriguez-Meira A, O’Sullivan J, Rahman H, Mead AJ. 2020. TARGET-Seq: a protocol for high-sensitivity single-cell mutational analysis and parallel RNA sequencing. STAR Protoc. 1(3):100125. eng.
- Romanov RA, Tretiakov EO, Kastriti ME, Zupancic M, Haring M, Korchynska S, Popadin K, Benevento M, Rebernik P, Lallemend F, et al. 2020. Molecular design of hypothalamus development. Nature. 582(7811):246–252.
- Rossi G, Manfrin A, Lutolf MP. 2018. Progress and potential in organoid research. Nat Rev Genet. 19(11):671–687.
- Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, Balgobind AV, Wind K, Gracanin A, Begthel H, et al. 2018. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 172(1-2):373–386 e310.
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, et al. 2009. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 459(7244):262–265.
- Setliff I, Shiakolas AR, Pilewski KA, Murji AA, Mapengo RE, Janowska K, Richardson S, Oosthuysen C, Raju N, Ronsard L, et al. 2019. High-Throughput mapping of B cell receptor sequences to antigen specificity. Cell. 179(7):1636–1646 e1615.
- Shao X, Lu X, Liao J, Chen H, Fan X. 2020. New avenues for systematically inferring cell-cell communication: through single-cell transcriptomics data. Protein Cell. 11(12):866–880. eng.
- Shen S, Wang R, Qiu H, Li C, Wang J, Xue J, Tang Q. 2021. Development of an autophagy-based and stemness-correlated prognostic model for hepatocellular carcinoma using bulk and single-cell RNA-sequencing. Front Cell Dev Biol. 9:743910.
- Sheng X, Guan Y, Ma Z, Wu J, Liu H, Qiu C, Vitale S, Miao Z, Seasock MJ, Palmer M, et al. 2021. Mapping the genetic architecture of human traits to cell types in the kidney identifies mechanisms of disease and potential treatments. Nat Genet. 53(9):1322–1333.
- Shirai H, Mandai M, Matsushita K, Kuwahara A, Yonemura S, Nakano T, Assawachananont J, Kimura T, Saito K, Terasaki H, et al. 2016. Transplantation of human embryonic stem cell-derived retinal tissue in two primate models of retinal degeneration. Proc Natl Acad Sci U S A. 113(1):E81–E90.
- Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, Smibert P. 2017. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 14(9):865–868. eng.
- Stuart T, Satija R. 2019. Integrative single-cell analysis. Nat Rev Genet. 20(5):257–272.
- Sun H, Wen X, Li H, Wu P, Gu M, Zhao X, Zhang Z, Hu S, Mao G, Ma R, et al. 2020. Single-cell RNA-Seq analysis identifies meniscus progenitors and reveals the progression of meniscus degeneration. Ann Rheum Dis. 79(3):408–417.
- Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, Zhang RR, Ueno Y, Zheng YW, Koike N, et al. 2013. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 499(7459):481–484.
- Tan T, Wu J, Si C, Dai S, Zhang Y, Sun N, Zhang E, Shao H, Si W, Yang P, et al. 2021. Chimeric contribution of human extended pluripotent stem cells to monkey embryos ex vivo. Cell. 184(13):3589.
- Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, Fisher JM, Rodman C, Mount C, Filbin MG, et al. 2016. Single-cell RNA-Seq supports a developmental hierarchy in human oligodendroglioma. Nature. 539(7628):309–313.
- Tosti L, Hang Y, Debnath O, Tiesmeyer S, Trefzer T, Steiger K, Ten FW, Lukassen S, Ballke S, Kuhl AA, et al. 2021. Single-nucleus and in situ RNA-sequencing reveal cell topographies in the human pancreas. Gastroenterology. 160(4):1330–1344 e1311.
- Van den Brink SC, Baillie-Johnson P, Balayo T, Hadjantonakis AK, Nowotschin S, Turner DA, Martinez Arias A. 2014. Symmetry breaking, germ layer specification and axial organisation in aggregates of mouse embryonic stem cells. Development. 141(22):4231–4242.
- Van de Sande B, Flerin C, Davie K, De Waegeneer M, Hulselmans G, Aibar S, Seurinck R, Saelens W, Cannoodt R, Rouchon Q, et al. 2020. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat Protoc. 15(7):2247–2276.
- Vishnubalaji R, Alajez NM. 2021. Transcriptional landscape associated with TNBC resistance to neoadjuvant chemotherapy revealed by single-cell RNA-Seq. Mol Ther Oncolytics. 23:151–162. eng.
- Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. 2019. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 30(1):36–50. .eng.
- Wang X, Ning Y, Zhang P, Poulet B, Huang R, Gong Y, Hu M, Li C, Zhou R, Lammi MJ, et al. 2021a. Comparison of the major cell populations among osteoarthritis, kashin-beck disease and healthy chondrocytes by single-cell RNA-Seq analysis. Cell Death Dis. 12(6):551.
- Wang Z, Li Z, Zhou K, Wang C, Jiang L, Zhang L, Yang Y, Luo W, Qiao W, Wang G, et al. 2021b. Deciphering cell lineage specification of human lung adenocarcinoma with single-cell RNA sequencing. Nat Commun. 12(1):6500. eng.
- Warmflash A, Sorre B, Etoc F, Siggia ED, Brivanlou AH. 2014. A method to recapitulate early embryonic spatial patterning in human embryonic stem cells. Nat Methods. 11(8):847–854.
- Wu F, Fan J, He Y, Xiong A, Yu J, Li Y, Zhang Y, Zhao W, Zhou F, Li W, et al. 2021. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat Commun. 12(1):2540.
- Wu SZ, Roden DL, Wang C, Holliday H, Harvey K, Cazet AS, Murphy KJ, Pereira B, Al-Eryani G, Bartonicek N, et al. 2020. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. Embo J. 39(19):e104063. eng.
- Xia J, Kang Z, Xue Y, Ding Y, Gao S, Zhang Y, Lv P, Wang X, Ma D, Wang L, et al. 2021. A single-cell resolution developmental atlas of hematopoietic stem and progenitor cell expansion in zebrafish. Proc Natl Acad Sci U S A. 118:14.
- Xie X, Liu M, Zhang Y, Wang B, Zhu C, Wang C, Li Q, Huo Y, Guo J, Xu C, et al. 2021. Single-cell transcriptomic landscape of human blood cells. Natl Sci Rev. 8(3):nwaa180.
- Yamanaka S. 2020. Pluripotent stem cell-based cell therapy—promise and challenges. Cell Stem Cell. 27(4):523–531.
- Yin Z, Lin J, Yan R, Liu R, Liu M, Zhou B, Zhou W, An C, Chen Y, Hu Y, et al. 2020. Atlas of musculoskeletal stem cells with the soft and hard tissue differentiation architecture. Adv Sci (Weinh). 7(23):2000938.
- Zeng Y, He J, Bai Z, Li Z, Gong Y, Liu C, Ni Y, Du J, Ma C, Bian L, et al. 2019a. Tracing the first hematopoietic stem cell generation in human embryo by single-cell RNA sequencing. Cell Res. 29(11):881–894.
- Zeng Y, Liu C, Gong Y, Bai Z, Hou S, He J, Bian Z, Li Z, Ni Y, Yan J, et al. 2019b. Single-Cell RNA sequencing resolves spatiotemporal development of Pre-thymic lymphoid progenitors and thymus organogenesis in human embryos. Immunity. 51(5):930–948.e6. e936.
- Zhang M, Hu S, Min M, Ni Y, Lu Z, Sun X, Wu J, Liu B, Ying X, Liu Y. 2021a. Dissecting transcriptional heterogeneity in primary gastric adenocarcinoma bysingle cell RNA sequencing. Gut. 70(3):464–475.
- Zhang M, Yang H, Wan L, Wang Z, Wang H, Ge C, Liu Y, Hao Y, Zhang D, Shi G, et al. 2020. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J Hepatol. 73(5):1118–1130.
- Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, Modak M, Carotta S, Haslinger C, Kind D, et al. 2019. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell. 179(4):829–845.e20. e820.
- Zhang Y, Han S, Kong M, Tu Q, Zhang L, Ma X. 2021b. Single-cell RNA-Seq analysis identifies unique chondrocyte subsets and reveals involvement of ferroptosis in human intervertebral disc degeneration. Osteoarthritis Cartilage. 29(9):1324–1334.
- Zhao T, Fu Y, Zhu J, Liu Y, Zhang Q, Yi Z, Chen S, Jiao Z, Xu X, Xu J, et al. 2018. Single-Cell RNA-Seq reveals dynamic early embryonic-like programs during chemical reprogramming. Cell Stem Cell. 23(1):31–45 e37.
- Zheng Y, Chen Z, Han Y, Han L, Zou X, Zhou B, Hu R, Hao J, Bai S, Xiao H, et al. 2020. Immune suppressive landscape in the human esophageal squamous cell carcinoma microenvironment. Nat Commun. 11(1):6268. eng.
- Zhou F, Li X, Wang W, Zhu P, Zhou J, He W, Ding M, Xiong F, Zheng X, Li Z, et al. 2016. Tracing haematopoietic stem cell formation at single-cell resolution. Nature. 533(7604):487–492.