
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Shape memory polymers (SMPs), particularly those influenced by entanglement effects, are gaining significant attention. These polymers exhibit notable shape-changing properties on a macroscopic scale and intricate structural characteristics on a microscopic level. Entanglements play a crucial role in the shape memory process of certain polymer systems, especially in physically crosslinked systems, and thus this mechanism offers potential for expanding the use of thermoplastic polymers in SMPs, which is particularly promising for applications requiring specific material properties, such as biomedical devices. Despite their promise, a comprehensive understanding of entanglement based SMPs remains elusive due to limited research and reviews in this area. This perspective endeavours to bridge this gap by delving into the intricate relationship between the shape memory characteristics of SMPs and their underlying structural properties, with a focus on entanglement-driven mechanisms. We attempt to conduct a comprehensive review of recent advancements on entanglement-driven shape memory polymers and analyse existing data to explore these complex interactions, and thus attempt to understand how these developments are contributing towards enhanced shape memory properties.
Graphical abstract
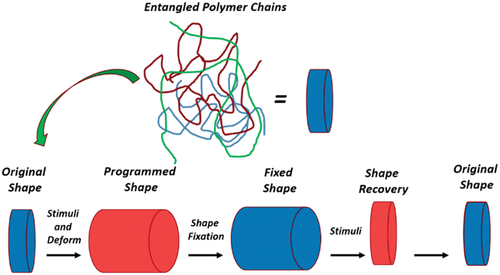
1. Introduction to shape memory polymers (SMPs)
Shape memory polymers (SMPs) represent an innovative class of smart polymeric materials revolutionizing various industries [Citation1–4]. These smart materials possess the remarkable ability to be manipulated into a temporary shape and then, upon exposure to an external stimulus such as heat [Citation6], light [Citation7], electromagnetic induction [Citation8], or solvents [Citation9], seamlessly revert to their original, memorized shape. Among these, thermal-responsive SMPs predominantly rely on Joule heating for activation, typically facilitated by hot gas or water immersion [Citation4]. However, direct activation using hot fluids like water or gas is often impractical in practical applications, limiting extensive exploration in this area. To circumvent this limitation, alternative strategies involve incorporating functional moieties within SMP matrices. These moieties enable activation via alternative stimuli such as electricity, light, magnetism, or moisture. Despite the varied triggering mechanisms, these methods fundamentally harness Joule heat generation albeit indirectly through the specific stimulus [Citation4]. For example, in the case of water or solvent-driven SMPs, the introduction of solvent molecules into the polymer structure acts as plasticizers upon water immersion, effectively reducing the transition temperature and facilitating shape recovery (the ability of the polymer to return from the deformed state to its original shape when stimulated is typically defined as the recovered strain relative to the programmed strain) [Citation6]. This mechanism represents an indirect form of thermal actuation. In contrast, light-induced SMPs integrate reversible photoreactive molecular switches [Citation6–11]. Unlike thermal methods, which rely on temperature changes for activation, light-induced activation operates independently of thermal effects. While a detailed discussion on the stimuli for shape-memory polymers is beyond the scope of this perspective, for reference, we have summarized several recent developments in shape-memory polymers that respond to various stimuli in Table S1 (supporting information).
Figure 1. Schematic representation of the mechanism of shape-memory effects for thermally induced shape-memory polymers (SMPs) based on a crystal-amorphous transition in a semicrystalline-based polymer network, reprinted with permission from [Citation5], copyright reserved Taylor and Francis 2015.
![Figure 1. Schematic representation of the mechanism of shape-memory effects for thermally induced shape-memory polymers (SMPs) based on a crystal-amorphous transition in a semicrystalline-based polymer network, reprinted with permission from [Citation5], copyright reserved Taylor and Francis 2015.](/cms/asset/26d42fea-fc3c-48c2-8aac-1685c96063ac/tsmm_a_2374342_f0001_oc.jpg)
In contrast to shape memory alloys (SMAs), SMPs offer a plethora of advantages, making them highly sought-after materials in diverse fields [Citation10]. One of their standout features is their ability to achieve high recoverable strains, providing unparalleled flexibility and adaptability in shape manipulation [Citation11]. Additionally, SMPs are advantageous in terms of their flexible transition temperatures, allowing for precise control over the activation of shape memory properties. Furthermore, their ease of processing and relatively low manufacturing cost contribute to their widespread appeal and adoption [Citation12,Citation13].
Expanding on the last point, polymeric materials inherently exhibit shape memory effects, though their mechanisms differ significantly from those in metal alloys. In shape memory alloys, the pseudoplastic fixing occurs through a martensitic de-twinning mechanism, with shape recovery triggered by a martensite-austenite phase transition. Temporary shape fixing in SMAs is typically achieved at a single temperature, usually just below room temperature, with recovery occurring upon heating beyond the martensitic transformation temperature [Citation12,Citation13]. In contrast, SMPs utilize a variety of physical mechanisms for temporary shape fixing and recovery, leveraging the intrinsic elasticity of polymeric networks. This elasticity provides the large extensibility characteristic of SMPs. Compared to SMAs, SMPs offer several advantages, including high elastic deformation (strain up to more than 200% for many materials), lower cost, lower density, and potential biocompatibility and biodegradability.
Additionally, SMPs have a wide range of application temperatures that can be tailored to specific needs, tunable stiffness, and ease of processing [Citation13]. The distinct mechanical, viscoelastic, and optical properties of polymers and metal alloys result in different applications for these two classes of shape memory materials. While SMAs are typically used in applications requiring high strength and thermal stability, SMPs are favoured in areas where high elasticity, biocompatibility, and versatility in processing and application temperatures are critical. This makes SMPs particularly suitable for biomedical applications, where specific material properties are required [Citation13].
At the molecular level, SMPs are engineered with two essential components: crosslinks and switching segments. These components work synergistically to enable the manifestation of shape memory effects. The crosslinks, constituting the fixed phase, dictate the permanent shape of the polymer. Meanwhile, the switching segments, characterized by transition temperatures (Tt), govern the fixation of the temporary shape [Citation6,Citation14,Citation15]. Notably, SMPs are categorized into two distinct groups based on the nature of their crosslinks: chemically crosslinked SMPs and physically crosslinked SMPs [Citation5,Citation6,Citation14,Citation15]. Moreover, the nature of the switching segments further classifies SMPs into two categories: those with amorphous switching segments, where Tt corresponds to the glass transition temperature (Tg), and those with crystalline switching segments, where Tt aligns with the melting temperature (Tm) [Citation16].
Figure 2. Mechanisms of shape memory effect in polymeric materials. (a) microscopic perspective: thermal programming induces shape memory by heating the material to deform into a temporary shape, which is fixed upon cooling and recovered upon reheating. (b) macroscopic perspective of the shape memory process (c) schematic illustrating key elements in the polymer network affecting shape memory: switching segments linking netpoints, side chains as switching segments, molecular switches forming reversible covalent bonds, and aba triblock segments linking netpoints, (a and c) reprinted with permission from [Citation16], copyright reserved MDPI 2023.
![Figure 2. Mechanisms of shape memory effect in polymeric materials. (a) microscopic perspective: thermal programming induces shape memory by heating the material to deform into a temporary shape, which is fixed upon cooling and recovered upon reheating. (b) macroscopic perspective of the shape memory process (c) schematic illustrating key elements in the polymer network affecting shape memory: switching segments linking netpoints, side chains as switching segments, molecular switches forming reversible covalent bonds, and aba triblock segments linking netpoints, (a and c) reprinted with permission from [Citation16], copyright reserved MDPI 2023.](/cms/asset/eae47c08-dd24-454c-863e-80211143924e/tsmm_a_2374342_f0002_oc.jpg)
As we discussed, SMPs consist of two distinct phases: a stable, permanent network and an unstable, temporary network [Citation5]. The permanent network remains unchanged by external stimuli, providing structural stability, while the temporary network can undergo a reversible phase change when triggered by an external stimulus [Citation16]. The temporary network enables the material to change shape temporarily, while the permanent network ensures it returns to its original shape after the stimulus is removed. Achieving the permanent phase involves creating chemical cross-links or interpenetrating networks, which provide stability. In contrast, the temporary phase utilizes mechanisms such as melting temperature, glass transition temperature, liquid crystalline phases, or reversible bonds . For instance, in a thermally stimulated SMP with a cross-linked permanent network and crystalline temporary domains, heating beyond the melting temperature of the crystals deactivates the temporary segment. If the material is deformed and then cooled below the crystallization temperature of the crystals, the deformed shape becomes fixed by the activated crystals. Subsequent heating enables the crystals to deactivate, restoring the material to its original shape due to the entropy-driven relaxation of the system .
Several factors influence SMP’s shape memory properties [Citation6–9]. Effective shape retention depends largely on the strength of the temporary network, which must securely hold the deformed shape. Meanwhile, the ability to perfectly recover the original shape is governed by properties like cross-link density, stress relaxation behaviour, and creep resistance of the permanent network . From a thermomechanical perspective, the elastic component of the SMP stores energy during deformation, requiring efficient energy storage without premature release [Citation9]. The temporary segment’s ability to undergo reversible deformation allows the material to recover its original shape by relieving constraints once the external stimulus is removed .
While there are numerous factors influencing the shape memory properties of polymers, entanglement is a significant aspect that motivates this perspective. In the realm of SMPs, entanglement plays a critical role in both the temporary and permanent networks. The temporary network, responsible for reversible deformation, relies on the flexibility and rearrangement of polymer chains facilitated by entanglements. Strong entanglements can improve the material’s ability to maintain a deformed shape until triggered to revert to its original form [Citation8]. Similarly, in the permanent network, entanglements contribute to the overall mechanical strength and stability of the SMP. Higher levels of entanglement can reinforce the network, affecting properties such as cross-link density and stress relaxation behaviour, which are essential for achieving consistent shape memory effects. Understanding and controlling entanglement in SMPs is crucial for optimizing their shape memory behaviour. Researchers and engineers investigate entanglement dynamics to tailor SMPs for specific applications, aiming to enhance performance metrics such as shape recovery efficiency, durability, and responsiveness to stimuli.
This perspective aims to reflect a phenomenon in the realm of shape memory polymers, which remains largely unexplored. While numerous reviews on SMPs exist [Citation1–3,Citation7,Citation10], focusing on specific themes, such as entanglements, can be particularly interesting. For example, in the case of amorphous polyurethanes, entanglements can significantly enhance shape memory properties compared to systems that do not incorporate entanglements [Citation10]. In amorphous SMPs, such as SMPU (Shape Memory Polyurethane), entanglements play a crucial role in defining the material’s shape memory behaviour. The mechanism is typically described using the concept of a confinement tube. Within this tube, the hard segments of the polymer can either move freely at high temperatures or be fixated at low temperatures. The presence of entanglements affects this mechanism, as well as the overall processing and chemical composition of the SMPU [Citation10]. As we can hypothesize, when the polymer network is highly entangled, the characteristic relaxation time of the chain segments within the confinement tube is longer. This results in a higher recovery temperature or, equivalently, a longer recovery time. This extended relaxation time means that the material can maintain its deformed shape for a longer period before returning to its original shape upon heating. Thus, entanglements enhance the stability of the temporary shape and the efficiency of the shape memory effect. Moreover, entanglements contribute to the overall mechanical strength and durability of SMPs [Citation13]. They create a more robust network that can withstand repeated cycles of deformation and recovery, essential for practical applications. In highly entangled systems, the polymer chains are more tightly interwoven, which can improve the material’s resistance to mechanical stress and fatigue. This makes entangled SMPs suitable for applications requiring high durability and long-term reliability, such as in biomedical devices, aerospace components, and smart textiles.
This perspective begins by introducing SMPs, typically the class of physically crosslinked polymers, and investigates the pivotal role of entanglements in governing their mechanical and shape memory properties. The perspective aims to elucidate recent advancements and empirical analyses of SMP behaviour to discern their practical implications. It is evident that entanglements significantly influence the viscoelastic behaviour of SMPs, impacting their ability to retain temporary shapes and resist mechanical deformation. Understanding the molecular interactions and chain dynamics within SMP networks is crucial for predicting and optimizing material performance across various applications.
By systematically examining the effects of entanglements on SMP properties, particularly in terms of recovery stress and shape fixity (he ability of the polymer to maintain its deformed state is typically defined as the fixed strain relative to the applied strain), this study aims to provide fundamental insights essential for engineering SMPs with tailored functionalities. Such advancements hold promise for applications in sectors requiring adaptive materials, including biomedical devices, aerospace components, and smart textiles. The pragmatic implications of this research lie in its potential to enhance the reliability and longevity of SMP-based technologies, thereby contributing to advancements in material science and enabling innovative solutions in diverse industrial applications.
2. Introduction to shape memory polymers comprising physically crosslinked block copolymers
Physically crosslinked block copolymers, particularly thermoplastic segmented polyurethanes, have emerged as prominent candidates in the realm of SMPs, drawing considerable attention and extensive research efforts [Citation17–21]. Traditionally, polyurethanes are structured as phase-separated multiblock copolymers, comprising alternating sequences of ‘hard’ and ‘soft’ segments [Citation17]. The ‘hard’ blocks within polyurethanes exhibit the highest thermal transition temperature (Th) and function as physical crosslinks through polar interaction, hydrogen bonding, and/or crystallization. These crosslinks play a pivotal role in defining the material’s ‘permanent’ shape. When the temperature surpasses Th, the polymer undergoes melting, effectively erasing the permanent shape and enabling conventional thermal processing techniques such as compression moulding, extrusion, or injection moulding [Citation17].
On the other hand, the ‘soft’ blocks encapsulate the thermally reversible phase. The vitrification or crystallization of these soft segments facilitates the fixation of the temporary shape. When the material is deformed above its transition temperature (Tt) but below Th, the polymer networks often exhibit ‘superelasticity’ [Citation22]. This phenomenon allows the polymer chains between crosslink points to deform freely. Subsequently, after undergoing elastic deformation, the temporary shape can be fixed by cooling the polymer below Tt, effectively storing the elastic energy exerted during the prior deformation [Citation22].
It’s commonly understood that when a polymer is cooled below its glass transition temperature (Tg), the movements of its molecular chains come to a halt. However, upon surpassing Tg, these motions resume significantly, marking a transition from a glassy state to a rubbery-elastic state [Citation23]. During this transition, if a uniaxial stress is applied briefly, the entanglement of polymer chains prevents extensive movement, leading to the storage of entropic energy. However, prolonged stress application triggers a relaxation process, causing chain slippage and bulk flow of polymer chains [Citation24].
This reversible deformation at the molecular level is facilitated by utilizing network chains as a form of molecular switch. At a certain level of external applied energy (stimulus), the chains become flexible (Step 1, ) at temperatures above a transitional temperature (Tt), while their flexibility diminishes below Tt. Consequently, freezing of molecular motion in the amorphous zone or crystallization in the crystalline zone impedes immediate reforming of coil-like structures and instinctive recovery of the original shape, thus fixing the programmed shape [Citation24,Citation25]. The stability of molecular orientation depends on the interaction strength between macromolecular segments and the conformations of the chains comprising a polymer. Stretching of molecular chains results (Step 2, ) in decreased entropy, which is compensated for by the cooling process (Step 3, ), leading to a reduction in internal energy. Conversely, upon heating (Step 4, ), oriented polymer chains soften from their glassy state or melt from crystals, allowing molecular chains to relax orientation and form more stable, coiled conformations. This relaxation or shrinkage of molecular chains drives shape recovery [Citation23–25]. Consequently, the elastic strain energy generated during the deformation process serves as the driving force for shape recovery in shape memory polyurethanes (SMPUs) (Step 4, ). The comprehensive molecular mechanism underlying the shape memory behaviour of SMPUs is depicted in .
Figure 3. (a) The molecular mechanism of the shape memory effect under different stimuli. Black dots: net points; blue lines: SMPU chains below Tt (low mobility); red lines: SMPU chains above Tt (high mobility), reprinted with permission from [Citation22], copyright reserved intech open 2017. (b) Entangled elastomer chains when responding to externally applied force in different ways.
![Figure 3. (a) The molecular mechanism of the shape memory effect under different stimuli. Black dots: net points; blue lines: SMPU chains below Tt (low mobility); red lines: SMPU chains above Tt (high mobility), reprinted with permission from [Citation22], copyright reserved intech open 2017. (b) Entangled elastomer chains when responding to externally applied force in different ways.](/cms/asset/b5cadfb9-b19d-415a-9c98-5857e7429554/tsmm_a_2374342_f0003_oc.jpg)
Here is where entanglements come into effect and play a pivotal role in driving the shape memory process. From the perspective of elastomer transition states, the movements of elastomer molecules are restricted in the glassy region (below the glass transition temperature) [Citation24]. When the temperature increases, molecular rotations around the elastomer bonds also increase, transitioning the material towards the rubbery region. In this state, elastomer molecules become highly entangled and adopt a coiled conformation with maximum entropy, as described by the Boltzmann equation [Citation24].
In the rubbery state, elastomers with sufficient molecular weight can be stretched along the direction of an applied external force. When the force is applied quickly or over short periods, local entanglements in the elastomer molecules obstruct molecular movement . This resistance to movement causes the elastomer to return to its original shape once the force is removed, displaying a viscoelastic response. This viscoelastic response, influenced by the entangled state of the molecules, forms the basis of the shape memory effect [Citation22]. The elastomer ‘remembers’ its initial randomly coiled state, which it returns to after deformation. In contrast, when a force is applied slowly or over extended periods, a relaxation process occurs. During this process, elastomer chains slip and disentangle, allowing the molecules to rearrange into new conformations with favourable entropy, as we already inferred . Crosslinking the molecules prevents the slipping or flow of elastomer chains under force. Chemically crosslinked elastomers form a network structure that becomes insoluble and maintains its shape after crosslinking, thereby enhancing the shape memory properties. Nevertheless, molecular mobility is a key factor in the SME of polymers [Citation22,Citation23]. During the transition of a shape memory elastomer from its original shape to a temporary shape and back, the system involves two types of phases or domains. These phases are related to phase diagrams, which graphically represent the stability of various material domains at equilibrium. Phase diagrams are typically constructed in temperature-pressure-composition space or Gibbs free energy-composition space. When a system is equilibrated in one domain and then moved to another with a different equilibrium structure, such as by changing its temperature, it undergoes microstructure transformations to reach the new equilibrium state [Citation19]. The microstructure with optimal properties can be selected by interrupting this process and quenching (rapid cooling) in the desired structure. This quenched state is where the shape memory elastomer exhibits its shape memory effect, and indeed regulated by the entanglements, as the entangled state of elastomer molecules, combined with their ability to undergo relaxation and rearrangement, regulates the shape memory effect in polymers, which we shall explore in the next section [Citation19].
3. Introduction to entanglements and the possibility of developing SMPs based on chain entanglements
Polymer, distinguished by its notably elongated and flexible macromolecules, inherently imposes topological constraints on each other due to the intertwining of these chains, resulting in entanglements. These entanglements, prevalent in the amorphous phase of solid polymers and in molten or dissolved states, significantly impact polymer properties, particularly toughness and strength, thereby granting polymers a distinct advantage over other materials and contributing to their wide-ranging applications [Citation26].
Structurally, entanglements can be categorized into topological and cohesional entanglements, as depicted in [Citation27]. The study of topological entanglements dates back to Treloar in the 1940s, with subsequent development of various models elucidating their essence [Citation28]. The tube model proposed by Edwards has emerged as the most successful, confining each polymer chain within a virtual tube, allowing free movement in one direction while constraining it in the transverse direction () [Citation29]. This classical model, widely accepted and utilized, effectively describes the dynamics of polymer entanglement.
Figure 4. Structure of entanglement: (a) topological entanglement and (b) cohesion entanglement, reprinted with permission from [Citation27], copyright reserved Wiley 2021.
![Figure 4. Structure of entanglement: (a) topological entanglement and (b) cohesion entanglement, reprinted with permission from [Citation27], copyright reserved Wiley 2021.](/cms/asset/132b235d-72a7-458d-9976-d7cfcaf7b6d0/tsmm_a_2374342_f0004_oc.jpg)
While there are no direct experimental methods to observe the topological features of molecular chains, the concept of entanglements serves as a valuable tool for explaining various phenomena related to the rheological behaviour and mechanical properties of polymers. Evidence supporting the existence of entanglements can be summarized as follows: (1) The shear modulus of vulcanized rubber exceeds the prediction of models solely considering chemical cross-links. This discrepancy arises because entanglements restrict the movement of molecular chains, consequently increasing the shear modulus of the entire network structure. In the latest theory of rubber elasticity, rubber networks must be treated with both physical cross-links and entanglement constraints [Citation30,Citation31]. (2) The melt viscosity (η) at zero shear rate exhibits an apparent dependence on molecular weight (M). The relationship between η and M for various polymers can be expressed by Equation 1 or Equation 2, where k1 is a coefficient. When the molecular weight surpasses the critical value (Mc), viscosity drastically increases with increasing M [Citation27] and (3) Polymer melt in shear flows demonstrates a lower viscosity compared to equilibrium, a phenomenon known as shear-thinning [Citation27].
Excessive entanglement among molecular chains can result in heightened polymer melt viscosity, presenting considerable challenges in polymer processing. Various methods have been explored to control entanglement, including the creation of dilute polymer solutions, adjustment of polymerization conditions, and application of external force fields [Citation32]. Dilute polymer solutions offer precise control over entanglement levels; however, their suitability for industrial production is limited due to their low efficiency. Controlled polymerization, although promising, faces inefficiencies necessitating specific conditions to suppress catalytic activities effectively [Citation27]. Furthermore, the disentanglement effect induced by external force fields requires refinement, as rapid re-entanglement of polymer chains often undermine the effectiveness of the process. This highlights the need for further research in entanglement control, which could potentially intersect with advancements in shape memory polymers, ultimately leading to the development of innovative shape memory properties. Such synergies between entanglement control and shape memory polymers hold promise for revolutionizing material science and engineering applications.
In addition to the conventional physically crosslinked SMPs, there exists another intriguing category characterized by amorphous polymers with ultra-high molecular weights exceeding 106g/mol. Despite lacking conventional physical crosslinking, these polymers exhibit remarkable shape memory properties attributed to their unique molecular structure [Citation33].
Among the most notable materials in this category are polynorbornene (PN, Norsorex®) [Citation34,Citation35] and high molecular weight poly(methyl methacrylate) (PMMA) [Citation36], both of which gained prominence in the late 1970s. What distinguishes these polymers is their substantial entanglement per chain, serving as functional analogs to physical crosslinks. This abundance of entanglements results in the formation of a dense three-dimensional network within the polymer matrix, imparting exceptional elasticity above the glass transition temperature (Tg). In addition to segmented polyurethanes, highly entangled polynorbornene has garnered attention for its remarkable shape recovery capabilities, attributed to its extended relaxation time slightly above its glass transition temperature (Tg), which conveniently aligns with room temperature (approximately Tg ≈ 35–50 °C).
A fascinating question arises: Could inert, instead of conventional ‘hard’ blocks, serve as the physical crosslink in this system, thereby inducing memory for multiblock polyurethanes? While numerous shape memory reviews have been published recently [Citation37–43], understanding shape memory polymers originating from entanglements presents a captivating and relatively unexplored territory. As already stated, this focused analysis delves into the advantages of the phenomenon of shape memory polymers achieved via entanglements, shedding light on the unique properties and performance relationships of these polymers compared to traditionally developed elastomers. By exploring this novel avenue, we aim to uncover new insights and potential applications in the field of materials science and engineering.
4. Developments in polymers exhibiting shape memory property due to chain entanglements
To understand why the concept of entanglement is crucial in regulating the shape memory properties of SMPs, it is important to delve into the mechanics of entropic energy and its impact on recovery stress. Achieving SMPs with high energy density that simultaneously exhibit high recovery stress and large recoverable strain is a significant challenge in materials science [Citation33]. The recovery stress in SMPs, which is the force exerted as the material returns to its original shape, is fundamentally determined by the entropic energy stored within the polymer network [Citation33–35]. This entropic energy is a measure of the disorder within the polymer chains and is controlled by the density and strength of network junctions. Network junctions are points within the polymer where chains are either chemically or physically bonded, creating a network structure.
In an entangled polymer network, the density of these junctions is determined by the entanglement molecular weight of the polymer. The entanglement molecular weight is the average length of a polymer chain segment between two entanglement points. As the molecular weight of these segments increases, the number of entanglement points decreases, resulting in lower junction density [Citation33]. The entropic energy stored in the polymer network is generally proportional to the entanglement plateau modulus, which is a measure of the material’s stiffness in the plateau region of its viscoelastic behaviour. For many polymers, this modulus is approximately 1 MPa, which sets an upper limit on the achievable recovery stress. This theoretical limit arises because the entropic energy, and thus the recovery stress, is directly related to the density of the entanglement points in the network [Citation34].
Adjusting the extent of entanglement can theoretically change the achievable recovery stress. Increasing the density of entanglements, by decreasing the entanglement molecular weight, can increase the stored entropic energy, thus enhancing the recovery stress. However, this must be balanced against the need for sufficient polymer chain mobility to allow large recoverable strains. If the network becomes too densely entangled, the material may become too rigid and unable to undergo significant deformation, thereby limiting its shape memory performance.
Recently, Mather et al. introduced a novel family of high molecular weight PCL – PEG thermoplastic polyurethanes (TPUs) synthesized through a one-step technique. The synthesis involved PCL diol (10 kg/mol) and PEG (10 kg/mol) chain-extended with lysine-diisocyanate (LDI), with variations in component ratios ranging from 0/100 to 100/0. X-ray measurements unveiled micro-phase separation between the hydrophilic PEG blocks and hydrophobic PCL blocks [Citation33]. To further elucidate the microstructure of our PCL – PEG TPUs post-processing, the authors conducted wide-angle X-ray scattering studies (WAXS) on hot-pressed films. The results, depicted in , shed light on the dependence of microstructure on PCL/PEG molar ratios.
Figure 5. WAXS patterns of PCL–PEG multiblock TPUs: (i) [PCL]100, (ii) [PCL]70–[PEG]30, (iii) [PCL]60–[PEG]40, (iv) [PCL]50–[PEG]50, (v) [PCL]40–[PEG]60, (vi) [PCL]30–[PEG]70, and (vii) [PEG]100. The X-ray wavelength (λ) is 1.54 Å (The subscript numbers represent the block weight %. The molecular weight for both PCL and PEG segments are 10 kg/mol), reprinted with permission from [Citation33], copyright reserved Elsevier 2012.
![Figure 5. WAXS patterns of PCL–PEG multiblock TPUs: (i) [PCL]100, (ii) [PCL]70–[PEG]30, (iii) [PCL]60–[PEG]40, (iv) [PCL]50–[PEG]50, (v) [PCL]40–[PEG]60, (vi) [PCL]30–[PEG]70, and (vii) [PEG]100. The X-ray wavelength (λ) is 1.54 Å (The subscript numbers represent the block weight %. The molecular weight for both PCL and PEG segments are 10 kg/mol), reprinted with permission from [Citation33], copyright reserved Elsevier 2012.](/cms/asset/764d8476-c530-480b-bd6c-b112add27668/tsmm_a_2374342_f0005_b.gif)
In the case of [PCL]100, the analysis revealed unoriented crystalline rings, indicative of the orthorhombic unit cell structure. These rings manifested as two strong reflection rings centred at d-spacing of 4.1 Å (2θ = 21.5°) and 3.7 Å (2θ = 23.8°), indexed as (110) and (200) respectively, along with weaker reflection rings at 5.6 Å (2θ = 15.8°) and 3.0 Å (2θ = 29.9°), indexed as (102) and (210) [Citation44]. Conversely, [PEG]100 exhibited two diffraction peaks centred at d-spacing of 4.6 Å (2θ = 19.3°) and 3.8 Å (2θ = 23.5°), attributed to the 120 and diverse planes of PEG monoclinic unit cell respectively [Citation45].
Across all five copolymers, both PCL and PEG components displayed well-developed crystalline structures. Notably, as depicted in , an increase in PEG content (ii → vi) corresponded to a decrease in the diffraction intensity of reflection peaks for PCL crystals at 4.1 Å, while those related to reflection from PEG crystal planes at 4.6 Å increased and sharpened. The peak overlap between PCL at 3.7 Å and PEG at 3.8 Å resulted in a broad peak. Additionally, beneath the diffraction peaks typical of PCL and PEG crystalline phases, a broad zone characteristic of the amorphous phase was observed.
Overall, these WAXS findings unveiled the independent existence of crystalline phases of both hydrophilic PEG blocks and hydrophobic PCL blocks in the multiblock TPUs, indicative of micro-phase separation driven by thermodynamic incompatibility [Citation33].
To gain insights into the entanglement of the polymer, the authors conducted dynamic mechanical analysis (DMA) on all compositions in tensile mode .
Figure 6. The evaluation of the tensile storage modulus for PCL-PEG multiblock TPUs was conducted at a heating rate of 3°C/min. The accompanying tan delta curve is provided as well, illustrating the proportion of the viscous to elastic contribution of the sample tested, reprinted with permission from [Citation33], copyright reserved Elsevier 2012.
![Figure 6. The evaluation of the tensile storage modulus for PCL-PEG multiblock TPUs was conducted at a heating rate of 3°C/min. The accompanying tan delta curve is provided as well, illustrating the proportion of the viscous to elastic contribution of the sample tested, reprinted with permission from [Citation33], copyright reserved Elsevier 2012.](/cms/asset/9e6b84bb-1770-4d99-8af2-4bde00b20c84/tsmm_a_2374342_f0006_oc.jpg)
Below Tg, E′ was at or above 1 GPa, gradually decreasing to approximately 100 MPa between Tg and Tm, before dramatically dropping to around 1 MPa upon heating above Tm. Notably, the observed rubbery modulus plateau above Tm spanned a broad temperature range for all TPUs, a critical factor for shape memory applications. This broad plateau indicates that the entanglements effectively function as physical crosslinks, contributing to the targeted elastic mechanical behaviour of the TPUs [Citation33].
The shape memory properties of robust semicrystalline multiblock TPUs were extensively investigated through Reversible Plasticity Shape Memory (RPSM) experiments. Utilizing the Linkam TST-350 tensile testing system, researchers subjected PCL – PEG TPUs to systematic analysis. Initially, samples were deformed up to 1240% at room temperature (RT) [Citation33], followed by the release of strain to approximately 800%, marking the establishment of the ‘stretched state’ [Citation33]. This experimental setup allowed for an evaluation of deformations between permanent and temporary shapes, reaching approximately 800%. Subsequently, samples were subjected to heating at 70°C for 1 minute to induce nearly complete shape recovery, referred to as the ‘recovered state’ [Citation33].
It is to be noted that based on DMA results , the chosen recovery temperature of 70°C fell within the rubbery plateau region, indicating elastomeric behaviour during recovery. Notably, in these systems, entanglements existing above the melting temperature served as physical crosslinks, facilitating recovery while impeding flow [Citation33].
Regarding the fixing ability, it was observed that the shape fixity (Rf %) remained independent of the overall degree of crystallinity. It is speculated that the fixing ability of SMPs gradually increased and eventually reached a plateau as the degree of crystallinity (χC) increased. The χC values of PCL – PEG TPUs ranged from 30% to 50%, deemed sufficiently high to ensure that the fixing ability of SMPs reached the plateau region, leading to invariance in the Rf % of the TPUs [Citation33].
Figure 7. (a) Shape memory properties of PCL–PEG TPUs with varying PCL weight content, original data sourced from [Citation33] (b) shape memory properties of PCL–SBS blends with varying PCL weight content, original data sourced from [Citation46].
![Figure 7. (a) Shape memory properties of PCL–PEG TPUs with varying PCL weight content, original data sourced from [Citation33] (b) shape memory properties of PCL–SBS blends with varying PCL weight content, original data sourced from [Citation46].](/cms/asset/c8c1fa95-e86f-4994-a5ab-d3b3ff459f21/tsmm_a_2374342_f0007_oc.jpg)
To gain further insights into the speculation regarding the plateau phenomenon, we investigated another system based on PCL and Styrene-Butadiene Styrene (SBS) blends. Our aim was to understand the variation of shape memory properties with PCL content, utilizing the published data available on these materials.
Our findings reinforce the significance of the presence of a plateau phase in PCL-based systems, as highlighted by the authors. In our secondary analysis, we also observe similar trends where shape fixity increases initially, followed by a plateau. We hypothesize that PCL domains form a percolating network, contributing to the consistent shape fixity of the materials, as evidenced by both datasets. This suggests a robust relationship between PCL content and the shape memory properties of the materials. Further investigations are warranted to elucidate the underlying mechanisms driving these observed trends and to optimize the performance of PCL-based systems for various applications.
An interesting observation we further derive from the same dataset is the variation of the shape memory property with the applied strain. While the shape recovery ratio remains almost constant, signifying that the recovery force generated during the programming process was sufficient to drive the shape recovery process, the shape fixity shows a steady increase with higher applied strain [Citation33].
Figure 8. Shape memory properties of PCL–PEG TPUs with varying deformation (%), original data sourced from [Citation33].
![Figure 8. Shape memory properties of PCL–PEG TPUs with varying deformation (%), original data sourced from [Citation33].](/cms/asset/503bf3ff-0aca-4a48-b7e0-de7bee9c6fcd/tsmm_a_2374342_f0008_oc.jpg)
Although various factors such as the chain pull out [Citation47], formation of new temporary domains [Citation48], stress relaxation, and chain alignment might affect the fixity ratio [Citation49], the entanglement factor may also play a pivotal role in engaging the shape fixity.
It is believed that in the initial stage of strain, the entanglement value notably increases with the rising strain [Citation50]. This phenomenon can be attributed to the elongation of the polymer along the stretch direction, causing a contraction in the cross-sectional area and thereby bringing polymer chains closer together, leading to increased aggregation. This notion finds support in the local configuration changes within the polymer network. Additionally, the straightening of chains during this phase increases their contact surface area with neighbouring chains, likely contributing to the rise in system entanglements. It is generally assumed that chain breakage is negligible during this phase [Citation50].
However, as the system progresses into the second stage, there is a notable reduction in the number of entanglements. This is because more bonds are subjected to force, causing gradual straightening of the polymer chains. During the initial stage, few chains experience breakage, and the fraction of broken chains devoid of entanglements remains low. Thus, entanglements remain at a minimal level. However, as strain increases and deformation enters the second stage, the proportion of broken chains without entanglements rises, resulting in a significant decrease in overall system entanglements [Citation50].
Moving into the third stage, the number of entanglements gradually decreases as a considerable number of chains break. Notably, there is a marked increase in the number of segments without entanglements as deformation transitions from the second to the third stage. This leads to a pronounced decline in the number of entanglements in this phase. Moreover, as strain increases in the third stage, the growth of chains lacking entanglements slows down, contributing to the tapering decrease in the system’s overall entanglement count [Citation50].
Further experimental investigations are warranted to comprehensively understand the complete viscoelastic profile and the entanglement profile of the SMP, as discussed. However, based on the observed recovery even at higher deformation levels, it can be speculated that during this process, the chains are brought closer together, resulting in increased aggregation. This closer proximity of polymer chains likely contributes to a higher value of entanglement, which may play a significant role in the shape fixity process. When the polymer chains are subjected to higher deformation levels, they experience increased stretching and reorientation. This stretching and reorientation bring the polymer chains into closer contact with each other, fostering stronger intermolecular interactions and aggregation [Citation36,Citation50]. As a result, the entanglement density within the polymer network may increase, as more polymer chains become intertwined with each other.
The higher entanglement density is expected to enhance the network’s resistance to deformation and promote shape fixation. Entanglements act as physical crosslinks within the polymer matrix, providing structural stability and preventing significant shape changes. Therefore, an increased entanglement density resulting from the closer proximity of polymer chains could contribute to a more efficient shape fixation process in the SMP.
However, it is important to note that this speculation requires further experimental validation. Detailed investigations into the viscoelastic behaviour and entanglement dynamics of the SMP at different deformation levels are essential to confirm the role of entanglements in the shape fixity process. Additionally, understanding the relationship between deformation-induced changes in polymer structure and entanglement density will provide valuable insights into the mechanisms underlying shape memory behaviour in polymers.
We hypothesize that aligning polymer chains significantly affects shape fixity by fundamentally altering the material’s behaviour during deformation and recovery processes [Citation33]. When polymer chains are aligned, the molecular structure becomes more ordered and stable. This enhanced structural stability minimizes the polymer’s tendency to revert to its original shape after deformation, thus improving shape fixity. Moreover, chain alignment restricts the polymer’s ability to elastically rebound to its initial configuration when external forces are removed. This restriction is crucial for maintaining the temporary shape programmed into the polymer, ensuring that it retains the desired form until intentionally triggered to recover its original shape.
Aligned polymer chains also provide better resistance against external disturbances, such as mechanical stress or thermal variations. This resilience helps preserve the shape memory effect over repeated cycles of deformation and recovery, making the material more reliable for applications requiring long-term shape retention. Furthermore, the controlled alignment of polymer chains enables a more predictable deformation mechanism, which is essential for applications needing precise and repeatable shape changes. This control over deformation behaviour allows for tailored functionalities, such as directional shape memory, where the polymer exhibits different memory behaviours depending on the direction of deformation.
Mather et al.‘s study utilized Wide-angle X-ray scattering (WAXS) and small-angle X-ray scattering (SAXS) to examine the microstructures of PCL – PEG TPUs in both deformed and recovered states [Citation33]. The WAXS patterns for three representative compositions in the stretched and recovered states are presented in respectively.
Figure 9. 2D WAXS patterns of representative PCL–PEG TPUs in the stretched state; (b) 2D WAXS patterns of PCL–PEG TPUs after recovery; (c) 2D SAXS patterns of PCL–PEG TPUs in the stretched state; and (d) 2D SAXS patterns of PCL–PEG TPUs after recovery. (i) [PCL]100, (ii) [PCL]50–[PEG]50, and (iii) [PEG]100. Stretch direction is horizontal, reprinted with permission from [Citation33], copyright reserved Elsevier 2012.
![Figure 9. 2D WAXS patterns of representative PCL–PEG TPUs in the stretched state; (b) 2D WAXS patterns of PCL–PEG TPUs after recovery; (c) 2D SAXS patterns of PCL–PEG TPUs in the stretched state; and (d) 2D SAXS patterns of PCL–PEG TPUs after recovery. (i) [PCL]100, (ii) [PCL]50–[PEG]50, and (iii) [PEG]100. Stretch direction is horizontal, reprinted with permission from [Citation33], copyright reserved Elsevier 2012.](/cms/asset/18d9f752-5e6b-4a1f-8994-b45a7f4c71f5/tsmm_a_2374342_f0009_oc.jpg)
Initially, the copolymers exhibited three strong isotropic rings, with the inner ring corresponding to the (120) reflection of the PEG monoclinic unit cell (d-spacing of 4.6 Å), the middle ring attributed to the (110) reflection of the PCL orthorhombic unit cell (d-spacing of 4.1 Å), and the outer ring representing a combination of (200) planes of PCL crystallites and various PEG reflections (d-spacing of 3.7–3.8 Å) [Citation33]. Upon stretching [PCL]100 to a fixed strain of 800%, the isotropic layers split into equatorial reflections . The WAXS pattern for [PCL]50–[PEG]50 displayed characteristics of both homopolymers at the stretched state, indicating high orientation of both PCL and PEG phases due to stretching. Upon recovery, the orientation of homopolymers and copolymers was largely eliminated . The SAXS patterns of the stretched samples () exhibited equatorial anisotropic diffuse scattering perpendicular to the stretching direction, suggesting the formation of elongated nano-voids or nano-fibrils along the stretching axis.
Studies have indicated that when polymers are stretched to high strains, the process of crystal reorientation leads to a more organized crystalline structure, thereby increasing the overall crystallinity of the material. This heightened level of crystallinity is often associated with improved shape fixity, especially at higher levels of deformation [Citation51]. The rationale behind this relationship lies in the fact that a higher degree of crystallinity provides a more robust framework for retaining the temporary shape during deformation, thereby enhancing the ability of the material to recover its original shape once the external force is removed.
Moreover, investigating the orientation factor, also known as the Hermans orientation factor, presents a promising avenue for further exploration [Citation51]. This factor quantifies the degree of molecular alignment within a polymer matrix, which directly influences its mechanical properties, including shape fixity. By analysing the correlation between the orientation factor, crystallinity, and shape fixity, researchers can gain deeper insights into the complex interplay between molecular structure and shape memory behaviour in polymers subjected to deformation. Such investigations could pave the way for the development of tailored polymer systems with enhanced shape memory properties for a wide range of applications.
We also examined the variation of the shape memory property with the elastic modulus of the PCL – PEG TPUs to determine if they correlate with the structural property relationships of the material. Interestingly, we found no such dependence on the shape memory property, which in turn can be linked to the plateau observed in .
The elastic modulus of these samples at room temperature is primarily dependent on the amount of crystalline fraction in the polymer. As discussed earlier, the amount of crystalline fraction, and thus the crystallinity, already reaches a plateau that eventually yields a plateaued shape memory property across increasing PCL weight fraction. It can be inferred that the elastic modulus at room temperature should also exhibit the same behaviour as observed from .
Figure 10. Shape memory properties of PCL–PEG TPUs with varying young’s modulus (MPa), original data sourced from [Citation33].
![Figure 10. Shape memory properties of PCL–PEG TPUs with varying young’s modulus (MPa), original data sourced from [Citation33].](/cms/asset/98151d7b-0a29-437e-af67-2c1e7695f15b/tsmm_a_2374342_f0010_oc.jpg)
Finally, the macroscopic shape memory properties of PCL – PEG TPUs are visually demonstrated through shape memory tests conducted on hot-pressed films . Starting with the samples on the left of each picture, we observe the original dogbone films, representing the initial state of the materials. Moving to the samples in the middle, we see the films stretched using the Linkam testing apparatus, illustrating the deformation process undergone by the materials . Finally, the samples on the right depict the films after being stretched at room temperature and subsequently subjected to heat recovery at 70°C for 1 minute. This final stage showcases the remarkable ability of the materials to recover their original shapes after undergoing significant deformation .
Figure 11. Shape memory tests of PCL–PEG TPUs hot-pressed film: (a) [PCL]100, (b) [PCL]50–[PEG]50, and (c) [PEG]100, reprinted with permission from [Citation33], copyright reserved Elsevier 2012.
![Figure 11. Shape memory tests of PCL–PEG TPUs hot-pressed film: (a) [PCL]100, (b) [PCL]50–[PEG]50, and (c) [PEG]100, reprinted with permission from [Citation33], copyright reserved Elsevier 2012.](/cms/asset/37017472-ef8d-4680-917a-45c98b8cfdc9/tsmm_a_2374342_f0011_oc.jpg)
Moreover, to compare the entanglement-based shape memory properties of these PCL/PEG-based thermoplastic polyurethanes with other PCL/PEG-based shape memory polymers that do not feature entanglements, we present a detailed comparison in . This table contrasts the key values and performance metrics from the existing literature, highlighting how the presence of entanglements affects the shape memory characteristics.
Table 1. Comparison of Shape Memory Properties in PCL/PEG-based Polymers in Entanglement-Based Systems [Citation33] as contrasted to conventional Systems [Citation52,Citation53].
Although the shape memory properties of PCL/PEG-based polymers in entanglement-based systems appear to be similar to those in conventional systems, we speculate that entanglements may significantly enhance other physical properties, leading to a more robust shape memory polymer, such as (a) Recovery Time: We speculate that the recovery time for entangled systems is faster. This is because the entangled network likely facilitates a quicker return to the original shape upon heating [Citation52]. Such rapid shape recovery is particularly advantageous for applications requiring immediate responsiveness, and (b) Maximum Recoverable Strain: We believe the maximum recoverable strain is substantially higher in entangled systems. Entanglements likely contribute to greater elasticity and flexibility, allowing the material to endure higher levels of deformation without permanent damage. This enhanced elasticity suggests that entangled systems can withstand more substantial mechanical stress compared to conventional systems [Citation51–54].
While the observed shape memory properties are similar, the potential benefits of entanglements on recovery time and maximum recoverable strain warrant further investigation. Understanding these aspects more thoroughly could confirm the advantages of entangled networks in producing more durable and efficient shape memory polymers.
In the same context, Guo and co-workers recently developed entanglement-based thermoplastic shape memory polymeric particles with photothermal actuation for biomedical applications [Citation54]. Triggering shape-memory functionality under clinical hyperthermia temperatures could enable the control and actuation of shape-memory systems in clinical practice. For this purpose, they developed light-inducible shape-memory microparticles composed of a poly(d,l-lactic acid) (PDLLA) matrix encapsulating gold nanoparticles (Au@PDLLA hybrid microparticles) [Citation54].
Interestingly, the development of shape-memory polymeric systems down to micron and submicron scales had been challenging until a film-stretching method recently enabled the facile programming of shape-memory functionality at such small scales [Citation55,Citation56]. Previously, SMPs of small sizes typically required either chemical cross-links or an additional set of polymer domains serving as physical net-points to determine the permanent shape of the material. However, polymer entanglements, a universal property of polymers with sufficient molecular weight, can also act as efficient physical net-points [Citation54].
On the macroscale, entanglement-based shape memory has been well-recognized and used industrially to produce shrink films. In typical use, commercial goods are loosely wrapped with a biaxially stretched plastic film that is subsequently heated to relax the film to a smaller, unoriented size, providing a close-fitting, high clarity wrapping. This application demonstrates the practicality of entanglement-based mechanisms in producing responsive materials [Citation57]. The potential to use polymer entanglements opens the door for a broader selection of thermoplastic polymers in shape-memory systems, particularly for applications such as biomedical devices that have restrictive requirements in terms of chemistry and materials modification. Despite the potential advantages, no study to date has successfully implemented such a strategy in miniature shape-memory systems, likely due to processing and handling difficulties.
Recent advancements have addressed these challenges. The development of a film-stretching method has enabled the programming of shape-memory functionality in microparticles. This method has facilitated the creation of entanglement-based shape-memory responses without the need for chemical cross-links in miniaturized systems. By incorporating biocompatible components, such as gold nanoparticles for photothermal actuation, researchers have demonstrated the feasibility of using these materials for biomedical applications, such as tuning macrophage phagocytosis.
This is where Guo et al.‘s work becomes interesting. This shape-memory polymeric system, for the first time, demonstrates the capability of maintaining an anisotropic shape at body temperature, with a triggered shape-memory effect returning to a spherical shape at a narrow temperature range above body temperature (37 < T < 45 °C) with an appropriate shape recovery speed. They applied a modified film-stretching processing method with carefully controlled stretching temperature to enable shape memory and anisotropy in these micron-sized particles. Consequently, they achieved a purely entanglement-based shape-memory response without chemical cross-links in the miniaturized shape-memory system [Citation54].
Furthermore, these shape-memory microparticles exhibited light-induced spatiotemporal control of their shape recovery using a laser to trigger the photothermal heating of the doped gold nanoparticles. This shape-memory system is composed of biocompatible components and demonstrates spatiotemporal controllability of its properties, showing potential for various biomedical applications, such as tuning macrophage phagocytosis, as demonstrated in their study.
To expand more on the theme, the choice of PDLLA was informed by its well-established biocompatibility and the presence of entanglements, as evidenced by rheological studies conducted by Petisco-Ferrero et al. These entanglements, characterized by an entanglement molecular weight of 5200 g/mol, serve as physical netpoints critical for driving the shape-memory effect. By leveraging these entanglements, the authors aimed to enhance biocompatibility and simplify the fabrication process, avoiding the need for chemical modifications [Citation54,Citation58].
The shape-memory response was triggered by encapsulating hydrophobically stabilized gold nanoparticles (AuNPs) within the PDLLA matrix, providing a photothermal trigger mechanism. The synthesis of PDLLA microparticles encapsulating AuNPs via bulk emulsion facilitated the formation of entanglements, essential for driving the SME. Scanning electron microscopy characterization confirmed the morphology of the microparticles, demonstrating their suitability for biomedical applications [Citation54].
Figure 12. (A) Shape-memory effect in polymeric particles driven by entropy. Poly(d,l-lactic acid) particles encapsulating hydrophobic lipid-stabilized gold nanoparticles form physical cross-links due to high molecular weight. Stretching under different temperatures results in anisotropic shapes. Low-temperature stretched particles revert to their original shape upon thermal stimuli, while high-temperature stretched particles retain their deformed shapes. (B) Polarized light optical microscopy (POM) shows greater polymer alignment in particles stretched at 65°C compared to 90°C. Reprinted with permission from [Citation54], copyright American Chemical Society 2018.
![Figure 12. (A) Shape-memory effect in polymeric particles driven by entropy. Poly(d,l-lactic acid) particles encapsulating hydrophobic lipid-stabilized gold nanoparticles form physical cross-links due to high molecular weight. Stretching under different temperatures results in anisotropic shapes. Low-temperature stretched particles revert to their original shape upon thermal stimuli, while high-temperature stretched particles retain their deformed shapes. (B) Polarized light optical microscopy (POM) shows greater polymer alignment in particles stretched at 65°C compared to 90°C. Reprinted with permission from [Citation54], copyright American Chemical Society 2018.](/cms/asset/d71ce465-e09d-4365-a225-37bdceef9f2e/tsmm_a_2374342_f0012_oc.jpg)
PDLLA’s amorphous thermoplastic nature, characterized by a glass transition temperature (Tg) of 41.3°C, rendered it ideal for triggering the SME near physiological temperatures. The broad glass transition range of PDLLA, spanning from 39 to 48°C, ensured a suitable temperature window for shape-memory activation, further enhancing its applicability in biomedical contexts [Citation54].
Stretching the PDLLA microparticles at different temperatures provided valuable insights into the SME mechanism. Stretching at low temperatures (e.g. 65 °C) preserved polymer entanglements, enabling subsequent shape-memory actuation upon heating. Conversely, stretching at high temperatures (e.g. 90 °C) led to the disentanglement of polymers, hindering the SME. Polarized light optical microscopy (POM) corroborated these findings, highlighting the importance of polymer alignment in triggering the SME [Citation54].
Moreover, in the realm of shape-memory-assisted self-healing (SMASH) systems, entanglement emerges as a fundamental element governing both the mechanical resilience and the self-healing capacity of polymers. At the core of this phenomenon lies the intricate interplay between the molecular structure of the polymer chains and the entanglements they form within the material matrix [Citation59].
Entanglements, characterized by the intertwining of polymer chains, exert a profound influence on the mechanical behaviour of polymers. These physical cross-links endow the material with elasticity and flexibility, allowing it to deform reversibly under stress. In SMASH systems, entanglements act as molecular anchors, storing elastic energy during deformation that can be harnessed to facilitate shape recovery and crack healing.
Crucially, entanglements play a pivotal role in the crack healing process outlined by Wool and OʹConnor [Citation60]. As cracks propagate within the polymer matrix, entangled polymer chains adjacent to the fracture surfaces undergo rearrangement and diffusion. This molecular mobility, facilitated by the presence of entanglements, enables the polymer chains to recombine across the fractured interface, ultimately leading to crack closure and material restoration. The entangled polymer chains effectively act as molecular bridges, bridging the gap between fractured surfaces and promoting healing [Citation61].
Furthermore, entanglements contribute significantly to the shape-memory effect observed in SMASH systems. During the deformation process, entangled polymer chains store elastic energy, which is subsequently released upon the application of external stimuli, such as heat. This stored energy drives the material to revert to its original shape, exerting mechanical forces that aid in crack closure and repair. The entangled polymer network thus serves as a reservoir of mechanical energy, enabling dynamic shape recovery and self-healing capabilities.
High molecular weight polymers with neat halos represent a compelling area of research. The development of amorphous thermoplastic poly(para-phenylene) (PPP) introduces it as a promising material for high-strength SMPs [Citation62]. Frick et al.‘s study fundamentally demonstrates the shape-memory behavior of PPP. Despite PPP being an amorphous thermoplastic lacking chemical crosslinks or crystalline segments typically associated with robust shape-recovery properties, their findings illustrate that PPP’s relatively rigid molecular structure fosters stable entanglements. These entanglements serve as crucial ‘hard points’ essential for facilitating shape recovery [Citation62].
To demonstrate practical application, relatively thick-walled heat-shrink PPP tubes were expanded to a maximum inner diameter of 13 mm at 175°C within 10 minutes, corresponding to approximately 30 seconds of relaxation time at 185°C – well within the rubbery modulus range. After programming, the tubes exhibited 99.7% fixity upon unloading, achieving a final programmed diameter of 12.96 mm, indicative of approximately 62% stored strain based on inner diameter changes. Subsequent recovery at 200°C revealed 96.3% recovery of the inner diameter, achieving a final recovered diameter of 8.18 mm. Despite potential for permanent deformation under constant high-temperature load, PPP retains structural integrity below 150°C due to its inherent mechanical strength [Citation62]. In the process of enhanced shape memory, these entanglements in PPP play a pivotal role. During the programming phase, when the material is deformed and fixed into a temporary shape, the stable entanglements act as anchoring points that maintain the deformed state. Upon application of the appropriate stimulus, such as heat, the entanglements enable the material to revert to its original shape by providing resistance to deformation. This mechanism ensures that PPP exhibits high fixity during the programming step and efficient recovery when the stimulus is applied, as demonstrated in the expansion and recovery of thick-walled heat-shrink PPP tubes.
Figure 13. (a) a hollow PPP tube was manufactured and (b) programmed to 62% circumferential strain at 175°C within 10 min. (c) The PPP tube demonstrated complete shape recovery when reheated to 200°C, reprinted with permission from [Citation62], copyright Wiley 2016.
![Figure 13. (a) a hollow PPP tube was manufactured and (b) programmed to 62% circumferential strain at 175°C within 10 min. (c) The PPP tube demonstrated complete shape recovery when reheated to 200°C, reprinted with permission from [Citation62], copyright Wiley 2016.](/cms/asset/665653b3-9a78-4e56-ab0a-3c99c99620d3/tsmm_a_2374342_f0013_oc.jpg)
This line of reasoning underscores the significance of chain entanglements in achieving thermally induced shape memory behaviour in polymers. It is widely acknowledged that two molecular components are fundamental for this behaviour: a fixing phase, typically comprised of physical or chemical cross-links providing dimensional stability, and a reversible phase imparting elastomeric properties. The poly(l-lactide, glycolide, TMC)s examined in this study exhibit a distinct glass-to-rubber transition in thermal analyses (DSC and DMTA), confirming their amorphous and single-phase nature [Citation63,Citation64]. Absent chemical cross-links or crystalline structures to anchor polymer chains, these materials rely on chain entanglements for shape recovery. The density and prevalence of these entanglements are heavily influenced by the polymer’s molecular weight, with their effectiveness during deformation shaped by both time and temperature. Therefore, achieving optimal shape memory properties requires careful control of both temperature and deformation time to prevent the disentanglement of polymer chains, which could otherwise result in irreversible deformation. This emphasis highlights the critical role of entanglements in dictating the shape memory characteristics of these polymers. This exploration of polymer entanglements provides valuable insights into the behaviour of shape-memory polyurethanes (SMPU), including both electrospun and bulk varieties. The observed differences in relaxation time constants between electrospun SMPU (8.3 minutes) and bulk SMPU (14.2 minutes) during the recovery isothermal state [Citation65] underscore the critical role played by entanglement density within polymer chains.
Electrospun SMPU, influenced by the extensional flow during its fabrication, experiences a reduction in chain entanglements. This results in longer segments between entanglements compared to bulk SMPU, where chains are more densely intertwined. Consequently, the mobility of polymer chains in electrospun SMPU is less restricted, leading to quicker relaxation times [Citation65]. Furthermore, the lower entanglement density in electrospun SMPU facilitates easier alignment of polymer chains and promotes the formation of strain-induced crystalline structures. This phenomenon enhances the presence of crystallinity in electrospun SMPU, a characteristic less prominent in bulk SMPU due to its higher entanglement density, and thus in terms of shape memory properties, electrospun SMPU demonstrates superior shape fixity, which can be attributed to its unique morphology, and chain entanglements.
illustrates a phenomenon where the SMPU in its amorphous state consists of polymer chains that are predominantly flexible but incorporate relatively long hard segments. These chains form a network through the creation of crosslinks (netpoints) between adjacent chains, which are primarily hydrogen bonds between the hard-soft and hard-hard segmental groups. During the programming phase, the network is heated to a temperature that allows segments of chains between crosslinks to become mobile, maintaining the integrity of the network as the crosslinks remain intact. Upon lowering the temperature for fixation (while remaining above the glass transition temperature, Tg), the long and rigid hard segments hinder the movement of chains relative to each other, thereby fixing the network in a stable state. During the recovery phase, when the temperature is raised again, the entrapment of the hard segments is relieved, enabling the chains to move more freely. Consequently, the network undergoes a recovery process back to its initial coiled state.
Figure 14. Illustration of the amorphous fixation mechanism of an SMPU in the state of the confinement tube during the programming or recovery steps, and in the state of the confinement tube after fixation.
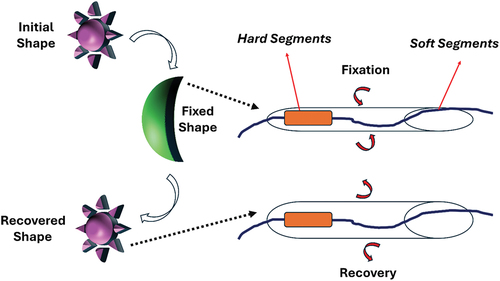
In general, the mobility of a chain segment constrained between two crosslinks depends significantly on the spatial constraints imposed by neighbouring chains, illustrated in as a tubular volume surrounding a typical constrained chain segment. This concept of a confinement tube is commonly employed in polymer physics to describe how neighbouring chains restrict the movement of a chain segment, allowing it to move through viscous reptation [Citation65]. In a densely entangled state, such as at lower temperatures, the confinement tube around the chain segment is narrow (), thereby hindering the mobility of bulky hard segments. In this state, the hard segments are effectively immobilized within the ‘walls’ of the confinement tube, resulting in the fixation of the SMPU shape. As the temperature increases, particularly during recovery or programming phases, the confinement around the chain segment becomes less restrictive. This expanded space enables the entire chain segment, including the hard segments, to move more freely (). When the mobility of hard segments increases, processes like strain recovery, relaxation, and creep require higher temperatures or longer durations to occur.
5. Conclusions and future outlook
In conclusion, the investigation into the shape-memory properties of PCL-based systems, particularly in relation to entanglement dynamics, has shed light on the underlying mechanisms governing their mechanical behaviour and self-healing capabilities. The perspective reinforces the pivotal role of entanglements in driving shape fixity, crack healing, and shape-memory responses.
By exploring the variation of shape memory properties with PCL content and applied strain, consistent trends were observed that underscore the significance of entanglements in shaping material behaviour. The presence of a plateau phase in PCL-based systems, as highlighted in the literature and supported by previous research, suggests a robust relationship between PCL content and shape memory properties. Further investigations are warranted to elucidate the precise mechanisms driving these observed trends and optimize PCL-based systems for diverse applications.
The analysis of entanglement dynamics during deformation stages provides valuable insights into the interplay between polymer structure and mechanical properties. The observed changes in entanglement density correlate with deformation stages, highlighting the role of entanglements in modulating material behaviour under stress. Moreover, the speculated relationship between entanglement density and shape fixity warrants further experimental validation to confirm the influence of entanglements on shape memory performance.
Looking ahead, future research should focus on comprehensive investigations into the viscoelastic profile and entanglement dynamics of shape memory polymers to deepen our understanding of their mechanical behaviour. Experimental validation of the speculated role of entanglements in promoting shape fixity and facilitating self-healing processes is essential for harnessing the full potential of SMPs in various applications. Moreover, the development of entanglement-based shape memory polymers marks a significant stride in polymer science and materials engineering. By leveraging entanglements as physical connections within polymer networks, researchers have the opportunity to finely tune SMPs for diverse applications, particularly in biomedicine.
The use of entanglements as physical connections offers clear advantages. Firstly, it simplifies the manufacturing process by eliminating the need for intricate chemical modifications or additional components. This simplification not only streamlines production but also improves the reproducibility and scalability of SMP manufacturing. Additionally, entanglement-based SMPs boast excellent biocompatibility, making them well-suited for applications like tissue engineering, drug delivery, and biomedical implants.
Moreover, entanglement-based SMPs exhibit impressive responsiveness, enabling swift and precise shape changes in response to external stimuli. Recent progress in fabrication techniques has facilitated the development of miniaturized shape-memory systems, opening up new possibilities for biomedical devices and responsive materials. These miniature SMPs hold potential for applications ranging from minimally invasive surgical tools to drug delivery microdevices, where precise control over shape change is crucial.
Furthermore, entanglements may impact factors beyond shape memory properties alone, such as recovery time and maximum recoverable strain, suggesting avenues for further exploration. Investigating the relationship between entanglement density, molecular alignment, and mechanical properties could yield valuable insights for designing SMPs with enhanced durability and efficiency [Citation66].
Finally, molecular entanglements, including topological entanglement and sub-entanglement, significantly influence the stored strain energy in amorphous polymers. Sub-entanglement refers to the physical and weak interactions between adjacent segments in macromolecular chains, resulting in temporary yet crucial interactions that facilitate the dynamic mechanical behaviour of polymers. These interactions play a pivotal role in the entropic conformation relaxation process, enabling the polymer to revert to its original shape when stress is removed. This behaviour is fundamental to the shape memory effect SME, where the material can ‘remember’ and return to its initial form after deformation. In contrast, topological entanglement arises from the chemical cross-linking between adjacent segments, creating a network that geometrically restricts the motion of the chains. This permanent and stable framework is essential for maintaining the polymer’s permanent conformation and providing the structural integrity required for various applications. The combination of topological and sub-entanglements results in a complex interplay where the hard, cross-linked network (topological entanglement) supports the permanent shape, while the softer, physically interacting segments (sub-entanglement) allow for reversible deformation and recovery [Citation66–72].
In terms of pragmatic applications, the coupling effect between these two types of entanglement is crucial in determining the mechanical properties and behaviour of polymers under stress. The cooperative role of sub-entanglements and topological entanglements ensures that the material can undergo significant deformation while still being able to recover its original shape. This understanding has practical implications for the design and development of advanced polymeric materials, particularly in fields requiring high-performance materials with specific mechanical and shape memory properties [Citation69–72].
For instance, in biomedical applications, such as stents and other implantable devices, polymers with well-balanced entanglement properties can ensure both flexibility and structural stability. In the automotive and aerospace industries, materials with finely tuned entanglement characteristics can lead to components that are lightweight yet durable, capable of withstanding varying stress conditions without permanent deformation. Furthermore, in consumer electronics, polymers exhibiting excellent shape memory properties can be utilized to create more durable and resilient devices. Thus, a comprehensive understanding of molecular entanglements in amorphous polymers provides a scientific foundation for developing materials with tailored properties for a wide range of applications. By manipulating the balance between topological and sub-entanglements, researchers and engineers can design polymers that meet specific performance criteria, ultimately leading to innovations in various technological and industrial fields.
We also speculate that entanglements will play a key role in developing humidity-responsive shape memory polymers in the near future [Citation41,Citation73]. Humidity-responsive shape memory effects SMEs occur because water absorbed by the polymer acts as a plasticizer and influences hydrogen bonding within the material. Specifically, absorbed water reduces the entanglement and intermolecular interactions of the polymer chains, increasing their mobility and facilitating shape recovery [Citation41].
By tuning the density and extent of entanglements within the polymer network, it is possible to create a more controlled and programmable shape recovery process [Citation73]. This approach enables the design of SMPs with complex and precise shape memory behaviours, which can be particularly beneficial for applications where traditional thermal-responsive SMPs fall short, such as in the biomedical field. In thermal-responsive SMPs, heat triggers the shape memory effect by causing a phase transition or relaxation of the polymer chains. While effective, these materials have limitations in biomedical applications due to the need for precise temperature control and potential thermal damage to surrounding tissues. Humidity-responsive SMPs, on the other hand, offer a safer and potentially more versatile alternative for biomedical applications [Citation41]. By adjusting the entanglement density, we can engineer humidity-responsive SMPs with tailored properties for specific applications. For example, a higher entanglement density might be used to achieve slower, more controlled shape recovery, while a lower density could enable quicker responses. This level of control allows for the programming of complex shape changes, enhancing the functionality and versatility of SMPs in practical applications.
Nevertheless, what we envision is that this study contributes significantly to our understanding of entanglement-based shape memory mechanisms and sets the stage for future advances in polymer science and materials engineering. By harnessing the unique properties of entangled polymer networks, researchers can unlock new opportunities for developing responsive materials with improved functionality and performance, driving innovation across various fields.
Abbreviations
| SMAs | = | Shape Memory Alloys |
| Tg | = | Glass Transition Temperature |
| Tm | = | Melting Temperature |
| SMPs | = | Shape Memory Polymers |
| Th | = | Thermal Transition Temperature |
| Tt | = | Transition Temperature |
| SME | = | Shape Memory Effect |
| SMPUs | = | Shape Memory Polyurethanes |
| DMA | = | Dynamic Mechanical Analysis |
| PCL | = | Polycaprolactone |
| PEG | = | Polyethylene Glycol |
| TPUs | = | Thermoplastic Polyurethanes |
| WAXS | = | Wide-Angle X-ray Scattering |
| PCL | = | Polycaprolactone |
| PEG | = | Polyethylene glycol |
| TPU | = | Thermoplastic polyurethane |
| RPSM | = | Reversible Plasticity Shape Memory |
| SAXS | = | Small-angle X-ray scattering |
| PDLLA | = | Poly(d,l-lactic acid) |
| AuNPs | = | Gold Nanoparticles |
| DSC | = | Differential Scanning Calorimetry |
| DMTA | = | Dynamic Mechanical Thermal Analysis |
| PPP | = | Poly(para-phenylene) |
| SMASH | = | Shape-Memory-Assisted Self-Healing |
| POM | = | Polarized Light Optical Microscopy |
| TMC | = | Trimethylene Carbonate |
Supporting Information_with name.docx
Download MS Word (21.2 KB)Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/29963176.2024.2374342.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Luo L, Zhang F, Wang L, et al. Recent advances in shape memory polymers: multifunctional materials, multiscale structures, and applications. Adv Funct Mater. 2024;34(14):2312036. doi: 10.1002/adfm.202312036
- Idumah CI. Multifunctional properties optimization and stimuli‐responsivity of shape memory polymeric nanoarchitectures and applications. Polym Eng Sci. 2023;63(7):1857–1873. doi: 10.1002/pen.26331
- Lendlein A, Kelch S. Shape‐memory polymers. Angewandte Chemie. 2002;41(12):2034–2057. doi: 10.1002/1521-3773(20020617)41:12<2034:AID-ANIE2034>3.0.CO;2-M
- Jayalath S, Herath M, Epaarachchi J, et al. Durability and long-term behaviour of shape memory polymers and composites for the space industry-A review of current status and future perspectives. Polym Degrad Stab query. 2023;211:110297. doi: 10.1016/j.polymdegradstab.2023.110297
- Ebara M. Shape-memory surfaces for cell mechanobiology. Sci Technol Adv Mater. 2015;16(1):014804. doi: 10.1088/1468-6996/16/1/014804
- Sun L, Huang WM. Mechanisms of the multi-shape memory effect and temperature memory effect in shape memory polymers. Soft Matter. 2010;6(18):4403–4406. doi: 10.1039/c0sm00236d
- Lendlein A, Jiang H, Jünger O, et al. Light-induced shape-memory polymers. Nature. 2005;434(7035):879–882. doi: 10.1038/nature03496
- Buckley PR, McKinley GH, Wilson TS, et al. Inductively heated shape memory polymer for the magnetic actuation of medical devices. IEEE Trans Biomed Eng. 2006;53(10):2075–2083. doi: 10.1109/TBME.2006.877113
- Du H, Zhang J. Solvent induced shape recovery of shape memory polymer based on chemically cross-linked poly (vinyl alcohol). Soft Matter. 2010;6(14):3370–3376. doi: 10.1039/b922220k
- Jose S, George JJ, Siengchin S, et al. Introduction to Shape-Memory Polymers, Polymer Blends and Composites: State of the Art, Opportunities, New Challenges and Future Outlook. In: Parameswaranpillai J, Siengchin S, George J, Jose S, editors. Shape Memory Polymers, Blends and Composites: Advanced Structured Materials. Vol. 115. Singapore: Springer; 2020. doi: 10.1007/978-981-13-8574-2_1
- Mather PT, Luo X, Rousseau IA. Shape memory polymer research. Annu Rev Mater Res. 2009;39(1):445–471. doi: 10.1146/annurev-matsci-082908-145419
- Mu T, Liu L, Lan X, et al. Shape memory polymers for composites. Compos Sci Technol query. 2018;160:169–198. doi: 10.1016/j.compscitech.2018.03.018
- O’Connor TC, Hopkins A, Robbins MO. Stress relaxation in highly oriented melts of entangled polymers. Macromol. 2019;52(22):8540–8550. doi: 10.1021/acs.macromol.9b01161
- Hu J, Zhu Y, Huang H, et al. Recent advances in shape–memory polymers: structure, mechanism, functionality, modeling and applications. Progress Polym Sci. 2012;37(12):1720–1763. doi: 10.1016/j.progpolymsci.2012.06.001
- Wu X, Huang WM, Zhao Y, et al. Mechanisms of the shape memory effect in polymeric materials. Polym. 2013;5(4):1169–1202. doi: 10.3390/polym5041169
- Rokaya D, Skallevold HE, Srimaneepong V, et al. Shape memory polymeric materials for biomedical applications: an update. J Compos Sci. 2023;7(1):24. doi: 10.3390/jcs7010024
- Ramezani M, Monroe MBB. Biostable segmented thermoplastic polyurethane shape memory polymers for smart biomedical applications. ACS Appl Polym Mater. 2022;4(3):1956–1965. doi: 10.1021/acsapm.1c01808
- Mondal S, Hu JL. Studies of shape memory property on thermoplastic segmented polyurethanes: Influence of PEG 3400. J Elastomers Plast. 2007;39(1):81–91. doi: 10.1177/0095244307067423
- Kim BK, Shin YJ, Cho SM, et al. Shape‐memory behavior of segmented polyurethanes with an amorphous reversible phase: The effect of block length and content. J Polym Sci B Polym Phys. 2000;38(20):2652–2657. doi: 10.1002/1099-0488(20001015)38:20<2652:AID-POLB50>3.0.CO;2-3
- Li F, Zhang X, Hou J, et al. Studies on thermally stimulated shape memory effect of segmented polyurethanes. J Appl Polym Sci. 1997;64(8):1511–1516. doi: 10.1002/(SICI)1097-4628(19970523)64:8<1511:AID-APP8>3.0.CO;2-K
- Ramezani M, Getya D, Gitsov I, et al. Solvent-free synthesis of biostable segmented polyurethane shape memory polymers for biomedical applications. J Mat Chem B. 2024;12(5):1217–1231. doi: 10.1039/D3TB02472E
- Thakur S, Hu J. Polyurethane: A Shape Memory Polymer (SMP)’. Aspects of Polyurethanes. InTech. 2017. doi: 10.5772/intechopen.69992
- Saleeb AF, Natsheh SH, Owusu-Danquah JS. A multi-mechanism model for large-strain thermomechanical behavior of polyurethane shape memory polymer. Polym. 2017;130:230–241. doi: 10.1016/j.polymer.2017.10.003
- Farzaneh S, Fitoussi J, Lucas A, et al. Shape memory effect and properties memory effect of polyurethane. J Appl Polym Sci. 2013;128(5):3240–3249. doi: 10.1002/app.38530
- Tobushi H, Okumura K, Endo M, et al. Thermomechanical properties of polyurethane-shape memory polymer foam. J Intell Mater Syst Struct. 2001;12(4):283–287. doi: 10.1106/FNSX-AP9V-QP1R-NMWV
- Pawlak A. The entanglements of macromolecules and their influence on the properties of polymers. Macromole Chem Phys. 2019;220(10):1900043. doi: 10.1002/macp.201900043
- Kong DC, Yang MH, Zhang XS, et al. Control of polymer properties by entanglement: a review. Macro Mater Eng. 2021;306(12):2100536. doi: 10.1002/mame.202100536
- Treloar LRG. Theory of large elastic deformations. Nature. 1943;151(3839):616–616. doi: 10.1038/151616a0
- Edwards SF. The statistical mechanics of polymerized material. Proc Phys Soc. 1967;92(1):9. doi: 10.1088/0370-1328/92/1/303
- Graessley WW, Pearson DS. Stress–strain behavior in polymer networks containing nonlocalized junctions. J Chem Phys. 1977;66(8):3363–3370. doi: 10.1063/1.434421
- Ball RC, Doi M, Edwards SF, et al. Elasticity of entangled networks. Polymer. 1981;22(8):1010–1018. doi: 10.1016/0032-3861(81)90284-6
- Drakopoulos SX, Psarras GC, Forte G, et al. Entanglement dynamics in ultra-high molecular weight polyethylene as revealed by dielectric spectroscopy. Polymer. 2018;150:35–43. doi: 10.1016/j.polymer.2018.07.021
- Gu X, Mather PT. Entanglement-based shape memory polyurethanes: synthesis and characterization. Polym. 2012;53(25):5924–5934. doi: 10.1016/j.polymer.2012.09.056
- Jeon HG, Mather PT, Haddad TS. Shape memory and nanostructure in poly (norbornyl‐POSS) copolymers. Poly Int. 2000;49(5):453–457. doi: 10.1002/(SICI)1097-0126(200005)49:5<453:AID-PI332>3.0.CO;2-H
- Sakurai K, Takahashi T. Strain‐induced crystallization in polynorbornene. J Appl Polym Sci. 1989;38(6):1191–1194. doi: 10.1002/app.1989.070380616
- Yang F, Zhang S, Li JC. Impression recovery of amorphous polymers. J Electron Mater. 1997;26(7):859–862. doi: 10.1007/s11664-997-0263-9
- Bhaskar J, Sharma AK, Bhattacharya B, et al. A review on shape memory alloy reinforced polymer composite materials and structures. Smart Mater Struct. 2020;29(7):073001. doi: 10.1088/1361-665X/ab8836
- Alshebly YS, Nafea M, Ali MSM, et al. Review on recent advances in 4D printing of shape memory polymers. Eur Polym J. 2021;159:110708. doi: 10.1016/j.eurpolymj.2021.110708
- van Vilsteren SJ, Yarmand H, Ghodrat S. Review of magnetic shape memory polymers and magnetic soft materials. Magnetochemistry. 2021;7(9):123. doi: 10.3390/magnetochemistry7090123
- Hornat CC, Urban MW. Shape memory effects in self-healing polymers. Prog In Polym Sci. 2020;102:101208. doi: 10.1016/j.progpolymsci.2020.101208
- Yang L, Lou J, Yuan J, et al. A review of shape memory polymers based on the intrinsic structures of their responsive switches. RSC Adv. 2021;11(46):28838–28850. doi: 10.1039/D1RA04434F
- Valvez S, Reis PN, Susmel L, et al. Fused filament fabrication-4D-printed shape memory polymers: a review. Polym. 2021;13(5):701. doi: 10.3390/polym13050701
- Idumah CI, Odera RS, Ezeani EO, et al. Construction, characterization, properties and multifunctional applications of stimuli-responsive shape memory polymeric nanoarchitectures: a review. Polym-Plast Technolo Mater. 2023;62(10):1247–1272. doi: 10.1080/25740881.2023.2204936
- Hu H, Dorset DL. Crystal structure of poly (iε-caprolactone). Macromolecules. 1990;23(21):4604–4607. doi: 10.1021/ma00223a017
- Takahashi Y, Tadokoro H. Structural studies of polyethers,(-(CH2) mO-) n. X. Crystal structure of poly (ethylene oxide). Macromol. 1973;6(5):672–675. doi: 10.1021/ma60035a005
- Kallel A, Abdallah AB, Gamaoun F, et al. Driving force for shape memory effect of polymers. J Polym Res. 2021;28(8):1–10. doi: 10.1007/s10965-021-02637-4
- Defize T, Riva R, Raquez JM, et al. Thermoreversibly crosslinked poly (ε‐caprolactone) as recyclable shape‐memory polymer network. Macromol Rapid Commun. 2011;32(16):1264–1269. doi: 10.1002/marc.201100250
- Behl M, Ridder U, Feng Y, et al. Shape-memory capability of binary multiblock copolymer blends with hard and switching domains provided by different components. Soft Matter. 2009;5(3):676–684. doi: 10.1039/B810583A
- Tobushi H, Shimada D, Hayashi S, et al. Shape fixity and shape recovery of polyurethane shape-memory polymer foams. Proc Inst Mech Eng, Part L: J Mater: Des Appl. 2003;217(2):135–143. doi: 10.1177/146442070321700205
- Liu Y, Xian W, He J, et al. Interplay between entanglement and crosslinking in determining mechanical behaviors of polymer networks. Int J Smart Nano Mater. 2023;14(4):474–495. doi: 10.1080/19475411.2023.2261777
- Peng J, Ellingham T, Sabo R, et al. Short cellulose nanofibrils as reinforcement in polyvinyl alcohol fiber. Cellul. 2014;21(6):4287–4298. doi: 10.1007/s10570-014-0411-3
- Mehrbakhsh E, Rezaei M, Lotfi Mayan Sofla R, et al. Physical and thermo-mechanical properties of PCL/PEG based shape memory polyurethane for orthodontic ligature application. Int J Polym Mater Polym Biomater. 2024;73(4):239–249. doi: 10.1080/00914037.2022.2155157
- Gu X, Mather PT. Water-triggered shape memory of multiblock thermoplastic polyurethanes (TPUs). RSC Adv. 2013;3(36):15783–15791. doi: 10.1039/c3ra41337c
- Guo Q, Bishop CJ, Meyer RA, et al. Entanglement-based thermoplastic shape memory polymeric particles with photothermal actuation for biomedical applications. ACS Appl Mater Inter. 2018;10(16):13333–13341. doi: 10.1021/acsami.8b01582
- Yoo JW, Mitragotri S. Polymer particles that switch shape in response to a stimulus. Proc Natl Acad Sci. 2010;107(25):11205–11210. doi: 10.1073/pnas.1000346107
- Kolhar P, Anselmo AC, Gupta V, et al. Using shape effects to target antibody-coated nanoparticles to lung and brain endothelium. Proc Natl Acad Sci. 2013;110(26):10753–10758. doi: 10.1073/pnas.1308345110
- Butler TI, Morris BA. Chapter 15—PE based multilayer film structures. Vol. USA. Boston (MA): Plastics Design Library; William Andrew Publishing; 2010. p 205–230.
- Petisco-Ferrero S, Fernández J, San Martín MF, et al. The relevance of molecular weight in the design of amorphous biodegradable polymers with optimized shape memory effect. J Mech Behav Biomed Mater. 2016;61:541–553. doi: 10.1016/j.jmbbm.2016.04.027
- Rodriguez ED, Luo X, Mather PT. Linear/network poly (ε-caprolactone) blends exhibiting shape memory assisted self-healing (SMASH). ACS Appl Mater Inter. 2011;3(2):152–161. doi: 10.1021/am101012c
- Wool R, O’connor KM. A theory crack healing in polymers. J Appl Phys. 1981;52(10):5953–5963. doi: 10.1063/1.328526
- Wang X, Xu J, Zhang Y, et al. A stretchable, mechanically robust polymer exhibiting shape-memory-assisted self-healing and clustering-triggered emission. Nat Commun. 2023;14(1):4712. doi: 10.1038/s41467-023-40340-8
- Collins DA, Yakacki CM, Lightbody D, et al. Shape‐memory behavior of high‐strength amorphous thermoplastic poly (para‐phenylene). J Appl Polym Sci. 2016;133(3). doi: 10.1002/app.42903
- Yang J, Liu F, Yang L, et al. Hydrolytic and enzymatic degradation of poly (trimethylene carbonate-co-D, L-lactide) random copolymers with shape memory behavior. Eur Polym J. 2010;46(4):783–791. doi: 10.1016/j.eurpolymj.2009.12.017
- Zini E, Scandola M, Dobrzynski P, et al. Shape memory behavior of novel (l-lactide− glycolide− trimethylene carbonate) terpolymers. Biomacromol. 2007;8(11):3661–3667. doi: 10.1021/bm700773s
- Nissenbaum A, Greenfeld I, Wagner HD. Shape memory polyurethane-Amorphous molecular mechanism during fixation and recovery. Polymer. 2020;190:122226. doi: 10.1016/j.polymer.2020.122226
- Liu J, Lu H, Fu YQ. Yielding mechanisms for mechano-chemo-thermal couplings in amorphous shape memory polymer undergoing molecular entanglement. J Phys D Appl Phys. 2021;54(41):415302. doi: 10.1088/1361-6463/ac0d29
- Boyce MC, Parks DM, Argon AS. Large inelastic deformation of glassy polymers. Part I: rate dependent constitutive model. Mech Mater. 1988;7(1):15–33. doi: 10.1016/0167-6636(88)90003-8
- Jiang H, Jiang C. Finite deformation constitutive model for macro-yield behavior of amorphous glassy polymers with a molecular entanglement-based internal-state variable. Int J Mech Sci. 2019;161:105064. doi: 10.1016/j.ijmecsci.2019.105064
- Bacova P, Lo Verso F, Arbe A, et al. The role of the topological constraints in the chain dynamics in all-polymer nanocomposites. Macromol. 2017;50(4):1719–1731. doi: 10.1021/acs.macromol.6b02340
- Wang X, Jian W, Lu H, et al. Selective entanglement coupling of nanoparticles in polymer nanocomposite with high shape recovery stress. Compos Sci Techno. 2021;207:108728. doi: 10.1016/j.compscitech.2021.108728
- Meng Y, Jiang J, Anthamatten M. Body temperature triggered shape‐memory polymers with high elastic energy storage capacity. J Polym Sci B Polym Phys. 2016;54(14):1397–1404. doi: 10.1002/polb.23990
- Cooper CB, Nikzad S, Yan H, et al. High energy density shape memory polymers using strain-induced supramolecular nanostructures. ACS Central Sci. 2021;7(10):1657–1667. doi: 10.1021/acscentsci.1c00829
- Xiao H, Ma C, Le X, et al. A multiple shape memory hydrogel induced by reversible physical interactions at ambient condition. Polym. 2017;9(4):138. doi: 10.3390/polym9040138