
Abstract
Skeletal muscle comprises the major organ system in our bodies and is the effector tissue for fundamental processes, such as movement, breathing and glucose homeostasis. However, muscle regeneration can be insufficient to repair major injuries or become exhausted by the ongoing fiber damage that occurs in muscular dystrophy. Cell therapy to provide a source of myogenic stem cells that repopulate the lost muscle fibers is an attractive option to treat skeletal muscle injury or dystrophy. Induced pluripotent stem cells (iPSCs) have the potential to be an ideal source of donor muscle cells. This review will discuss major research applications of iPSCs to facilitate skeletal muscle repair, which involve myogenic cell transplantation, in vitro modeling of normal and dystrophic muscle tissue, and drug screening protocols. Recent advances in these fields will be discussed in this review, such as the production of neuromuscular junctions between muscle fibers and motor neurons, along with an overview of the problems that currently prevent iPSCs from being a clinically valid source of transplantable skeletal muscle stem cells. Potential future applications of iPSCs to enhance skeletal muscle regeneration will also be discussed.
Introduction: skeletal muscle structure, function and regenerative capacity
Skeletal muscle is a highly specialized, post-mitotic tissue and the largest organ system in the body, comprising around 40% of body weight (Kim et al. Citation2016). The structure of skeletal muscle is shown in . The muscle consists of bundles of aligned, multinucleate contractile cells termed myofibers (Mukund and Subramaniam Citation2020). Each myofiber is composed of thousands of myofibrils, which are the contractile units of skeletal muscle, being made up of thick myosin and thin actin myofilaments. The myofilaments are organized into structural units termed sarcomeres. The sarcolemma (also known as the myolemma) is a specialized cell membrane system that surrounds each myofiber. Muscle contraction is initiated by synaptic input from the motor neuron and muscle fiber at the neuromuscular junction. Contraction takes place via the process of excitation–contraction coupling (Sandow Citation1952), in which the signal is carried along muscle fibers by a network of transverse tubules (t-tubules). Depolarization of the inner portion of the myofiber activates dihydropyridine receptors in enlarged areas of the sarcoplasmic reticulum surrounding the t-tubules, known as the terminal cisternae. The activated dihydropyridine receptors are in close proximity to ryanodine receptors in the adjacent sarcoplasmic reticulum, and physically interact with the ryanodine receptors to activate them. The ryanodine receptors open, and calcium (Ca2+) is released from the sarcoplasmic reticulum into the local junctional space. Ca2+ diffuses into the cytoplasm to produce a Ca2+ spark (Niggli Citation1999). The released Ca2+ in the cytosol binds troponin C by the actin filaments that results in cross-bridge cycling and the production of force (Kuo and Ehrlich Citation2015).
Figure 1. Highly organized structure of skeletal muscle. Individual muscles are surrounded by a layer of connective tissue called the perimysium. Each muscle is composed of bundles of contractile cells termed muscle fibers. These fibers contain sarcomeres, which are repeating units of myofibrils that give skeletal muscle its banding pattern. Two contractile proteins, myosin and actin, form the thick and thin filaments of the sarcomere, respectively. The interaction between myosin and actin produces muscle contraction.
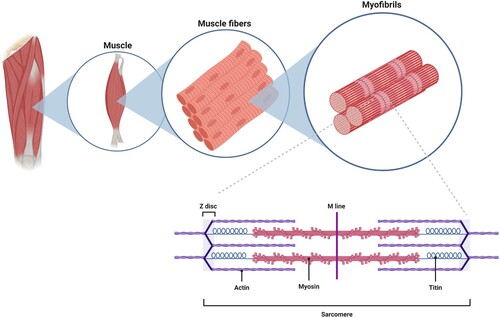
Skeletal muscle is also highly vascularized due to the metabolic demands required for contraction (Latroche et al. Citation2015). Numerous primary arteries run parallel to the myofibers. These branch into arterioles and capillaries that carry blood to the myofibers. Connective tissue plays an important role in maintaining the structure of skeletal muscle (Purslow Citation2010). Individual myofibers are surrounded by a thin layer of connective tissue termed the endomysium, containing nerves and capillaries. Bundles of myofibers are grouped together into fascicles surrounded by the perimysium connective tissue. Finally, the entire muscle is surrounded by a fibrous connective tissue termed the epimysium.
Myofibers can be subdivided into two major subtypes, termed slow (type 1) or fast (type 2), which is dependent on their functional properties and the expression of ATPase or myosin heavy chain isoforms (Cretoiu et al. Citation2018). Type 1 myofibers are slow twitch, utilize oxidative metabolism and possess relatively greater endurance than type 2 myofibers, whereas type 2 myofibers are fast twitch and designed for strength. In humans, type 2 fibers can be further subdivided into type 2A (fast oxidative/glycolytic) and type 2X (fast glycolytic). Additional fiber types exist in other mammals, for example type 2b, based on myosin heavy chain expression. It should be noted that myofibers are capable of fiber type switching in response to workload or disease (Pette and Staron Citation1997; Lee et al. Citation2018). For example, heart failure induces type 1 myofibers to switch to type 2 (Philippou et al. Citation2020). Individual muscles vary in their proportion of type 1 and type 2 myofibers that also reflects their function. For example, the soleus muscle, which is involved in standing, contains around 70% type 1 myofibers, whereas the vastus lateralis, which is the most powerful part of the quadriceps femoris, contains around 70% type 2 myofibers (Edgerton et al. Citation1975). A separate group are the craniofacial muscles, which are evolutionarily, morphologically and molecularly distinct from those of the trunk muscle and account for over 10% of the skeletal muscle system (Sambasivan et al. Citation2011a; Kim et al. Citation2020). For example, the extraocular muscles are spared from pathology in Duchenne muscular dystrophy (Khurana et al. Citation1995).
Skeletal muscle possesses an impressive capacity for regeneration (Carlson and Faulkner Citation1983). This is due to the presence of a population of quiescent muscle stem cells (termed satellite cells) that reside beneath the basal lamina and above the sarcolemma of myofibers (Dhawan and Rando Citation2005). Satellite cells (also termed myosatellite cells) are mitotically quiescent in mature muscle, but retain the potential to provide myoblasts for muscle hypertrophy and repair (Scharner and Zammit Citation2011). In response to muscle injury, satellite cells become activated, re-enter the cell cycle and proliferate to supply a population of myoblasts (the precursors to skeletal muscle cells), which can repair or replace damaged myofibers by differentiation and fusion with existing myofibers or other myoblasts (Kadi et al. Citation2004; Siegel et al. Citation2011). Alternatively, a portion of the activated satellite cells may return to a quiescent state (Kuang et al. Citation2007).
The regenerative capacity of skeletal muscle is not unlimited. Myopathies that produce cycles of myofiber degeneration, such as Duchenne muscular dystrophy, eventually exhaust these regeneration mechanisms and lead to fibrosis and adipose tissue infiltration (Mah Citation2018). In addition, aging is associated with a decline in skeletal muscle function that becomes noticeable from the fifth decade of life and can progress to severe muscle loss in old age, which is termed sarcopenia (Vicenti et al. Citation2020). Injuries that produce major skeletal muscle tissue injuries, for example those caused by firearms and automobile collisions, can also overwhelm the regenerative response if the connective tissue scaffold is destroyed.
Satellite cell activity during the process of muscle regeneration is tightly controlled by intrinsic factors within the satellite cells and extrinsic factors produced by the local microenvironment (also known as the muscle stem cell niche) (Yin et al. Citation2013). For example, although satellite cells are a source muscle stem cells for skeletal muscle regeneration, this process is also regulated by diffusible molecules released from other cell types in the muscle tissue, such as motor neurons (e.g. nerve growth factor (Yin et al. Citation2013), endothelial cells (e.g. vascular endothelial growth factor (Prior et al. Citation2003), fibro-adipogenic progenitors (e.g. interleukin-6 (Joe et al. Citation2010), immune cells (e.g. tumor necrosis factor-α secreted by macrophages (Koike et al. Citation2022), and connective tissue cells (e.g. angiopoietin 1 secreted by pericytes Kostallari et al. Citation2015. The extracellular matrix can also be a source of factors that influence the efficiency of regeneration.
It should be noted that laboratory studies have indicated that some non-satellite cell populations in skeletal muscle possess the potential for myogenic differentiation. For example, pericytes and ‘side population’ cells (harvested by cell sorting) can commit to myogenesis after muscle injury or co-culture with myoblasts (Tanaka et al. Citation2009; Dellavalle et al. Citation2011; Wang and Rudnicki Citation2011). However, satellite cell ablation experiments have indicated that these cells do not effectively contribute to skeletal muscle regeneration or replenish the satellite cell population (Lepper et al. Citation2011; Wang and Rudnicki Citation2011; Sambasivan et al. Citation2011b).
Cell therapy is an attractive option for replenishing or complimenting the endogenous satellite cell population when their capacity for regeneration is insufficient to maintain muscle mass and function. Autologous muscle stem cell therapy would be an ideal approach, in which the patient’s satellite cells are obtained from a muscle biopsy, propagated by culture in vitro, and then transplanted back into the muscle tissue to enhance regeneration capacity. Studies in mice have shown that the transplantation of as few as 900 purified satellite cells into the tibialis anterior muscle can improve function in models of Duchenne muscular dystrophy and sarcopenia (Cosgrove et al. Citation2014; Judson and Rossi Citation2020). However, this success was not translated to human trials and it became apparent that the need to expand the satellite cell population by culture causes their conversion into a muscle precursor cell termed myoblasts, which have much reduced myogenic potential and survival post-transplantation. Research efforts have adopted a number of approaches to attempt to circumvent this problem, including; 1) small molecule compounds and/or growth factor cocktails that promote myoblast engraftment and differentiation, 2) biophysical factors that model the muscle ECM during satellite cell culture, and 3) inclusion of biomaterials that mimic the structure of muscle to enhance transplantation (reviewed in (Judson and Rossi Citation2020).
Due to the inability of satellite cells to be sufficiently expanded in vitro, alternative sources of myogenic cells have been considered. One appealing approach is to utilize stem cells that are pluripotent (the ability to differentiate into all of the structures in the adult body) and induce myogenic commitment prior to transplantation. Theoretically, embryonic stem cells (ESCs) derived from the inner cell mass of the blastocyst before implantation into the uterus can provide a reliable source of high quality pluripotent cells. A major stumbling block for this approach is the ethical issues associated with ESC research, such as the reliance on discarded embryos to produce ESC lines (Jung et al. Citation2014). Embryos can also be created for therapeutic cloning using somatic cell nuclear transfer, although this still involves the destruction of the donor egg cell nucleus (de Wert and Mummery Citation2003). These issues necessitated the need to develop alternative sources of pluripotent cells for skeletal muscle cell therapy and disease modeling.
iPSCs: history and applicability to skeletal muscle regeneration
A major breakthrough in the generation of patient-derived cells with a similar differentiation potential to ECSs was the development of mouse induced pluripotent stem cells (iPSCs) in 2006. Readily available somatic cells can be reprogramed into iPSCs by the forced expression of transcription factors linked to dedifferentiation or the maintenance of stem cell behavior, such as the classic ‘Yamanaka’ factors: Oct3/4, Sox2, Klf4, c-Myc (Takahashi and Yamanaka Citation2006). In 2007, the iPSC protocol had been modified to use Nanog as the reporter gene for pluripotency (instead of Fbx15), generating iPSCs that achieved the ‘gold standard’ for pluripotency: production of chimeric mice and contribution to the germ line (Maherali et al. Citation2007; Okita et al. Citation2007; Wernig et al. Citation2007). By late 2007, it was reported that iPSC technology had been successfully applied to human cells, using the same four transcription factors or Oct4, Sox2, Nanog, and Lin28 (Takahashi et al. Citation2007; Yu et al. Citation2007). iPSC reprograming methods have been optimized to reduce reliance on potentially oncogenic transcription factors and improve the efficiency of iPSC generation, such as the substitution of one or more transcription factors with small molecules or utilizing microRNAs to inhibit repressors of the reprograming process (Bao et al. Citation2013; Hou et al. Citation2013).
The potential for iPSC-based technology to produce large numbers of myogenic cells was of great interest to researchers specializing in skeletal muscle degenerative disorders, because it would circumvent problems associated with utilizing ESCs or non-muscle cells with myogenic potential. These cells could be utilized for cell therapy studies or in vitro modeling of the disease phenotype, for applications such as drug screening or the generation of 3D cultures of human skeletal muscle tissue. In the next section of this review, we will discuss recent progress in two major therapeutic applications of iPSC-derived myogenic cells: Duchenne muscular dystrophy and volumetric muscle loss.
iPSC-based therapeutic approaches for Duchenne muscular dystrophy
Disease course and current treatment options
Duchenne muscular dystrophy (DMD) is a severe form of X-chromosome linked muscular dystrophy with an incidence of approximately 1 in 5,000 male births (Emery Citation2002). Much more rarely, females with compound heterozygous mutations may also be affected, with an incidence of approximately 1:50,000,000 births (Nozoe et al. Citation2016). Muscle weakness presents at around 4 years of age with symptoms such as Gowers's sign (difficulty standing up) and increased frequency of falls (Takeshita et al. Citation2017; Hong et al. Citation2019). Walking ability usually disappears by 13 years of age and patients become essentially paralyzed from the neck down by age 21 (Rowland Citation1985). With high quality health care, patients may survive until the fourth or even fifth decade, although the mean life expectancy is estimated to be around 28 years (Landfeldt et al. Citation2020; Broomfield et al. Citation2021). The most common cause of death is cardio-respiratory failure (Van Ruiten et al. Citation2016). DMD is caused by mutations in the dystrophin gene, resulting in much reduced or absent expression of the dystrophin protein. Dystrophin maintains the structural integrity of muscle fibers by connecting actin filaments to the ECM via the dystrophin-dystroglycan protein complex at the fiber sarcolemma (Matsumura et al. Citation1999). The absence of dystrophin allows unregulated calcium entry through the sarcolemma, producing mitochondrial dysfunction, reactive oxygen species (ROS) production and fiber death in a process that is not yet completely understood (Zablocka et al. Citation2021).
The standard drug regime for DMD is treatment with the glucocorticoids prednisone, prednisolone, or deflazacort. Even though glucocorticoids are known to cause skeletal muscle atrophy, they are thought to prolong patient survival by dampening the inflammatory response observed in degenerating muscles (Werneck et al. Citation2019). Recent research progress in the development of exon-skipping oligonucleotides has provided new, U.S. Food and Drug Administration approved therapies for DMD, although they are still in their infancy and their long term effects on DMD progression are not fully known (Dzierlega and Yokota Citation2020). Examples include eteplirsen (brand name Exondys 51), which targets mutations found in around 14% of DMD patients, and viltolarsen (Viltepso), which targets mutations found in around 8% of patients (Iftikhar et al. Citation2021). The European Medicines Agency approved the oxadiazole, ataluren (Translarna), which is applicable for the premature stop codon mutation found in around 10% of patients (Hoffman Citation2020). Current research for new treatments include in vivo gene editing techniques, such as CRISPR/Cas9-mediated genome editing, which have the potential to repair the genetic defect in the dystrophin gene in situ (reviewed in (Lim et al. Citation2018). However, CRISPR/Cas9 technology is not yet applicable to humans. Consequently, there is an urgent need to develop clinically effective therapies for DMD.
Development of cell therapy approaches for DMD
Cell therapy has been investigated as a treatment for DMD since the demonstration that genetically normal mouse myoblasts transplanted into dystrophin-deficient muscles of the mdx model persisted and gave rise to dystrophin positive muscle fibers (Partridge et al. Citation1989). As discussed above, a major caveat to this approach is the generation of sufficient numbers of myogenic cells for intramuscular transplantation. Although iPSCs have the potential to form all of the tissues in the body, some tissue types are simpler than others to produce in the laboratory. This applies to muscle, with the differentiation of iPSCs into cardiac cells proving to be relatively straightforward compared to skeletal muscle cells (Pourquie et al. Citation2018). In the next section, we will describe approaches to enhance myogenesis in iPSCs, firstly focusing on mouse iPSCs and then describing progress using human iPSCs (hiPSCs).
Mouse iPSC-based cell therapy and validation in animal models
Due to the difficulty in producing myogenic cells from iPSC cultures in vitro, an alternative approach is the utilization of iPSCs at an earlier stage of potency, followed by in vivo intramuscular delivery and reliance on the host myogenic environment for inducing differentiation into muscle tissues. This was recently demonstrated by Chan, et al., who generated teratomas (tumors containing immature and/or fully differentiated tissues, including muscle) in mice by intramuscular transplantation of undifferentiated mouse iPSCs (Chan et al. Citation2018). Muscle-derived teratoma cells were shown to contain myogenic cells, as defined α7-integrin and vascular cell adhesion molecule-1 (VCAM-1) expression. These cells could be purified by FACS and showed relatively high regeneration potential, compared to myogenic cells derived from iPSCs using standard protocols, and regenerated approximately 80% of the tibialis anterior muscle volume (Chan et al. Citation2018). Remarkably, the authors showed that 1.5 × 103 teratoma-derived myogenic cells were functionally equivalent to 400 freshly isolated Pax7 expressing satellite cells in their ability to produce muscle fibers with resident satellite cells (Chan et al. Citation2018).
Another area of DMD research where iPSCs are facilitating progress is the development of in vivo gene editing strategies based on clustered regularly interspaced short palindromic repeats/CRISPR-associated 9 (CRISPR/Cas9) technology. For example, Jin, et al., used CRISPR/Cas9 in vitro to delete exon 23 of the dystrophin gene in mouse iPSCs derived from the mdx model of DMD (Jin et al. Citation2020). This deletion is sufficient to restore dystrophin expression, but is traditionally very inefficient to achieve. This was overcome using a homology-directed repair donor vector, which carries floxed fluorescent protein and antibiotic selection genes. These mouse iPSCs were differentiated into myogenic cells via upregulated expression of MyoD, and shown to form dystrophin expressing fibers in vivo after transplantation into the tibialis anterior muscle of mdx mice. More recently, Domenig, et al., applied CRISPR/Cas9 editing of the dystrophin gene in vitro to mouse iPSCs derived from mdx mouse fibroblasts, which were converted into myogenic precursors via MyoD overexpression in concert with small molecule treatment (Domenig et al. Citation2022). In vivo transplantation into the TA muscle restored dystrophin expression and the level of engraftment was not affected by the dystrophin gene editing protocol.
Development of iPSC-based therapies using human cells
Some early efforts to potentiate the myogenic differentiation of hiPSCs focused on viral-mediated delivery of key transcription factors, such (a regulator of muscle precursor cell proliferation that binds target genes as a heterodimer with PAX3) (Darabi et al. Citation2012), and MYOD1 (one of the first markers of commitment to myogenic differentiation) (Buckingham and Relaix Citation2007; Young et al. Citation2016; Judson and Rossi Citation2020). This increased the numbers of myogenic cells that produce muscle tissue in vitro and in vivo, although doubts remained over the degree to which these cells recapitulate myogenesis during embryonic development (Hicks and Pyle Citation2015; Judson and Rossi Citation2020). Nonetheless, it should also be noted that adult myogenesis in satellite cells does not fully recapitulate myogenesis during embryonic development. For example, Lepper et al demonstrated a differential requirement for Pax7 expression during embryonic, juvenile and adult myogenesis (Lepper et al. Citation2009; Wang and Conboy Citation2010).
Additionally, the use of viral vectors that produce genomic integrations raises concerns about their use in the clinical setting and suitability for disease modeling (Kaiser et al. Citation2015; Judson and Rossi Citation2020). To circumvent these issues, more recent methodologies have relied upon manipulating the iPSC culture environment in vitro with growth factors and bioactive small molecules that induce the myogenic program (Judson and Rossi Citation2020). A large number of protocols have been developed based on the developmental signals that guide skeletal muscle formation from precursor cells in the somites of the embryo paraxial mesoderm (for a review, see (Chal et al. Citation2018). Based on these methods, cultures of PAX7-expressing myogenic cells, myosin heavy chain positive myofibers and other cell types can be produced from hiPSCs in vitro within 30–50 days (Chal et al. Citation2016). Subsequent protocols have aimed to reduce the time required to generate myogenic cells from hiPSCs in vitro and enrich their presence in the cell population, for example by cell sorting using the markers ERBB3+/NGFR+ or CD10+/CD24- (Shelton et al. Citation2016; Wu et al. Citation2018). These optimizations have produced myogenic precursor cells with the ability to generate large numbers of dystrophin expressing myofibers in vivo in the mdx/nsg immunocompromised mouse model of DMD. Further studies have shown that hiPSC-derived myogenic cells can form 3D contractile skeletal muscle in vivo in immunocompromised mice that fuses with adjacent, damaged tissue (Rao et al. Citation2018). These cells also showed enhanced in vitro differentiation into myotubes, indicating their usefulness for skeletal muscle disease modeling (Wu et al. Citation2018). Other modifications to induce myogenic cell formation from hiPSCs in vitro include the generation of EZ spheres (cell aggregates to derive pre-rosette multipotent stem cells) by treatment with high concentrations of basic fibroblast growth factor (FGF2) and epidermal growth factor (EGF), or activation of the Wnt signaling pathway (Hosoyama et al. Citation2014; Sakai-Takemura et al. Citation2018) (see (Sato Citation2020) for a review of these methodologies). A summary of the various protocols to enhance myogenesis in iPSC cultures is shown in .
Table 1. Culture strategies to enhance iPSC differentiation into myogenic precursor cells.
Due to a large proportion of DMD patients succumbing to respiratory failure, cell therapy strategies focusing on the diaphragm have the potential to increase average lifespan. To assess the feasibility of this approach, Miura, et al., developed a protocol to study in vivo cell transplantation in the diaphragm of mdx mice, by injection at the edge of the muscle via the abdomen using stereomicroscopy (Miura et al. Citation2022). Muscle stem cells derived from hiPSCs were shown to survive the injection process and produce dystrophin-expressing myofibers. Moreover, suspending these iPSC-derived cells in a polymer solution enhanced their transplantation into the rapidly contracting diaphragm muscle (Miura et al. Citation2022).
A novel strategy to enhance myogenic cell differentiation from iPSCs in vitro was recently reported by Mashinchian, et al. (Mashinchian et al. Citation2022). Utilizing heterotypic three dimensional embryoid cultures, the cell types that were most effective for directing myogenesis in hiPSCs were identified by screening. Embryonic endothelial cells and fibroblasts were found to be highly permissive for myogenesis, and triple in vitro culture with hiPSCs plus Wnt and FGF pathway activation enhanced differentiation into Pax7-positive myogenic cells. In a DMD model, these cells outperformed human myoblasts in terms of re-population of the satellite cell niche in vivo and muscle repair over repeated cycles of degeneration. This study demonstrates the importance of fully characterizing the stem cell niche to realize the full therapeutic potential of iPSCs.
In addition to being a robust source of myogenic cells for in vivo transplantation, iPSCs are being utilized in vitro to improve our understanding of DMD disease progression, with the aim of providing novel therapeutic targets or treatment modalities. For example, in vitro myogenesis in iPSCs derived from normal or skeletal muscle disease conditions can be compared to elucidate differences in the molecular framework regulating muscle development and maintenance (Lim et al. Citation2021). This approach has identified signaling pathways that are differentially activated during the stages of in vitro myogenesis in hiPSCs derived from normal and DMD patient tissues, such as aberrant activation of the bone morphogenetic protein (BMP) and transforming growth factor (TGF)-β signaling pathways (Lim et al. Citation2021). Myogenesis was inhibited in the DMD iPSCs in vitro, as shown by reduced myotube formation. As a validation of this analysis, pharmacological inhibition of the SMAD activity, the downstream effector of TGF-β signaling, restored myotube formation in the DMD iPSCs (Lim et al. Citation2021). Recently, a multi-omics strategy based on published transcriptomic and proteomic datasets was used to investigate early onset abnormalities in the in vitro myogenesis of hiPSCs obtained from DMD patients (Mournetas et al. Citation2021). It was observed that DMD iPSCs follow the same developmental steps of myogenesis in vitro as normal iPSCs (mesoderm, somite, and skeletal muscle), but abnormalities in the transcriptome occur as early as the somite stage. Approximately 10% of the expressed genes were affected, including mitochondrial genes which showed increased transcriptome abnormalities as myogenesis progressed (Mournetas et al. Citation2021). Significantly, fibrosis-related pathways were identified as an intrinsic feature of early stage myogenesis in DMD iPSCs in vitro, even though fibrosis is generally thought of as a complication of mature muscle fiber degeneration in DMD patients. These fibrosis pathways were also independent of TGF-β signaling, which is thought to be a major driver of fibrosis in DMD muscle (Ismaeel et al. Citation2019). This use of cutting-edge technologies has provided new insights into the molecular pathogenesis of DMD, as a disease that also affects the early steps of myogenesis.
The role of TGF-β signaling in DMD progression was also investigated in skeletal muscle cells differentiated in vitro from DMD patient-derived iPSCs. Caputo, et al., utilized a rapid myogenesis protocol for DMD hiPSCs based on the forced expression of MyoD and BAF60C, a protein the functions in chromatin remodeling (Caputo et al. Citation2020). Myotubes derived by this in vitro method showed high levels of TGF-β- SMAD2/3 signaling, which was further exacerbated by electrical pacing-induced contraction. Using this approach, the authors demonstrated that in vitro-based models of DMD skeletal muscle could recapitulate defects in ECM-derived signals or mechanical cues and may be suitable for the identification of new therapeutic targets (Caputo et al. Citation2020).
Development of hiPSC-based therapies for cardiomyopathy in DMD
The use of iPSC-derived cardiomyocytes to study the pathogenesis of DMD is also gaining research prominence, because heart failure is becoming an increasingly common cause of mortality (D’Amario et al. Citation2017). It has previously been shown that heart failure and skeletal muscle wasting are linked (Ding et al. Citation2018). Yasutake, et al., investigated the role of yes-associated protein (YAP) signaling in hiPSC-derived cardiomyocytes from DMD patients in vitro (Yasutake et al. Citation2021). YAP activity was compared to hiPSC-derived cardiomyocytes with the DMD mutation corrected using CRISPR/Cas9. It was observed that YAP signaling and proliferation was reduced in cardiomyocytes with the DMD mutation, which was due to disorganization of the actin filament network, suggesting that the activation of YAP signaling could be a target for preventing cardiac disease in DMD. A recent report by Willi, et al., demonstrated in vitro bioenergetic and metabolic impairments in hiPSC-derived cardiomyocytes from DMD patients, such as a 75% decrease in the mitochondrial ATP production rate (Willi et al. Citation2022).
hiPSCs for the study of drug mechanisms
iPSC-based models of DMD are also providing novel insights into the mechanisms of prescribed therapeutics. Tanoury, et al., generated late stage myogenic cells from hiPSCs with a DMD mutation (Al Tanoury et al. Citation2021). Compared to normal iPSC derived muscle cells, the DMD mutation produced numerous abnormalities, such as increased fusion and myofiber branching, and defective contraction force. Treatment with the prescribed glucocorticoid drug, prednisolone, reduced these abnormalities in the DMD mutated myofibers. These findings shed new light on the action mechanism of prednisolone, because it was previously thought that this drug functioned by immunosuppressant effects, rather than directly acting on the myofibers (Al Tanoury et al. Citation2021).
Increasing access to hiPSC lines from DMD patients
New protocols are also being developed to increase the availability of hiPSCs from DMD patients. Recently, Ghori and Wahid demonstrated that hiPSCs could be generated from urine-derived cells harvested from DMD patients using an integration-free episomal vector system (Ghori and Wahid Citation2021). This can provide a low-cost, non-invasive, painless, and repeatable source of hiPSCs. To reduce the need to obtain hiPSCs from donors with DMD, Valetdinova, et al., have generated three hiPSC lines from a DMD patient using non-integrating episomal plasmid vectors (Valetdinova et al. Citation2020). The unique stem cell lines identifiers are ICGi002-A, ICGi002-B, and ICGi002-C and have been deposited in a cell line repository. These hiPSC lines were validated by their ESC-like morphology, expression of pluripotency markers, normal karyotype and tri-lineage differentiation potential. Similarly, Guan, et al., have generated an hiPSC line (SDQLCHi007-A) from a patient with deletions in exons 49–50 of the dystrophin gene, via reprograming blood mononuclear cells using non-integrating vectors (Guan et al. Citation2020). More recently, Ahamd, et al., reported the generation of two hiPSC lines (IGIBi006-A and IGIBi008-A) from the lateral collateral ligament of two patients of Indian origin carrying exonic deletions of the dystrophin gene (Ahmad et al. Citation2022).
CRISPR/Cas editing in hiPSCs and targeting long noncoding RNAs
In addition to the CRISPR/Cas9 based editing of mouse mdx iPSCs, described above, Morisaka, et al., tested the multi-component Class I Cas 3 CRISPR system as a tool to induce exon skipping of the DMD mutation in patient-derived hiPSCs. This approach provided a unique editing system and alternative to the more widely used single-component Class 2 CRISPR systems (Morisaka et al. Citation2019).
Long noncoding RNAs (lncRNA) regulate gene expression at multiple levels (epigenetic, transcriptional, and post-transcriptional) (Wei et al. Citation2018). iPSC-based production of skeletal muscle cells from patients has allowed the investigation of lncRNAs in the pathogenesis of DMD. Zhang, et al., demonstrated that lncRNA H19 interacts with dystrophin and inhibits E3-ligase-dependent degradation using hiPSC-derived muscle cells from patients with Becker muscular dystrophy, which also results from mutations in the dystrophin gene (Zhang et al. Citation2020). Exon skipping approaches that circumvented a silent mutation that was responsible for preventing the H19-dystrophin interaction were shown to be effective at preventing muscle atrophy in the mdx model of DMD.
hiPSC-based drug screening protocols
An important application of iPSC-based technologies is the development of disease- and patient-specific drug screening protocols. In the case of DMD, Sun, et al. produced myoblasts from DMD patient hiPSCs for a screening platform based on high-content imaging parameters, such as morphological changes during differentiation and myogenic marker protein expression (Sun et al. Citation2020). This approach led to the identification of two compounds (ginsenoside Rd and fenofibrate), that improved disease phenotype in the mdx mouse model of DMD. Similarly, Xuan, et al., reported a drug discovery advance using hiPSCs (Xuan et al. Citation2021). In this study, a candidate compound that promoted iPSC differentiation into skeletal muscle progenitor cells was tested. The histone deacetylase inhibitor givinostat was shown to enhance differentiation compared to the standard protocol, and produce progenitor cells that effectively repaired damaged muscle and restored dystrophin expression in the mdx model of DMD.
In summary, iPSC-based methods have significantly contributed to DMD research by enhancing our understanding of the disease process, providing a robust source of myogenic cells for transplantation, and indicating candidate compounds for drug development. Due to constraints related to the pathogenesis of DMD, as a severe myopathy that affects all muscles of the body, cell therapy approaches may be limited by the poor migration of transplanted myogenic cells from the site of delivery, and the lack of widespread engraftment after vascular delivery. In the next section of this review, we describe a cell therapy-based application of iPSC technology that may be suitable for a skeletal muscle disorder: volumetric muscle loss.
iPSC-based therapeutic approaches for volumetric muscle loss (VML)
Socioeconomic impact of VML
As described above, skeletal muscle possesses an impressive capacity for regeneration. However, injuries or insults (such as trauma, surgery or degeneration) may occur that exceed this capacity, resulting in connective tissue infiltration, abnormal morphology, and loss of muscle function (Shayan and Huang Citation2020). This occurs when the bio-scaffold of the muscle is damaged to such an extent that it cannot support satellite cell proliferation and myogenesis (Wu et al. Citation2021). Unfortunately, the incidence of VML is not well documented, because it is not tracked by civilian trauma data (Langridge et al. Citation2021). Some estimation can be gained from records of open bone fractures: approximately 150, 000 occur annually in the United States with the majority having soft tissue loss, and approximately 58% of severe open tibial fractures have significant muscle damage (Court-Brown and McBirnie Citation1995; Bosse et al. Citation2002; Langridge et al. Citation2021). Military data shows that VML accounts for 65% of disability following severe open tibial fractures and produces a lifetime disability cost of around $400,000 per patient, in addition to medical fees (Corona et al. Citation2015; Langridge et al. Citation2021). VML lacks a standard treatment protocol to replace lost muscle tissue and restore strength (Shayan and Huang Citation2020). Surgical approaches aim to restore tissue defects and involve autologous free flap grafting, scar tissue debridement or minced skeletal tissue transfer, although they are limited by factors such as tissue availability, high level of surgical skill, morbidity at the donor tissue site, and a lack of demonstrable clinical efficacy (Lin et al. Citation2007; Corona et al. Citation2016; Shayan and Huang Citation2020).
Experimental therapies for VML
Multiple potential cell types could be utilized to enhance muscle regeneration for VML repair, such as patient-derived muscle stem cells (Shayan and Huang Citation2020). Patient-derived hiPSCs can be advantageous for this approach, because they are obtained from any tissue source, expanded to provide sufficient donor cells in vitro, and differentiated into the various cell types that comprise skeletal muscle tissue (Shayan and Huang Citation2020). However, it should be noted that a number of alternative strategies for treating VML are also being investigated in clinical and pre-clinical trials. For example, in patients acellular biological scaffolds composed of ECM proteins known to promote muscle regeneration have undergone clinical investigation and shown promising results (Dziki et al. Citation2016). Protocols for physical therapy in patients are also undergoing continuous modification in response to new clinical data (Washington and Wolchok Citation2023). In animal models, the local delivery of regeneration-promoting cytokines, such as interleukin-10, has also been shown to promote muscle growth after VML in rats (Huynh et al. Citation2023). FDA-approved drug repositioning has also been applied to a mouse model of VML, with the demonstration that the β2 adrenergic receptor agonist, formoterol, is a translational option (Raymond-Pope et al. Citation2023). These approaches are interesting, depending on their efficacy, because they do not rely on the production of stem cell populations. Due to iPSCs not being currently approved for clinical use, other sources of progenitor cells with the potential for muscle repair are under investigated in animal models, such as mesenchymal stem cells and mesoangioblasts (Shayan and Huang Citation2020). An interesting alternative to treat VML is skeletal muscle transplantation. As a source of donor tissue (because post-mortem human muscle is not viable), humanized skeletal muscle is being developed in pigs using a combination of gene editing, somatic cell nuclear transfer and blastocyst complementation (Greising et al. Citation2022). If successful, this strategy could provide large quantities of donor muscle tissue for VML patients.
iPSC-based studies for the treatment of VML
Studies of iPSCs as a source of myogenic cells for VML have utilized numerous approaches, such in vitro myogenesis enhanced using scaffolds constructed from biomaterials and 3D cell culture systems, followed by validation in mouse models of VML. It has been shown that myotubes differentiated from hiPSCs can be matured in 3D cell culture, integrate into sites of muscle damage and induce vascularization (Rao et al. Citation2018; Langridge et al. Citation2021). Using a mouse model of VML, which involves tibialis anterior muscle excision to produce 30-40% muscle loss, Wu, et al., recently demonstrated that hiPSCs differentiated into myogenic cells by activation of Wnt signaling, followed by inhibition of BMP and TGF-β signaling, were transplanted into the area of muscle resection within a fibrin hydrogel (Wu et al. Citation2021). The authors reported abundant formation of donor-derived myofibers and satellite cells, and restoration of the lost bio-scaffold (as shown by laminin staining). Moreover, donor myofibers expressed components of the neuromuscular junction, indicating functional coupling with motor neurons. This was supported by the demonstration of increased in situ contraction of muscles that were transplanted with myogenic cells from hiPSCs. A recent preprint by Jodat, et al., describes the use of 3D bioprinting to create hiPSC-derived skeletal muscle implants for VML (Jodat et al. Citation2021). Using tailored hydrogels with chemical, structural and mechanical properties based on skeletal muscle ECM, hiPSC-derived myoblasts differentiated into linear, contractile immature myofibers in the millimeter length range. Implantation of these pre-vascularized constructs into a mouse model of VML produced successful graft-host interaction and the de novo production of muscle tissue.
iPSC-based methodologies for the effective formation of neuromuscular junctions
As mentioned above, skeletal muscle is a complex tissue composed of highly structured contractile elements, neuromuscular junctions with motor neurons, and a rich capillary network. Some tissue engineering approaches have specifically focused on effectively re-building these structures after injury or disease to ensure effective functional recovery. The integrity of the regenerated structures has been assessed using in vitro and in vivo approaches. Much research attention has focused on the neuromuscular junction (NMJ), because it is critical for initiating muscle contraction and iPSCs have the potential to differentiate into both muscle fibers and motor neurons.
In vitro studies of NMJ formation from mouse and hiPSCs
NMJ formation using iPSC cultures has been investigated in vitro, which can also increase understanding of the molecular mechanisms underlying this process (see (Luttrell et al. Citation2021) for a dedicated review). Recently, 3D in vitro culture of mouse skeletal muscle and motor neurons (derived from mouse iPSCs) were constructed using magnetic force-based tissue engineering (Mag-TE) (Yoshioka et al. Citation2020). This produced functional NMJs with excitation-contraction coupling and potential applicability for ALS drug screening.
Co-cultures of hiPSCs and mouse skeletal muscle primary cells also results in NMJ formation with neurite processes associated with acetylcholine receptor clustering and muscle twitching (Liu et al. Citation2016; Barbeau et al. Citation2020). Lin, et al., demonstrated that single hiPSC cultures can be differentiated into motor neurons and myotubes with mature functioning NMJs containing pre- and post- synaptic components, and Schwann cells (Lin et al. Citation2020). This was achieved by switching between neurogenic and myogenic induction media during a one month differentiation period. The development of robust, reproducible, step-wise protocols for the development of NMJs from hiPSCs in vitro can be useful for characterizing the physiology and pathology of human NMJs (Lin et al. Citation2019). In addition, such simplified protocols may facilitate drug development for neuromuscular disease, via their adoption by laboratories which specialize in medicinal chemistry, rapid high throughput screening, and/or target druggability.
3D- and co-culture techniques to enhance NMJ formation
The optimization of culture protocols for differentiating hiPSCs into muscle precursor cells (discussed above) has made possible the co-culture of healthy and disease iPSC-derived motor neurons and muscle from the same human donor (Mazaleyrat et al. Citation2020). NMJ formation showed acceptable functionality, as determined by AChR clustering, muscle contractions and calcium imaging. To identify new drug targets and therapies for patients with nervous system disorders affecting NMJ function, it is important to develop a system that allows objective quantitative methods to assess NMJ formation and degeneration. Recently, Vila, et al., combined the direct differentiation of myotubes and motor neurons from hiPSCs in a 3D model with optogenetics, microfabrication, optoelectronics and video processing to allow the automated stimulation and recording of photosensitive NMJ function (Vila et al. Citation2021). The adjustable analysis parameters related to light-induced electrical depolarization were baseline time, peak threshold, and minimum minima height and width, with sufficient sensitivity for drug development applications.
To further recreate the skeletal muscle tissue milieu, culture strategies have moved towards three dimensional systems rather than traditional two dimensional environments. In addition, increasing culture time and including trophic factors for different cell fates have been used to improve iPSC differentiation into the various cell types present in skeletal muscle. Complex isogenic multi-lineage 3D cultures have been developed that allow hiPSCs differentiation into skeletal muscle cells, vascular endothelial cells, pericytes, and motor neurons (Maffioletti et al. Citation2018). 3D culture systems have also been combined with microfluidics, which allows human muscle cells and human motor neurons to be compartmentalised while promoting spatial guidance and integration using bioactive hydrogels (Osaki et al. Citation2018; Vila et al. Citation2019). These advances could facilitate the development of mature ‘all human’ NMJs between muscle and motor neurons in vitro, with applications such as drug development for neuromuscular diseases involving NMJ degeneration (e.g. amyotrophic lateral sclerosis (ALS)). A detailed review of research progress for NMJ formation in vitro is provided by Barbeau, et al. (Barbeau et al. Citation2020).
Alongside the approaches to improve NMJ formation described above, research has also focused on driving myogenesis in iPSCs towards fully differentiated myofibers, rather than immature myotubes. These approaches are discussed in the next section of this review.
Approaches to improve iPSC differentiation into mature skeletal muscle tissue
In vitro myogenesis of primary skeletal muscle cells produces cell fusion into multinucleate syncytia termed myotubes, which can be striated and undergo spontaneous contractions in the culture dish. However, skeletal muscle tissue is composed of myofibers, which are much larger and more highly structured than myotubes. In addition, myofibers are further sub-divided into fast- and slow-twitch fibers, and individual muscles may be predominantly composed of fast, slow or a mixture of fiber types depending on their function in the body, e.g. contraction with high force versus the maintenance of posture (Mukund and Subramaniam Citation2020). Protocols than can enhance myogenesis in iPSCs to generate more mature myofibers in vitro would facilitate neuromuscular disease research and drug screening.
hiPSC differentiation into craniofacial muscles in vitro
The MyoD-induced microRNA-494-3p has been shown to inhibit the formation of fast oxidative fibers during hiPSC myogenesis (Iwasaki et al. Citation2015). Iwasaki, et al., demonstrated that this microRNA targets the histone acetyltransferase, E1A-binding protein p300, which down-regulates expression of myosin heavy chain 2 (Iwasaki et al. Citation2021). Thus, strategies to suppress the activity of microRNA-494-3p during hiPSC myogenesis may promote the development of fast oxidative fibers in the muscle cultures. The craniofacial muscles, which control food consumption, facial expressions, and eye movement, have a distinct embryonal lineage from the limb and trunk muscles (Buckingham et al. Citation2003). The formation of craniofacial muscles is also regulated by specific combinations of transcription factors and these muscles are preferentially affected by some myopathies, such as myotonic, facioscapulohumeral and oculopharyngeal muscular dystrophy (Kim et al. Citation2020). However, myogenesis protocols for hiPSCs are generally based on observations of limb and trunk muscle formation. To address this issue, Kim, et al., developed an optimized protocol mimicking the key signaling pathways during craniofacial embryonic myogenesis with pharyngeal mesoderm induction media, which controlled BMP signaling via notch inhibition (Kim et al. Citation2020). Transcriptomic profiling indicated that the muscle tissue generated by this protocol had a gene expression pattern that was highly similar to craniofacial myoblasts and distinct from primary myoblasts obtained from the limb muscle.
hiPSC differentiation into muscle cells modeling adult myofibers
As mentioned above, it is difficult to fully recapitulate the myogenesis program in vitro to produce mature myofibers. A recent study by Chen, et al., utilized a 3D tissue-engineered model of human skeletal muscle, termed ‘myobundle’ that can be composed of primary myoblasts or iPSC-derived myogenic progenitors (Rao et al. Citation2018; Chen et al. Citation2021). The myobundles show enhanced myotube differentiation compared to 2D cultures, electrically or chemically induced twitch and tetanic contractions, positive force-frequency relationship, strong calcium transients, and physiological responses to different drugs. Moreover, this culture system was validated as a model of adult skeletal muscle by the discovery that exercise-mimetic electrical stimulation or Janus kinase pathway inhibition can prevent myobundle atrophy mediated by interferon-γ, which has been linked to aging, infection-related skeletal muscle atrophy (Chen et al. Citation2021).
Future challenges
This review has described the multiple applications of iPSC technology for facilitating research into skeletal muscle biology and neuromuscular disorders. iPSCs are also being studied as a source of therapeutic cells for treating myopathies or muscle injury. Although impressive progress has been made, there remain a number of technical challenges that should be addressed before the full potential of iPSCs can be realized.
From the standpoint of iPSC-based cell therapy for myopathies, a major problem is the effective delivery of therapeutic cells to the sites of muscle damage throughout the body. This issue also applies to other types of cell therapy, such as myoblast transplantation. One option under investigation is to develop a delivery system that utilizes the vasculature. For example, Darabi, et al., showed that myogenic cells derived from ESCs can be delivered to degenerating skeletal muscle via intra-arterial delivery and contribute to muscle repair (Darabi et al. Citation2011). The potential of intra-arterial delivery was also demonstrated by Tedesco, et al., using genetically corrected iPSC-derived mesoangioblasts (myogenic progenitor cells derived from pericytes located in skeletal muscle adjacent to blood vessels) in a model of limb-girdle muscular dystrophy 2D (Tedesco et al. Citation2012). Alternatively, iPSC-derived muscle precursor cells have been used to identify factors that can enhance their migration from the transplantation site. Using a combination of bioinformatics and tissue engineering studies, Choi, et al., recently demonstrated the treatment with Delta-like 4 and platelet-derived growth factor-BB can increase the migration and therapeutic effect of transplanted cells (Choi et al. Citation2022). However, it appears that the majority of studies concerning iPSC-based cell therapy for myopathies still test the therapeutic potential of transplanted cells using intramuscular delivery to a single muscle site of interest. Due to these constraints on achieving the widespread delivery of myogenic cells to the major muscle groups, a more achievable option for iPSC-based cell therapy is VML that involves one or a few muscles. It may also be noted that, while most studies of iPSC-based approaches for myopathies have focused on generating myogenic cells, it may be preferable to utilize other stem/precursor cell types, such as mesoangioblasts. This is because of their potential to into different mesodermal lineages (including muscle) and their capacity for efficient extravasation across blood vessels into muscle tissue, making them more suitable for systemic delivery (Biressi et al. Citation2020).
Although this review has discussed research approaches to develop an iPSC-based cell therapy for DMD, a number of major obstacles remain before this treatment can be successfully applied in the clinical setting. One issue is the safety of the transplanted cells, because of the genetic recombination events used to produce iPSCs, along with the overexpression of myogenic factors employed in some protocols (Darabi et al. Citation2012; Young et al. Citation2016; Danisovic et al. Citation2018; Jin et al. Citation2020). In addition, although these protocols report a high level of terminal differentiation, the possible occurrence of residual undifferentiated cells could potentially lead to teratoma formation (Suman et al. Citation2019). Thus, there is a need to develop methods that do not rely on stable genetic integration, coupled with methodologies that can remove undifferentiated cells from the myogenic cultures. Due to these limitations, iPSC-based cell therapies are not currently approved for patients (Dahlke et al. Citation2021; Kim et al. Citation2022b). An additional obstacle to the development of iPSC-based cell therapies for DMD is the problem of reproducing the later stages of the disease process in animal models. As discussed above, the mdx mouse is the most commonly used model of DMD for cell therapy research. However, this model fails to present the disease phenotype observed in the muscle of patients with advanced DMD (extensive fibrosis, fatty infiltration, severe cardiomyopathy and physical disability) (Zaynitdinova et al. Citation2021). An exception is the mdx diaphragm, which develops extensive fibrosis but is technically difficult to study (Henry et al. Citation2017). An alternative model may be the dystrophin-utrophin (which has high homology to dystrophin and is upregulated during fetal development) double knockout (dKO) mouse that presents a more severe disease progression more representative of a late-stage phenotype (Deconinck et al. Citation1997; Grady et al. Citation1997). However, the dKO model is not commonly used in cell therapy research compared to the mdx model. Another major stumbling block for iPSC-based cell therapy for DMD is the uniform lack of success of myoblast transplantation clinical trials in humans (Piga et al. Citation2019). Transplanted myoblasts showed limited engraftment ability and efficacy of muscle regeneration. Differences in disease progression and underlying molecular and cellular mechanisms between DMD patients and the mdx model may have contributed to the failure of these clinical trials, similar to numerous drug clinical trials that showed efficacy in the mdx model but lacked efficacy and produced previously unreported collateral effects in patients (De Luca Citation2012).
The ability of transplanted hiPSC-derived myogenic cells to contribute to skeletal muscle regeneration and contribute to the satellite cell pool has been repeatedly demonstrated in animal models. However, issues remain concerning how effectively the transplanted cells could migrate into the skeletal muscle niche areas, their capacity for long-term engraftment and transition to dormant/quiescent status, and cellular maturation from progenitor cells into the adult cell phenotype (Lim et al. Citation2021). Thus, a more rewarding approach for future studies with hiPSCs for muscular dystrophies may be transcriptional profiling, such as the subsets skeletal muscle precursor/stem cells produced during the step-wise myogenesis. This may reveal abnormalities in the signaling or transcriptional networks of patient-derived iPSCs and indicate novel targets for drug development (Lim et al. Citation2021).
As mentioned above, a major stumbling block in the use of iPSCs for cell therapy is the risk of teratoma formation after transplantation, which currently precludes the approval of iPSC-based therapies by the FDA, and mice derived from iPSCs have an increased rate of cancer-related mortality (Aoi et al. Citation2008; Jarrige et al. Citation2021). There is also debate about the potential immunogenicity of iPSCs (Zhao et al. Citation2011; Araki et al. Citation2013). Consequently, there are a number of technical, regulatory and ethical issues currently preventing the clinical translation of this technology (Moradi et al. Citation2019). Nevertheless, a number of clinical trials of iPSC transplantation are ongoing (Kim et al. Citation2022b). The majority are nontherapeutic and aim to evaluate tolerance to the iPSCs. Therapeutic trials are mainly focused on circulatory and visual disorders. The outcomes of these trials will shed further light on the applicability of iPSCs as a cell therapy for skeletal muscular disorders.
The majority of iPSC-based cell therapy studies for myopathies have focused on DMD due to its prevalence in society. However, these studies can also be applied to less common myopathies, such as nemaline rod myopathy, for which iPSC lines have recently become available (Ma et al. Citation2020; Houweling et al. Citation2021; Suleski et al. Citation2022). As discussed above, iPSC lines from DMD patients are becoming more widely available in cell repositories. A larger amount of lines with a greater range of mutations in the dystrophin gene will be required, because assessing therapeutic approaches such as exon skipping will vary in their methodology depending on the mutation type (Eser and Topaloglu Citation2022). In addition to genetic muscle disorders, iPSCs can be source of myogenic cells for other prominent muscle disorders. For example, aging-related loss of muscle mass and function (sarcopenia) is a major health problem in developed countries with demographic aging (Ding et al. Citation2018; Kim et al. Citation2022a). Although the potential application of iPSCs for treating/preventing sarcopenia has been discussed (Pareja-Galeano et al. Citation2016a; Pareja-Galeano et al. Citation2016b), there have been few published studies. A recent study has demonstrated the effectiveness of extracellular vesicles purified from myogenic cells derived from iPSCs for inhibiting muscle aging (Xuan et al. Citation2022), which may indicated renewed interest in this disease application.
Although myogenic cell therapy is a major application of iPSC technology for muscle disorders, products derived from iPSCs are also being investigated as potential treatment options. These so-called ‘acellular therapies’ are based on the two types of iPSC-derived extracellular vesicles: exosomes and microvesicles (Jarrige et al. Citation2021). The rationale for this methodology is based on the findings that beneficial effects of stem cell transplantation may arise from paracrine effects from the donor cells, rather than a direct functional contribution to the diseased tissue. Extracellular vesicles can cross biological barriers and can carry therapeutic molecules to target cells or tissues via fusion with the plasma membrane (Jarrige et al. Citation2021). Recently, cardiomyocytes derived from normal iPSCs were utilized to show that exosomes can produce beneficial effects in DMD cardiomyocytes, whereas exosomes purified from cardiomyocytes derived from DMD iPSCs were detrimental (Gartz et al. Citation2020). However, from a clinical perspective, there are challenges due to the lack of development of robust procedures to isolate and purify high yields of extracellular vesicles with sufficient purity while maintaining their structure and activity. Currently, there is no consensus on a ‘gold standard’ protocol for the isolation of extracellular vesicles for therapeutic applications or basic research (Jarrige et al. Citation2021).
Due to the large distances between the cell body of the motor neuron and muscle fiber in humans, it is currently not possible to entirely replicate the NMJ in vitro. Some studies of NMJ formation in vitro have reported the observation of myotube contractions as supporting evidence of the construction of fully functional NMJs. However, it should be noted that spontaneous contractions have also been observed in cultures of hiPSCs undergoing myogenesis in the absence of motor neurons (for example (Fujiwara et al. Citation2022) or primary myoblast cultures (Vaughan and Lamia Citation2019). This may be due to factors such as the presence of calcium ions in the culture media. Congruent with this issue, it has been shown that contractions in cultured cardiomyocytes can be prevented by the calcium ion chelator EGTA (Neco et al. Citation2010; Koh et al. Citation2017).
Although much research has focused on producing in vitro models of the NMJ from stem cells, the role of the muscle spindle (which detects changes in muscle length) is also important and undervalued in neuromuscular disease (Badiola-Mateos et al. Citation2022). To address this issue, protocols have been developed that allow the efficient differentiation of proprioceptors, which are mechanosensory neurons contained in muscle spindles and develop synaptic bouton-like structures with co-cultured skeletal muscle cells (Badiola-Mateos et al. Citation2022).
Concerning the use of biomaterials-based constructs for de novo skeletal muscle formation, there are issues regarding the adequate diffusion of nutrients and oxygen into the constructs (Shayan and Huang Citation2020). Additionally, poor levels of innervation remains a main problem for achieving full functional recovery after VML and re-establishing the myotendinous junction is still an unmet challenge (Shayan and Huang Citation2020). Another challenge will be culture media formulation for the multiple cell types derived from iPSCs that are required to re-build contractile muscle, such as endothelial cells, tenocytes, motor neurons, and connective tissue cells (Shayan and Huang Citation2020; Jarrige et al. Citation2021). Recent technological advances, such as Slide-seq, which allows mapping of the multiple cell types patterned in the biomaterials scaffold, may facilitate the correct arrangement of these diverse cell types (Rodriques et al. Citation2019; Shayan and Huang Citation2020). The use of patient-derived iPSC cells for the clinical application of biomaterials may also be hampered by the highly inefficient process of cellular reprograming (Langridge et al. Citation2021).
Figure 2. Schematic of iPSC-based cell therapy for DMD patients. iPSCs are generated from a tissue biopsy, and the genetic mutation in dystrophin corrected. After expansion in culture to generate sufficient cell numbers for transplantation, the cells are then induced to enter the myogenic pathway to yield cultures of muscle precursor cells. These cells are then transplanted into the patient’s skeletal muscle and produce dystrophin positive myofibers and satellite cells.
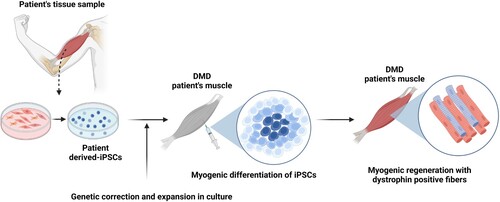
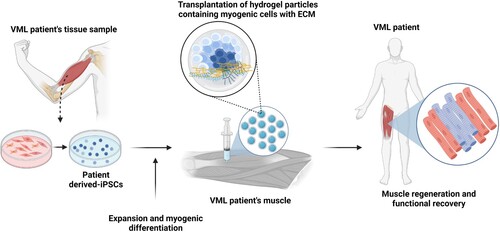
Figure 3. Schematic of iPSC-based cell therapy for VML patients. Similar to DMD patients, a tissue biopsy is used to generate iPSCs, which are then expanded in culture and induced to enter the myogenic pathway to produce muscle precursor cells. Alternatively, culture conditions may be modified to yield cultures containing multiple cells types found in skeletal muscle, such as motor neurons and immature myotubes. Due to the loss of the connective tissue scaffold in VML, a biomaterials-based matrix is transplanted with the iPSC-derived cells to support skeletal muscle regeneration. The transplanted cells utilize the artificial matrix enter myogenesis and effectively replace the loss muscle tissue.
Concluding remarks
Since their discovery in 2006, iPSCs have made a tremendous contribution to the field of regenerative medicine. Skeletal muscle possesses impressive inherent regenerative capacity, but this can be hampered by widespread injury (VML) or become exhausted in muscular dystrophies, such as DMD. iPSCs can be expanded and differentiated in vitro to provide a large pool of myogenic cells for transplantation studies. Dissecting the individual steps and culture conditions required for myogenesis in iPSC cultures has facilitated our understanding of this process during embryogenic development. Mapping the trajectory of myogenesis in patient-derived iPSCs has shed new light on disease mechanisms and possible new drug targets, such as TGF-β signaling in DMD muscle stem cells (Caputo et al. Citation2020). These cultures protocols have been advanced to the stage where specific subtypes of skeletal muscle cells can be produced, such as the craniofacial muscles. 3D culture techniques, combined with advances in the production of biomaterials scaffolds, allows the patterned culture of the multiple cell types found in skeletal muscle to produce tissues with greater resemblance to mature muscle, such as contractile myobundles or myotubes with functional innervation by motor neurons (Maffioletti et al. Citation2018; Chen et al. Citation2021). The advances in gene-editing techniques, such as CRISPR/Cas9, allow the study of gene editing in muscle cells differentiated from patient-derived iPSCs. Moreover, these muscle cultures can be utilized for disease-specific drug screening. From the perspective of cell therapy, iPSC-derived myogenic cells may be most suited for VML, which involves delivery to one/few affected muscles. It should be noted that the majority of research publications have focused on the DMD application, even though VML could be a more achievable aim. The development of acellular therapies relying on extracellular vesicles isolated from iPSC-derived myogenic cells may broaden cell therapy applications to the muscular dystrophies, because they can be applicable for systemic delivery to reach all muscles in the body. Overall, iPSC technology has greatly increased our understanding of skeletal muscle biology in health and disease, and the further advancement of this technology may yield effective treatments for VML, genetic disorders, or defective regeneration.
Author contributions
Kim, H. carried out the literature search, designed the figures, produced the table, and drafted the paper. Jung, D-W. and Williams, D.R. carried out the literature search, drafted the paper, revised it critically for intellectual content, and gave the final approval of the version to be published. All authors agree to be accountable for all aspects of the work.
Acknowledgements
Figures were created with BioRender.com.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Additional information
Funding
References
- Ahmad I, Goel D, Ghosh A, Kapoor H, Kumar D, Ramesh K, Ashley B, Deepika K, Shastry A, Faruq M. 2022. Generation of two induced pluripotent stem cell (iPSC) lines from patients with Duchenne muscular dystrophy (IGIBi006-A and IGIBi008-A) carrying exonic deletions in the dystrophin gene. Stem Cell Res. 64:102927.
- Al Tanoury Z, Rao J, Tassy O, Gobert B, Gapon S, Garnier JM, Wagner E, Hick A, Hall A, Gussoni E, Pourquié O. 2020. Differentiation of the human PAX7-positive myogenic precursors/satellite cell lineage in vitro. Development. 147(12).
- Al Tanoury Z, Zimmerman JF, Rao J, Sieiro D, McNamara HM, Cherrier T, Rodríguez-delaRosa A, Hick-Colin A, Bousson F, Fugier-Schmucker C, et al. 2021. Prednisolone rescues Duchenne muscular dystrophy phenotypes in human pluripotent stem cell-derived skeletal muscle in vitro. Proc Natl Acad Sci U S A. 118(28).
- Aoi T, Yae K, Nakagawa M, Ichisaka T, Okita K, Takahashi K, Chiba T, Yamanaka S. 2008. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science. 321(5889):699–702.
- Araki R, Uda M, Hoki Y, Sunayama M, Nakamura M, Ando S, Sugiura M, Ideno H, Shimada A, Nifuji A, Abe M. 2013. Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature. 494(7435):100–104.
- Baci D, Chirivì M, Pace V, Maiullari F, Milan M, Rampin A, Somma P, Presutti D, Garavelli S, Bruno A, Cannata S. 2020. Extracellular vesicles from skeletal muscle cells efficiently promote myogenesis in induced pluripotent stem cells. Cells. 9(6).
- Badiola-Mateos M, Osaki T, Kamm RD, Samitier J. 2022. In vitro modelling of human proprioceptive sensory neurons in the neuromuscular system. Sci Rep. 12(1):21318.
- Bao X, Zhu X, Liao B, Benda C, Zhuang Q, Pei D, Qin B, Esteban MA. 2013. MicroRNAs in somatic cell reprogramming. Curr Opin Cell Biol. 25(2):208–214.
- Barbeau S, Tahraoui-Bories J, Legay C, Martinat C. 2020. Building neuromuscular junctions in vitro. Development. 147(22).
- Biressi S, Filareto A, Rando TA. 2020. Stem cell therapy for muscular dystrophies. J Clin Invest. 130(11):5652–5664.
- Bosse MJ, MacKenzie EJ, Kellam JF, Burgess AR, Webb LX, Swiontkowski MF, Sanders RW, Jones AL, McAndrew MP, Patterson BM, et al. 2002. An analysis of outcomes of reconstruction or amputation after leg-threatening injuries. N Engl J Med. 347(24):1924–1931.
- Broomfield J, Hill M, Guglieri M, Crowther M, Abrams K. 2021. Life expectancy in duchenne muscular dystrophy. Neurology. 97(23):e2304–e2314.
- Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S, Montarras D, Rocancourt D, Relaix F. 2003. The formation of skeletal muscle: from somite to limb. J Anat. 202(1):59–68.
- Buckingham M, Relaix F. 2007. The role of Pax genes in the development of tissues and organs: Pax3 and Pax7 regulate muscle progenitor cell functions. Annu Rev Cell Dev Biol. 23:645–673.
- Caputo L, Granados A, Lenzi J, Rosa A, Ait-Si-Ali S, Puri PL, Albini S. 2020. Acute conversion of patient-derived Duchenne muscular dystrophy iPSC into myotubes reveals constitutive and inducible over-activation of TGFbeta-dependent pro-fibrotic signaling. Skelet Muscle. 10(1):13.
- Carlson BM, Faulkner JA. 1983. The regeneration of skeletal muscle fibers following injury: a review. Med Sci Sports Exerc. 15(3):187–198.
- Chal J, Al Tanoury Z, Hestin M, Gobert B, Aivio S, Hick A, Cherrier T, Nesmith AP, Parker KK, Pourquié O. 2016. Generation of human muscle fibers and satellite-like cells from human pluripotent stem cells in vitro. Nat Protoc. 11(10):1833–1850.
- Chal J, Al Tanoury Z, Oginuma M, Moncuquet P, Gobert B, Miyanari A, Tassy O, Guevara G, Hubaud A, Bera A, Sumara O. 2018. Recapitulating early development of mouse musculoskeletal precursors of the paraxial mesoderm in vitro. Development. 145(6).
- Chan SSK, Arpke RW, Filareto A, Xie N, Pappas MP, Penaloza JS, Perlingeiro RC, Kyba M. 2018. Skeletal muscle stem cells from PSC-derived teratomas have functional regenerative capacity. Cell Stem Cell. 23(1):74–85e6.
- Chen Z, Li B, Zhan RZ, Rao L, Bursac N. 2021. Exercise mimetics and JAK inhibition attenuate IFN-gamma-induced wasting in engineered human skeletal muscle. Sci Adv. 7(4).
- Choi S, Ferrari G, Moyle LA, Mackinlay K, Naouar N, Jalal S, Benedetti S, Wells C, Muntoni F, Tedesco FS. 2022. Assessing and enhancing migration of human myogenic progenitors using directed iPS cell differentiation and advanced tissue modelling. EMBO Mol Med. e14526.
- Corona BT, Rivera JC, Owens JG, Wenke JC, Rathbone CR. 2015. Volumetric muscle loss leads to permanent disability following extremity trauma. J Rehabil Res Dev. 52(7):785–792.
- Corona BT, Wenke JC, Ward CL. 2016. Pathophysiology of volumetric muscle loss injury. Cells Tissues Organs. 202(3-4):180–188.
- Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, Llewellyn ME, Delp SL, Blau HM. 2014. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med. 20(3):255–264.
- Court-Brown CM, McBirnie J. 1995. The epidemiology of tibial fractures. J Bone Joint Surg Br. 77(3):417–421.
- Cretoiu D, Pavelescu L, Duica F, Radu M, Suciu N, Cretoiu SM. 2018. Myofibers. Adv Exp Med Biol. 1088:23–46.
- Dahlke J, Schott JW, Vollmer Barbosa P, Klatt D, Selich A, Lachmann N, Morgan M, Moritz T, Schambach A. 2021. Efficient genetic safety switches for future application of iPSC-derived cell transplants. J Pers Med. 11(6).
- D’Amario D, Amodeo A, Adorisio R, Tiziano FD, Leone AM, Perri G, Bruno P, Massetti M, Ferlini A, Pane M, et al. 2017. A current approach to heart failure in Duchenne muscular dystrophy. Heart. 103(22):1770–1779.
- Danisovic L, Culenova M, Csobonyeiova M. 2018. Induced pluripotent stem cells for duchenne muscular dystrophy modeling and therapy. Cells. 7(12).
- Darabi R, Arpke R, Irion S, Dimos J, Grskovic M, Kyba M, Perlingeiro RR. 2012. Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell. 10(5):610–619.
- Darabi R, Santos FNC, Filareto A, Pan W, Koene R, Rudnicki MA, Kyba M, Perlingeiro RCR. 2011. Assessment of the myogenic stem cell compartment following transplantation of Pax3/Pax7-induced embryonic stem cell-derived progenitors. Stem Cells. 29(5):777–790.
- De Luca A. 2012. Pre-clinical drug tests in the mdx mouse as a model of dystrophinopathies: an overview. Acta Myol. 31(1):40–47.
- de Wert G, Mummery C. 2003. Human embryonic stem cells: research, ethics and policy. Hum Reprod. 18(4):672–682.
- Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L, Watt DJ, Dickson JG, Tinsley JM, Davies KE. 1997. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 90(4):717–727.
- Dellavalle A, Maroli G, Covarello D, Azzoni E, Innocenzi A, Perani L, Antonini S, Sambasivan R, Brunelli S, Tajbakhsh S, Cossu G. 2011. Pericytes resident in postnatal skeletal muscle differentiate into muscle fibres and generate satellite cells. Nat Commun. 2:499.
- Dhawan J, Rando TA. 2005. Stem cells in postnatal myogenesis: molecular mechanisms of satellite cell quiescence, activation and replenishment. Trends Cell Biol. 15(12):666–673.
- Ding S, Dai Q, Huang H, Xu Y, Zhong C. 2018. An Overview of Muscle Atrophy. Adv Exp Med Biol. 1088:3–19.
- Domenig SA, Bundschuh N, Lenardič A, Ghosh A, Kim I, Qabrati X, D'Hulst G, Bar-Nur O. 2022. CRISPR/Cas9 editing of directly reprogrammed myogenic progenitors restores dystrophin expression in a mouse model of muscular dystrophy. Stem Cell Rep. 17(2):321–336.
- Dzierlega K, Yokota T. 2020. Optimization of antisense-mediated exon skipping for Duchenne muscular dystrophy. Gene Ther. 27(9):407–416.
- Dziki J, Badylak S, Yabroudi M, Sicari B, Ambrosio F, Stearns K, Turner N, Wyse A, Boninger ML, Brown EH, Rubin JP. 2016. An acellular biologic scaffold treatment for volumetric muscle loss: results of a 13-patient cohort study. NPJ Regen Med. 1:16008.
- Edgerton VR, Smith JL, Simpson DR. 1975. Muscle fibre type populations of human leg muscles. Histochem J. 7(3):259–266.
- Emery AE. 2002. The muscular dystrophies. Lancet. 359(9307):687–695.
- Eser G, Topaloglu H. 2022. Current outline of exon skipping trials in duchenne muscular dystrophy. Genes (Basel). 13(7).
- Fujiwara K, Yamamoto R, Kubota T, Tazumi A, Sabuta T, Takahashi MP, Sakurai H. 2022. Mature myotubes generated from human-induced pluripotent stem cells without forced gene expression. Front Cell Dev Biol. 10:886879.
- Gartz M, Lin CW, Sussman MA, Lawlor MW, Strande JL. 2020. Duchenne muscular dystrophy (DMD) cardiomyocyte-secreted exosomes promote the pathogenesis of DMD-associated cardiomyopathy. Dis Model Mech. 13(11).
- Ghori FF, Wahid M. 2021. Induced pluripotent stem cells from urine of Duchenne muscular dystrophy patients. Pediatr Int. 63(9):1038–1047.
- Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR. 1997. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 90(4):729–738.
- Greising SM, Weiner JI, Garry DJ, Sachs DH, Garry MG. 2022. Human muscle in gene edited pigs for treatment of volumetric muscle loss. Front Genet. 13:948496.
- Guan J, Liu X, Zhang H, Yang X, Ma Y, Li Y, Gai Z, Liu Y. 2020. Reprogramming of human Peripheral Blood Mononuclear Cell (PBMC) from a Chinese patient suffering Duchenne muscular dystrophy to iPSC line (SDQLCHi007-A) carrying deletion of 49-50 exons in the DMD gene. Stem Cell Res. 42:101666.
- Henry CC, Martin KS, Ward BB, Handsfield GG, Peirce SM, Blemker SS. 2017. Spatial and age-related changes in the microstructure of dystrophic and healthy diaphragms. PLoS One. 12(9):e0183853.
- Hicks M, Pyle A. 2015. The path from pluripotency to skeletal muscle: developmental myogenesis guides the Way. Cell Stem Cell. 17(3):255–257.
- Hoffman EP. 2020. Pharmacotherapy of duchenne muscular dystrophy. Handb Exp Pharmacol. 261:25–37.
- Hong Y, Wang Y, Zhang Y, Royer JA, Cai B, Mann JR, McDermott S. 2019. Risk factors for falls among boys under 18 years with muscular dystrophy. J Pediatr Rehabil Med. 12(1):3–10.
- Hosoyama T, McGivern JV, Van Dyke JM, Ebert AD, Suzuki M. 2014. Derivation of myogenic progenitors directly from human pluripotent stem cells using a sphere-based culture. Stem Cells Transl Med. 3(5):564–574.
- Hou P, Li Y, Zhang X, Liu C, Guan J, Li H, Zhao T, Ye J, Yang W, Liu K, et al. 2013. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science. 341(6146):651–654.
- Houweling PJ, Coles CA, Tiong CF, Nielsen B, Graham A, McDonald P, Suter A, Piers AT, Forbes R, Ryan MM, et al. 2021. Generating an iPSC line (with isogenic control) from the PBMCs of an ACTA1 (p.Gly148Asp) nemaline myopathy patient. Stem Cell Res. 54:102429.
- Huynh T, Reed C, Blackwell Z, Phelps P, Herrera LCP, Almodovar J, Zaharoff DA, Wolchok J. 2023. Local IL-10 delivery modulates the immune response and enhances repair of volumetric muscle loss muscle injury. Sci Rep. 13(1):1983.
- Iftikhar M, Frey J, Shohan MJ, Malek S, Mousa SA. 2021. Current and emerging therapies for Duchenne muscular dystrophy and spinal muscular atrophy. Pharmacol Ther. 220:107719.
- Ismaeel A, Kim JS, Kirk JS, Smith RS, Bohannon WT, Koutakis P. 2019. Role of transforming growth factor-beta in skeletal muscle fibrosis: A review. Int J Mol Sci. 20(10).
- Iwasaki H, Ichihara Y, Morino K, Lemecha M, Sugawara L, Sawano T, Miake J, Sakurai H, Nishi E, Maegawa H, Imamura T. 2021. MicroRNA-494-3p inhibits formation of fast oxidative muscle fibres by targeting E1A-binding protein p300 in human-induced pluripotent stem cells. Sci Rep. 11(1):1161.
- Iwasaki H, Imamura T, Morino K, Shimosato T, Tawa M, Ugi S, Sakurai H, Maegawa H, Okamura T. 2015. MicroRNA-494 plays a role in fiber type-specific skeletal myogenesis in human induced pluripotent stem cells. Biochem Biophys Res Commun. 468(1-2):208–213.
- Jarrige M, Frank E, Herardot E, Martineau S, Darle A, Benabides M, Domingues S, Chose O, Habeler W, Lorant J, Baldeschi C. 2021. The future of regenerative medicine: cell therapy using pluripotent stem cells and acellular therapies based on extracellular vesicles. Cells. 10(2).
- Jin Y, Shen Y, Su X, Weintraub NL, Tang Y. 2020. Effective restoration of dystrophin expression in iPSC (Mdx)-derived muscle progenitor cells using the CRISPR/Cas9 system and homology-directed repair technology. Comput Struct Biotechnol J. 18:765–773.
- Jodat YA, Zhang T, Al Tanoury Z, Kamperman T, Shi K, Huang Y, Panayi A, Endo Y, Wang X, Quint J, Arnaout A. 2021. hiPSC-derived 3D bioprinted skeletal muscle tissue implants regenerate skeletal muscle following volumetric muscle loss. Res Sq. doi:10.21203/rs.3.rs-146091/v1.
- Joe AWB, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, Rossi FMV. 2010. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol. 12(2):153–163.
- Judson RN, Rossi FMV. 2020. Towards stem cell therapies for skeletal muscle repair. NPJ Regen Med. 5:10.
- Jung DW, Kim WH, Williams DR. 2014. Reprogram or reboot: small molecule approaches for the production of induced pluripotent stem cells and direct cell reprogramming. ACS Chem Biol. 9(1):80–95.
- Kadi F, Schjerling P, Andersen LL, Charifi N, Madsen JL, Christensen LR, Andersen JL. 2004. The effects of heavy resistance training and detraining on satellite cells in human skeletal muscles. J Physiol. 558(Pt 3):1005–1012.
- Kaiser AD, Assenmacher M, Schröder B, Meyer M, Orentas R, Bethke U, Dropulic B. 2015. Towards a commercial process for the manufacture of genetically modified T cells for therapy. Cancer Gene Ther. 22(2):72–78.
- Khurana TS, Prendergast RA, Alameddine HS, Tomé FM, Fardeau M, Arahata K, Sugita H, Kunkel LM. 1995. Absence of extraocular muscle pathology in Duchenne's muscular dystrophy: role for calcium homeostasis in extraocular muscle sparing. J Exp Med. 182(2):467–475.
- Kim E, Wu F, Wu X, Choo HJ. 2020. Generation of craniofacial myogenic progenitor cells from human induced pluripotent stem cells for skeletal muscle tissue regeneration. Biomaterials. 248:119995.
- Kim HJ, Kim SW, Lee SH, Jung DW, Williams DR. 2022a. Inhibiting 5-lipoxygenase prevents skeletal muscle atrophy by targeting organogenesis signalling and insulin-like growth factor-1. J Cachexia Sarcopenia Muscle. n/a(n/a).
- Kim JY, Nam Y, Rim YA, Ju JH. 2022b. Review of the current trends in clinical trials involving induced pluripotent stem cells. Stem Cell Rev Rep. 18(1):142–154.
- Kim KM, Jang HC, Lim S. 2016. Differences among skeletal muscle mass indices derived from height-, weight-, and body mass index-adjusted models in assessing sarcopenia. Korean J Intern Med. 31(4):643–650.
- Koh CH, Wu J, Chung YY, Liu Z, Zhang R-R, Chong K, Korzh V, Ting S, Oh S, Shim W, et al. 2017. Identification of Na+/K+-ATPase inhibition-independent proarrhythmic ionic mechanisms of cardiac glycosides. Sci Rep. 7(1):2465.
- Koike H, Manabe I, Oishi Y. 2022. Mechanisms of cooperative cell-cell interactions in skeletal muscle regeneration. Inflamm Regen. 42(1):48.
- Kostallari E, Baba-Amer Y, Alonso-Martin S, Ngoh P, Relaix F, Lafuste P, Gherardi RK. 2015. Pericytes in the myovascular niche promote post-natal myofiber growth and satellite cell quiescence. Development. 142(7):1242–1253.
- Kuang S, Kuroda K, Le Grand F, Rudnicki MA. 2007. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 129(5):999–1010.
- Kuo IY, Ehrlich BE. 2015. Signaling in muscle contraction. Cold Spring Harb Perspect Biol. 7(2):a006023.
- Landfeldt E, Thompson R, Sejersen T, McMillan HJ, Kirschner J, Lochmüller H. 2020. Life expectancy at birth in Duchenne muscular dystrophy: a systematic review and meta-analysis. Eur J Epidemiol. 35(7):643–653.
- Langridge B, Griffin M, Butler PE. 2021. Regenerative medicine for skeletal muscle loss: a review of current tissue engineering approaches. J Mater Sci Mater Med. 32(1):15.
- Latroche C, Gitiaux C, Chrétien F, Desguerre I, Mounier R, Chazaud B. 2015. Skeletal muscle microvasculature: A highly dynamic lifeline. Physiology (Bethesda). 30(6):417–427.
- Lee SH, Kim BJ, Park DR, Kim UH. 2018. Exercise induces muscle fiber type switching via transient receptor potential melastatin 2-dependent Ca(2+) signaling. J Appl Physiol (1985). 124(2):364–373.
- Lepper C, Conway SJ, Fan CM. 2009. Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature. 460(7255):627–631.
- Lepper C, Partridge TA, Fan CM. 2011. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development. 138(17):3639–3646.
- Lim H, Choi IY, Hyun S-H, Kim H, Lee G. 2021. Approaches to characterize the transcriptional trajectory of human myogenesis. Cell Mol Life Sci. 78(9):4221–4234.
- Lim KRQ, Yoon C, Yokota T. 2018. Applications of CRISPR/Cas9 for the treatment of duchenne muscular dystrophy. J Pers Med. 8(4).
- Lin C-H, Lin Y-T, Yeh J-T, Chen C-T. 2007. Free functioning muscle transfer for lower extremity posttraumatic composite structure and functional defect. Plast Reconstr Surg. 119(7):2118–2126.
- Lin CY, Yoshida M, Li LT, Ikenaka A, Oshima S, Nakagawa K, Sakurai H, Matsui E, Nakahata T, Saito MK. 2019. iPSC-derived functional human neuromuscular junctions model the pathophysiology of neuromuscular diseases. JCI Insight. 4(18).
- Lin CY, Yoshida M, Li LT, Saito MK. 2020. In vitro neuromuscular junction induced from human induced pluripotent stem cells. J Vis Exp. 166.
- Liu ML, Zang T, Zhang CL. 2016. Direct lineage reprogramming reveals disease-specific phenotypes of motor neurons from human ALS patients. Cell Rep. 14(1):115–128.
- Luttrell SM, Smith AST, Mack DL. 2021. Creating stem cell-derived neuromuscular junctions in vitro. Muscle Nerve. 64(4):388–403.
- Ma Y, Zhang H, Li X, Yang X, Li Y, Guan J, Lv Y, Gai Z, Liu Y. 2020. An integration-free iPSC line (SDQLCHi017-A) derived from a patient with nemaline myopathy-2 disease carrying compound heterozygote mutations in NEB gene. Stem Cell Res. 43:101729.
- Maffioletti SM, Sarcar S, Henderson ABH, Mannhardt I, Pinton L, Moyle LA, Steele-Stallard H, Cappellari O, Wells KE, Ferrari G, et al. 2018. Three-Dimensional human iPSC-derived artificial skeletal muscles model muscular dystrophies and enable multilineage tissue engineering. Cell Rep. 23(3):899–908.
- Mah JK. 2018. An Overview of Recent Therapeutics Advances for Duchenne Muscular Dystrophy. Methods Mol Biol. 1687:3–17.
- Maherali N, Sridharan R, Xie W, Utikal J, Eminli S, Arnold K, Stadtfeld M, Yachechko R, Tchieu J, Jaenisch R, et al. 2007. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 1(1):55–70.
- Mashinchian O, De Franceschi F, Nassiri S, Michaud J, Migliavacca E, Aouad P, Metairon S, Pruvost S, Karaz S, Fabre P, Molina T. 2022. An engineered multicellular stem cell niche for the 3D derivation of human myogenic progenitors from iPSCs. EMBO J. 41(14):e110655.
- Matsumura K, Saito F, Yamada H, Hase A, Sunada Y, Shimizu T. 1999. Sarcoglycan complex: a muscular supporter of dystroglycan-dystrophin interplay? Cell Mol Biol (Noisy-le-grand). 45(6):751–762.
- Mazaleyrat K, Badja C, Broucqsault N, Chevalier R, Laberthonnière C, Dion C, Baldasseroni L, El-Yazidi C, Thomas M, Bachelier R, Altié A. 2020. Multilineage differentiation for formation of innervated skeletal muscle fibers from healthy and diseased human pluripotent stem cells. Cells. 9(6).
- Miura Y, Sato M, Kuwahara T, Ebata T, Tabata Y, Sakurai H. 2022. Transplantation of human iPSC-derived muscle stem cells in the diaphragm of Duchenne muscular dystrophy model mice. PLoS One. 17(4):e0266391.
- Moradi S, Mahdizadeh H, Šarić T, Kim J, Harati J, Shahsavarani H, Greber B, Moore JB. 2019. Research and therapy with induced pluripotent stem cells (iPSCs): social, legal, and ethical considerations. Stem Cell Res Ther. 10(1):341.
- Morisaka H, Yoshimi K, Okuzaki Y, Gee P, Kunihiro Y, Sonpho E, Xu H, Sasakawa N, Naito Y, Nakada S, et al. 2019. CRISPR-Cas3 induces broad and unidirectional genome editing in human cells. Nat Commun. 10(1):5302.
- Mournetas V, Massouridès E, Dupont J, Kornobis E, Polvèche H, Jarrige M, Dorval ARL, Gosselin MRF, Manousopoulou A, Garbis SD, et al. 2021. Myogenesis modelled by human pluripotent stem cells: a multi-omic study of Duchenne myopathy early onset. J Cachexia Sarcopenia Muscle. 12(1):209–232.
- Mukund K, Subramaniam S. 2020. Skeletal muscle: A review of molecular structure and function, in health and disease. Wiley Interdiscip Rev Syst Biol Med. 12(1):e1462.
- Neco P, Rose B, Huynh N, Zhang R, Bridge JHB, Philipson KD, Goldhaber J. 2010. Sodium-calcium exchange is essential for effective triggering of calcium release in mouse heart. Biophys J. 99(3):755–764.
- Niggli E. 1999. Localized intracellular calcium signaling in muscle: calcium sparks and calcium quarks. Annu Rev Physiol. 61:311–335.
- Nozoe KT, Akamine RT, Mazzotti DR, Polesel DN, Grossklauss LF, Tufik S, Andersen ML, Moreira GA. 2016. Phenotypic contrasts of Duchenne Muscular Dystrophy in women: Two case reports. Sleep Sci. 9(3):129–133.
- Okita K, Ichisaka T, Yamanaka S. 2007. Generation of germline-competent induced pluripotent stem cells. Nature. 448(7151):313–317.
- Osaki T, Uzel SGM, Kamm RD. 2018. Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci Adv. 4(10):eaat5847.
- Pareja-Galeano H, Sanchis-Gomar F, Emanuele E, Gallardo ME, Lucia A. 2016a. Ipscs, a promising tool to restore muscle atrophy. J Cell Physiol. 231(2):259–260.
- Pareja-Galeano H, Sanchis-Gomar F, Pérez LM, Emanuele E, Lucia A, Gálvez BG, Gallardo ME. 2016b. iPSCs-based anti-aging therapies: Recent discoveries and future challenges. Ageing Res Rev. 27:37–41.
- Partridge TA, Morgan JE, Coulton GR, Hoffman EP, Kunkel LM. 1989. Conversion of mdx myofibres from dystrophin-negative to -positive by injection of normal myoblasts. Nature. 337(6203):176–179.
- Pette D, Staron RS. 1997. Mammalian skeletal muscle fiber type transitions. Int Rev Cytol. 170:143–223.
- Philippou A, Xanthis D, Chryssanthopοulos C, Maridaki M, Koutsilieris M. 2020. Heart failure-induced skeletal muscle wasting. Curr Heart Fail Rep. 17(5):299–308.
- Piga D, Salani S, Magri F, Brusa R, Mauri E, Comi GP, Bresolin N, Corti S. 2019. Human induced pluripotent stem cell models for the study and treatment of Duchenne and Becker muscular dystrophies. Ther Adv Neurol Disord. 12:1756286419833478.
- Pourquie O, Tanoury ZA, Chal J. 2018. The Long Road to Making Muscle In Vitro. Curr Top Dev Biol. 129:123–142.
- Prior BM, Lloyd PG, Yang HT, Terjung RL. 2003. Exercise-induced vascular remodeling. Exerc Sport Sci Rev. 31(1):26–33.
- Purslow PP. 2010. Muscle fascia and force transmission. J Bodyw Mov Ther. 14(4):411–417.
- Rao L, Qian Y, Khodabukus A, Ribar T, Bursac N. 2018. Engineering human pluripotent stem cells into a functional skeletal muscle tissue. Nat Commun. 9(1):126.
- Raymond-Pope CJ, Basten AM, Bruzina AS, McFaline-Figueroa J, Lillquist TJ, Call JA, Greising SM. 2023. Restricted physical activity after volumetric muscle loss alters whole-body and local muscle metabolism. J Physiol. 601(4):743–761.
- Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, Welch J, Chen LM, Chen F, Macosko EZ. 2019. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science. 363(6434):1463–1467.
- Rowland LP. 1985. Gene expression in muscle. In: Strohman C, Wolf S, editors. Advances in Experimental Medicine and Biology. Springer Nature City: London; p. 3–5.
- Sakai-Takemura F, Narita A, Masuda S, Wakamatsu T, Watanabe N, Nishiyama T, Nogami K, Blanc M, Takeda S, Miyagoe-Suzuki Y. 2018. Premyogenic progenitors derived from human pluripotent stem cells expand in floating culture and differentiate into transplantable myogenic progenitors. Sci Rep. 8(1):6555.
- Sambasivan R, Kuratani S, Tajbakhsh S. 2011a. An eye on the head: the development and evolution of craniofacial muscles. Development. 138(12):2401–2415.
- Sambasivan R, Yao R, Kissenpfennig A, Van Wittenberghe L, Paldi A, Gayraud-Morel B, Guenou H, Malissen B, Tajbakhsh S, Galy A. 2011b. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development. 138(17):3647–3656.
- Sandow A. 1952. Excitation-contraction coupling in muscular response. Yale J Biol Med. 25(3):176–201.
- Sato T. 2020. Induction of skeletal muscle progenitors and stem cells from human induced pluripotent stem cells. J Neuromuscul Dis. 7(4):395–405.
- Scharner J, Zammit PS. 2011. The muscle satellite cell at 50: the formative years. Skelet Muscle. 1(1):28.
- Selvaraj S, Mondragon-Gonzalez R, Xu B, Magli A, Kim H, Lainé J, Kiley J, Mckee H, Rinaldi F, Aho J, Tabti N. 2019. Screening identifies small molecules that enhance the maturation of human pluripotent stem cell-derived myotubes. Elife. 8.
- Shayan M, Huang NF. 2020. Pre-Clinical cell therapeutic approaches for repair of volumetric muscle loss. Bioengineering (Basel). 7(3).
- Shelton M, Kocharyan A, Liu J, Skerjanc IS, Stanford WL. 2016. Robust generation and expansion of skeletal muscle progenitors and myocytes from human pluripotent stem cells. Methods. 101:73–84.
- Siegel AL, Kuhlmann PK, Cornelison DD. 2011. Muscle satellite cell proliferation and association: new insights from myofiber time-lapse imaging. Skelet Muscle. 1(1):7.
- Suleski IS, Smith R, Vo C, Scriba CK, Saker S, Larmonier T, Malfatti E, Romero NB, Houweling PJ, Nowak KJ, et al. 2022. Generation of two isogenic induced pluripotent stem cell lines from a 1-month-old nemaline myopathy patient harbouring a homozygous recessive c.121C > T (p.Arg39Ter) variant in the ACTA1 gene. Stem Cell Res. 63:102830.
- Suman S, Domingues A, Ratajczak J, Ratajczak MZ. 2019. Potential Clinical Applications of Stem Cells in Regenerative Medicine. Adv Exp Med Biol. 1201:1–22.
- Sun C, Choi IY, Gonzalez YIR, Andersen P, Talbot Jr CC, Iyer SR, Lovering RM, Wagner KR, Lee G. 2020. Duchenne muscular dystrophy hiPSC-derived myoblast drug screen identifies compounds that ameliorate disease in mdx mice. JCI Insight. 5(11).
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 131(5):861–872.
- Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 126(4):663–676.
- Takeshita E, Minami N, Minami K, Suzuki M, Awashima T, Ishiyama A, Komaki H, Nishino I, Sasaki M. 2017. Duchenne muscular dystrophy in a female with compound heterozygous contiguous exon deletions. Neuromuscul Disord. 27(6):569–573.
- Tanaka KK, Hall JK, Troy AA, Cornelison DDW, Majka SM, Olwin BB. 2009. Syndecan-4-expressing muscle progenitor cells in the SP engraft as satellite cells during muscle regeneration. Cell Stem Cell. 4(3):217–225.
- Tedesco FS, Gerli MF, Perani L, Benedetti S, Ungaro F, Cassano M, Antonini S, Tagliafico E, Artusi V, Longa E, Tonlorenzi R. 2012. Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci Transl Med. 4(140):140ra89.
- Uchimura T, Otomo J, Sato M, Sakurai H. 2017. A human iPS cell myogenic differentiation system permitting high-throughput drug screening. Stem Cell Res. 25:98–106.
- Uchimura T, Otomo J, Sato M, Sakurai H. 2020. Engraftment of human induced pluripotent stem cell-derived myogenic progenitors restores dystrophin in mice with duchenne muscular dystrophy. Biol Res. 53(1):22.
- Valetdinova KR, Maretina MA, Vyatkin YV, Perepelkina MP, Egorova AA, Baranov VS, Kiselev AV, Gershovich PM, Zakian SM. 2020. Generation of three Duchenne muscular dystrophy patient-derived induced pluripotent stem cell (iPSC) lines ICGi002-A, ICGi002-B and ICGi002-C. Stem Cell Res. 48:101941.
- van der Wal E, Herrero-Hernandez P, Wan R, Broeders M, in 't Groen SLM, van Gestel TJM, van IJcken WFJ, Cheung TH, van der Ploeg AT, Schaaf GJ, Pijnappel WWMP. 2018. Large-Scale expansion of human iPSC-derived skeletal muscle cells for disease modeling and cell-based therapeutic strategies. Stem Cell Rep. 10(6):1975–1990.
- Van Ruiten HJA, Marini Bettolo C, Cheetham T, Eagle M, Lochmuller H, Straub V, Bushby K, Guglieri M. 2016. Why are some patients with Duchenne muscular dystrophy dying young: An analysis of causes of death in North East England. Eur J Paediatr Neurol. 20(6):904–909.
- Vaughan M, Lamia KA. 2019. Isolation and differentiation of primary myoblasts from mouse skeletal muscle explants. J Vis Exp. 152.
- Vicenti G, Bortone I, Bizzoca D, Sardone R, Belluati A, Solarino G, Moretti B. 2020. Bridging the gap between serum biomarkers and biomechanical tests in musculoskeletal ageing. J Biol Regul Homeost Agents. 34(4 Suppl. 3):263–274. Congress of the Italian Orthopaedic Research Society.
- Vila OF, Chavez M, Ma SP, Yeager K, Zholudeva LV, Colón-Mercado JM, Qu Y, Nash TR, Lai C, Feliciano CM, et al. 2021. Bioengineered optogenetic model of human neuromuscular junction. Biomaterials. 276:121033.
- Vila OF, Uzel SGM, Ma SP, Williams D, Pak J, Kamm RD, Vunjak-Novakovic G. 2019. Quantification of human neuromuscular function through optogenetics. Theranostics. 9(5):1232–1246.
- Wang J, Conboy I. 2010. Embryonic vs. adult myogenesis: challenging the ‘regeneration recapitulates development’ paradigm. J Mol Cell Biol. 2(1):1–4.
- Wang YX, Rudnicki MA. 2011. Satellite cells, the engines of muscle repair. Nat Rev Mol Cell Biol. 13(2):127–133.
- Washington TA, Wolchok JC. 2023. Prescribing reduced physical activity following volumetric muscle loss not really a good idea. J Physiol. 601(7):1157–1158.
- Wei CW, Luo T, Zou SS, Wu AS. 2018. The role of long noncoding RNAs in central nervous system and neurodegenerative diseases. Front Behav Neurosci. 12:175.
- Werneck LC, Lorenzoni PJ, Ducci RD-P, Fustes OH, Kay CSK, Scola RH. 2019. Duchenne muscular dystrophy: an historical treatment review. Arq Neuropsiquiatr. 77(8):579–589.
- Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, Bernstein BE, Jaenisch R. 2007. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 448(7151):318–324.
- Willi L, Abramovich I, Fernandez-Garcia J, Agranovich B, Shulman M, Milman H, Baskin P, Eisen B, Michele DE, Arad M, Binah O. 2022. Bioenergetic and metabolic impairments in induced pluripotent stem cell-derived cardiomyocytes generated from duchenne muscular dystrophy patients. Int J Mol Sci. 23(17).
- Wu J, Matthias N, Bhalla S, Darabi R. 2021. Evaluation of the therapeutic potential of human iPSCs in a murine model of VML. Mol Ther. 29(1):121–131.
- Wu J, Matthias N, Lo J, Ortiz-Vitali JL, Shieh AW, Wang SH, Darabi R. 2018. A myogenic double-reporter human pluripotent stem cell line allows prospective isolation of skeletal muscle progenitors. Cell Rep. 25(7):1966–1981e4.
- Xuan W, Khan M, Ashraf M. 2021. Pluripotent stem cell-induced skeletal muscle progenitor cells with givinostat promote myoangiogenesis and restore dystrophin in injured Duchenne dystrophic muscle. Stem Cell Res Ther. 12(1):131.
- Xuan W, Tipparaju SM, Ashraf M. 2022. Extracellular vesicles from iPSC derived muscle progenitor cells rejuvenate the dysfunctional muscle stem cells during aging. FASEB J. 36(S1).
- Yasutake H, Lee J-K, Hashimoto A, Masuyama K, Li J, Kuramoto Y, Higo S, Hikoso S, Hidaka K, Naito AT, et al. 2021. Decreased YAP activity reduces proliferative ability in human induced pluripotent stem cell of duchenne muscular dystrophy derived cardiomyocytes. Sci Rep. 11(1):10351.
- Yin H, Price F, Rudnicki MA. 2013. Satellite cells and the muscle stem cell niche. Physiol Rev. 93(1):23–67.
- Yoshioka K, Ito A, Kawabe Y, Kamihira M. 2020. Novel neuromuscular junction model in 2D and 3D myotubes co-cultured with induced pluripotent stem cell-derived motor neurons. J Biosci Bioeng. 129(4):486–493.
- Young C, Hicks M, Ermolova N, Nakano H, Jan M, Younesi S, Karumbayaram S, Kumagai-Cresse C, Wang D, Zack J, et al. 2016. A single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell. 18(4):533–540.
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. 2007. Induced pluripotent stem cell lines derived from human somatic cells. Science. 318(5858):1917–1920.
- Zablocka B, Gorecki DC, Zablocki K. 2021. Disrupted calcium homeostasis in duchenne muscular dystrophy: A common mechanism behind diverse consequences. Int J Mol Sci. 22(20).
- Zaynitdinova MI, Lavrov AV, Smirnikhina SA. 2021. Animal models for researching approaches to therapy of Duchenne muscular dystrophy. Transgenic Res. 30(6):709–725.
- Zhang Y, Li Y, Hu Q, Xi Y, Xing Z, Zhang Z, Huang L, Wu J, Liang K, Nguyen TK, et al. 2020. The lncRNA H19 alleviates muscular dystrophy by stabilizing dystrophin. Nat Cell Biol. 22(11):1332–1345.
- Zhao T, Zhang Z-N, Rong Z, Xu Y. 2011. Immunogenicity of induced pluripotent stem cells. Nature. 474(7350):212–215.