Abstract
We studied the use of Prussian blue nanoparticles (PBNPs) as novel nanocarriers for sending DNA drugs into cancer cells. 11-mercaptoundecanoic acid (MUA) was used to functionalize the surfaces of PBNPs (nanocubes with an average dimension of 75 nm) for subsequent covalent grafting of a 33-mer DNA drug with a FAM reporter at the 3′ end. The PBNPs synthesis and DNA drug conjugation were characterized by transmission electron microscopy (TEM) and Fourier-transform infrared absorption (FTIR), respectively. The drug was a decoy oligodeoxynucleotide (dODN) that inhibits the signal transducer and activator of transcription 3 (STAT3). The DNA–PBNPs drug (dODN@MUA–PBNPs) was delivered into human prostate carcinoma 22rv1 cells by endocytosis in vitro as confirmed by confocal fluorescence microscopy. MTT cell viability assays were carried out to assess the effect of the DNA–PBNPs drug. The results showed that the dODN molecules were successfully conjugated to the MUA modified PBNPs via amide and/or disulfide bond formation and could thus be successfully delivered into the cancer cells. The control experiments showed that the unconjugated dODN molecules were not able to enter the cancer cells no matter whether non-functionalized PBNPs were present or not. It was also found that the DNA-PBNPs drugs were internalized and then distributed homogeneously throughout the cell, including cytoplasmic and nucleic regions, after endocytosis. The cancer cell-killing ability increased with the amount of dODN conjugated on PBNPs and the dosage of DNA–PBNPs drug internalized.
1. Introduction
The drug delivery systems (DDS) based on biocompatible nanoparticles have promised a great future of nanomedicine. Having the advantages of functionalization and biocompatibility, the nanoparticles have been demonstrated as ideal carriers of both diagnostic and therapeutic molecules, which not only increases the efficacy of drug delivery in targeted therapies but also allows in situ theranostics of malignant tumors [Citation1–Citation3]. In principle, the efficiency and efficacy of a DDS nanoparticle highly depend on its specific composition, surface property and morphology. Hence, the preparations of various nanoparticles [Citation4], especially with different morphologies like nanocubes, spheres, octahedra and mesoporous frameworks [Citation5] are widely discussed. How different nanoparticles make contribution in DDS have also drawn much attention and discussion in recent years [Citation6–Citation8].
Among the DDS nanoparticles reported, we consider that Prussian blue nanoparticles (PBNPs) are novel and potential nanocargoes for drug delivery owing to their multiple advantages, including biocompatibility, biodegradability, low cost, easy preparation and controllable morphology [Citation9]. Prussian blue (iron hexacyanoferrate) is a known, non-toxic blue pigment as well as a widely investigated functional material for sensors and electrochromics [Citation10, Citation11]. The advent of nanomedicine has rendered this old dyeing material a new life in the DDS field. Prussian blue nanoparticles are iron-based nanoparticles and are capable of mass production with low-temperature facile methods [Citation12]. Several studies on DDS and cell imaging applications have revealed the potential of PBNPs for nanomedicine [Citation7, Citation13, Citation14]. Similar to iron oxide nanoparticles, PBNPs have been demonstrated as a potent magnetic resonance imaging contrast agent along with its DDS application, and their chemical stability, cytotoxicity and cellular penetrating ability have also been proved [Citation6]. Moreover, PBNPs can be made with controllable structures [Citation15]. For examples, hollow or mesoporous PBNPs can be synthesized based on their coordination framework structures to enhance the specific surface area and thus the amount of drug loading. From the aforementioned evidences, we think that PBNPs can be rivals for iron oxide nanoparticles and other inorganic nanoparticles in future nanomedicine. Although PBNPs show niches and potentials in DDS and cellular imaging, they have still not been widely studied and engineered for the nanomedicine aspects as compared to their well-known applications in electrochromics and biosensors.
To date, it has been already demonstrated that PBNPs can successfully deliver small-molecule medicines, like cisplatin, in to cancer cells in vitro [Citation13]. However, proteins, peptides and DNA drugs are still frontiers to explore for the PBNP-based DDS. As a state of the art, DNA drugs have been thought to be an emerging class of therapeutic molecules for disease treatments such as infectious disease and cancer therapies [Citation15, Citation16]. Take a cancer therapy as an example, when carrying out a DNA drug therapy, a short synthetic functional DNA sequences are brought to the nuclei of targeted cells to regulate the gene expression, and then finally cause the apoptosis of the cells [Citation17]. Despite having an appreciable working principle, the DNA drugs without appropriate modifications are prone to degradation inside the body by extracellular deoxyribonucleases [Citation18] and are hindered from the cellular uptake [Citation19]. There are many workable strategies to overcome this problem [Citation20]. The DDS nanoparticles are also promising nuclease-resistance strategies in addition to their cargo roles [Citation21–Citation23] and are thus considered well suited to work as a counter part for DNA drugs. For examples, the Fe3O4 and γ-Fe2O3 nanoparticles are appropriate for DNA drug delivery [Citation24]. Besides, magnetite and maghemite nanoparticles are known for their low cytotoxicity approved by FDA [Citation25], intrinsic magnetic features [Citation26], and facile cellular uptake. But the iron oxide nanoparticles usually lack significant latitude of controllable morphology [Citation27]. The above reasons have come together to drive this work toward the study of PBNPs as nanocargoes for delivery of DNA drugs into cancer cells. In particular, there is no literature reporting a method for preparing a DNA drug–PBNPs conjugate and conducting the relevant DDS research.
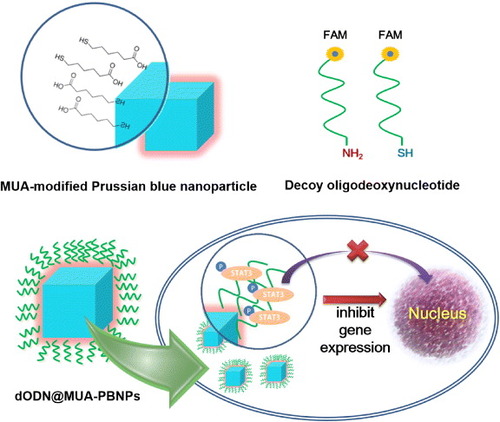
This work is a proof-of-principle study that describes a workable method for grafting DNA drugs onto chemically modified PBNPs and subsequent delivery of the DNA drugs into cancer cells with the aid of PBNPs in vitro. The DNA drug used is a class of decoy oligodeoxynucleotides (dODN) that inhibits the gene expression of STAT3 [Citation24], and two kinds of FAM-labeled dODNs with an amino and a thiol linker are grafted onto 11-mercaptoundecanoic acid (MUA)-functionalized PBNPs separately or together via covalent linkages that result in a DNA drug–PBNPs conjugate (abbreviated as dODN@MUA–PBNPs). The model cancer cells used are human prostate carcinoma 22rv1cells [Citation19]. Cellular uptakes of dODN@MUA–PBNPs into the 22rv1 cancer cells are successfully observed that demonstrates the first attempt of using PBNPs as nanocargoes for delivering DNA drugs to cancer cells. Figure illustrates the above ideas and the potential cancer cell-killing effect.
Figure 1. Illustration summarizing the concepts of the MUA modification of cubic PBNPs, the dODN drugs end-labeled with a FAM reporter and a linker, and the final configuration of the DNA nano drug (dODN@MUA–PBNPs) and its potential cancer cell-killing effect.
2. Experimental
2.1. Materials
Potassium ferricyanide (III) (K3[Fe(CN)6]), polyvinylpyrrolidone (PVP, K30), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), N-hydroxysuccinimide (NHS), 11-mercaptoundecanoic acid (MUA, 95%), RPMI-1640 medium, fetal bovine serum, dimethyl sulfoxide (DMSO, 99.9%), MTT assay kit, 4, 6-diamidino-2-phenylindole dihydrochloride (DAPI, 98%) and hydrochloric acid were all obtained from Sigma Aldrich Taiwan and used as received. The 33-mer DNA drug (dODN) with a sequence 5′CAT TTC CCG TAA ATC GAA GAT TTA CGG GAA ATG 3′ [Citation24], and two kinds of 3′ FAM-labeled dODNs with an 5′ amino and a 5′ thiol linker were synthesized by PURIGO biotechnology Co., Ltd. The human prostate carcinoma 22rv1 cells were obtained from the bioresource collection and research center (BCRC, Taiwan).
2.2. Synthesis of Prussian blue nanoparticles (PBNPs)
The synthesis of PBNPs was referred to a previous research [Citation28]. In the typical procedure, 1 g of PVP was mixed with 30 mM of K3[Fe(CN)6] solution under magnetic stirring. The mixture was adjusted to pH 2 by 12 M HCl solution. After 30 min of continuous stirring, the clear yellow color solution was obtained and aged at 80 °C for 20 h. Following, the well-dispersed PBNPs were collected by adding acetone with a volume of 2.5 times of PBNPs dispersion. The precipitate collection was then washed to remove the residual PVP and K3[Fe(CN)6]. The wash step was carried out by adding the mixture of DI water and acetone into the collection and vortex the collection. Then the collection was centrifuged and the precipitate was obtained. The wash step was carried out three times. In the final step, the precipitate was dried in an oven under vacuum at 40 °C for 12 h to obtain the PBNPs powder. The as-prepared PBNPs were characterized by transmission electron microscopy (JEM-1200EX II, JEOL, Japan). The hydrodynamic radius and zeta potential of the PBNPs were determined by a dynamic light scattering (DLS) instrument (Zetasizer 3000HS, Marlven, UK).
2.3. The dODN grafting onto the MUA-PBNPs
Firstly, the MUA modification of PBNPs was done to decorate the carboxyl and thiol groups on the surfaces of PBNPs, so that 5′ NH2-dODN-FAM and/or 5′ SH-dODN-FAM could be grafted onto the surfaces of PBNPs through EDC/NHS chemistry and/or oxidative disulfide bond linkage. In the MUA modification step, 50 mg of PBNPs were mixed with 5 mM MUA by using 20 ml of 50% ethanol solution. After 16 h of sonication at 25 °C, the mixture was then washed and centrifuged with DI water and ethanol several times using the same method mentioned above to remove the free reagents. For activating the surfaces of the PBNPs prior to 5′ NH2-dODN-FAM covalent grafting, 5 mg of MUA-decorated PBNPs (MUA–PBNPs) were dispersed into a 20 ml mixture with 20 mg of both EDC and NHS, which was followed by sonication at 4 °C for 25 min. The activated MUA–PBNPs were then reacted with 3 μM of 5′ NH2-dODN-FAM at 4 °C for 16 h. In the 5′ SH-dODN-FAM grafting procedure, 3 μM of the oligodeoxynucleotide was simply mixed with the MUA–PBNPs and reacted at 4 °C for 16 h. Both the two kinds of grafting process were followed by centrifugation and washing with the method mentioned above to obtain the dODN-grafted MUA–PBNPs (dODN@MUA–PBNPs).
2.4. Spectroscopic characterization of dODN@MUA–PBNPs
The amount of the dODN molecules grafted on the PBNPs was estimated by the UV–Vis absorption measurement (V-650 UV–Vis spectrophotometer, JASCO, USA) of dODN@MUA–PBNPs, in which the DNA oligodeoxynucleotide and FAM reporter had absorption maxima at 260 and 490 nm, respectively. Fourier-transform infrared (FTIR) spectroscopy (Nicolet 6700, Thermo Scientific, USA) was also applied to confirm the covalent conjugation between dODNs and MUA–PBNPs. Desorption of the MUA on the PBNPs was observed through the FTIR spectra by comparing the spectra taken from the MUA–PBNPs which underwent the different sonication time interval in phosphate-buffered saline (PBS).
2.5. Characterization of subcellular localization of dODN@MUA–PBNPs
The human prostate carcinoma 22rv1 cells were grown in a PRMI 1640 medium with 10% fetal calf serum and humidified atmosphere of 95% air and 5% CO2 at 37 °C [Citation29]. For the confocal fluorescence microscopy imaging of cellular uptake of dODN@MUA–PBNPs, the 22rv1 cancer cells were firstly cultured on a coverslip. Incubated with the medium containing the specified concentration of dODN@MUA–PBNPs for 16 h, the incubated cells were washed with PBS to remove the residual dODN@MUA–PBNPs and the cell nuclei were stained with DAPI. The protocol of DAPI staining is that a 5 mg ml−1 DAPI solution was first diluted to a 300 nM solution in PBS. A 300 μl of the diluted DAPI staining solution was used to the coverslip preparation, making certain that the cells were completely covered and incubated for 5 min. Rinsed the sample several times in PBS and drain excess buffer from the coverslip and mount with a nail polish. Finally, the cellular uptake of FAM-labeled dODN@MUA–PBNPs was visualized and recorded by a two-channel confocal fluorescence microscope (TCS SP5 II, Leica, Germany). The recorded bright-field cell images and those contrasted by the FAM emission (from the DNA drug) and DAPI emission (from the nuclei) were compared and merged to be considered as the proof of dODN@MUA–PBNPs internalization.
2.6. Viability evaluation of dODN@MUA–PBNPs treated 22rv1 cell
The cytotoxicity effects of different cellular uptake conditions with differently prepared dODN@MUA–PBNPs and control samples were characterized by analyzing the cell viability with standard MTT assays [Citation30]. Generally, the cells were cultured in the 96-well plate to a concentration of 105 cells per well. The specified concentration of nanoparticles and chemicals were incubated with the cells overnight, which was followed by a buffer-exchanging step with 100 μl of fresh culture medium to remove the residual of nanoparticles and chemicals. To prepare a 12 mM MTT stock solution, a 1 ml of PBS and one 5 mg vial of MTT were mixed by vortexing and sonication until dissolved. Using 10 μl of the stock solution per well, the cells were incubated with MTT solution at 37 °C for 4 h. After labeling the cells with MTT, 75 μl of medium was removed from the wells and 50 μl of DMSO was added to each well and mixed thoroughly with a pipette. Finally, after 10 min of incubation with DMSO under 37 °C, each mixture of the sample was measured with a UV–Vis spectrometer to read the absorbance at 540 nm. The absorbance of untreated cell was taken as the 100% cell viability and used as the basis to calculate the other cell's viability.
3. Results and discussion
3.1. Characteristics of PBNPs and MUA-PBNPs
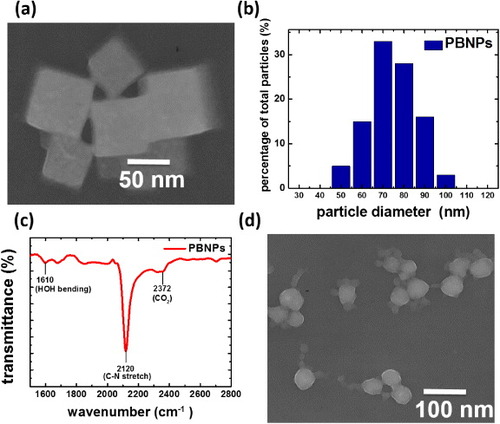
Typically, the nanoparticles used in DDS are synthesized with an average size ranging from 10 to 100 nm. The appropriate size helps DDS nanoparticles to deliver the therapeutic or diagnostic molecules into the target cell [Citation31]. The morphology and uniformity of the PBNPs prepared in this work were characterized by transmission electron microscopy (TEM) (figure (a)) and DLS (figure (b)). The PBNPs are nanocubes with distinct boundaries, and their average size is about 75 nm. This typical cubic shape of PBNPs comes from the inherent face-centered cubic crystal structure. The DLS result suggests that the size distribution of the PBNPs is uniform, and sizes of all PBNPs are well suited for DDS (between 50 and 100 nm). The composition of the PBNPs was verified by the FTIR spectrum (figure (c)). The absorption peak at 2120 cm−1 for the synthesized PBNPs is consistent with the FTIR characteristics of PB crystal [Citation30]. In addition to cubic PBNPs, spherical PBNPs (figure (d)) were also achieved by changing the synthetic acidity from pH 2 to pH 1. This result reveals a controllable morphology and provides a potential for further investigation of the morphologic effect of the PBNPs in DDS. Here we focus on the DDS application of cubic PBNPs.
Figure 2. (a) TEM image of cubic PBNPs, (b) DLS size distribution analysis of cubic PBNPs, (c) FTIR characterization of cubic PBNPs and (d) TEM image of spherical PBNPs prepared under a different condition.
To covalently graft synthetic DNA drugs that can be readily end-labeled with an amino or a thiol linker, the surfaces of PBNPs must be functionalized. In this work, we applied a bifunctional linkage molecule MUA, which bears a thiol group at one end and a carboxylic acid at the other end, to modify the PBNPs. MUA has been used as the self-assembly reagent for preparing carboxylated noble metal nanoparticles [Citation32] and is suitable for preparing other carboxylated nanoparticles. A previous research suggested that MUA forms a shell-layer around a non-noble-metal nanoparticle [Citation33], the MUA might be attached to PBNPs by an electrostatic linkage. The negative charged functional groups of MUA could be linked to the exposed Fe2+ or Fe3+ on the PBNPs. Since MUA molecules might surround PBNPs in a random orientation, we expect that both the carboxyl group and thiol group were formed on the surfaces of MUA decorated PBNPs (MUA–PBNPs). The existence of two surface functional groups enables covalently grafting of the DNA drugs with different end-labeled linkers.
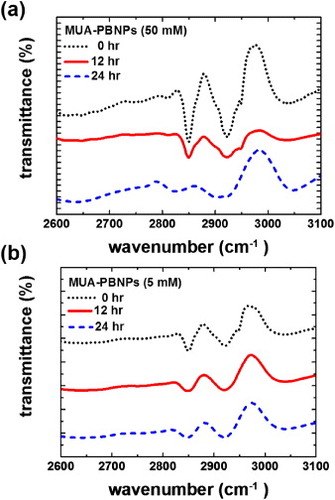
The decoration of PBNPs with MUA is not based on covalent bonding, so MUA desorption might occur. To minimize this adverse effect for grafting of the DNA drugs, we compared two PBNPs decoration conditions (one with 5 mM MUA and the other with 50 mM MUA) and see if MUA desorption occurred when being immersed in a PBS solution for a long time. We utilized FTIR to characterize the spectral feature of MUA of a MUA–PBNPs preparation. In figure , it is shown that the as-prepared MUA–PBNPs under both conditions show the absorption bands at 2840 and 2920 cm−1, which are the MUA characteristics [Citation34]. After immersing the MUA–PBNPs in PBS for a given time, a decrease in the FTIR characteristic absorption indicates desorption of MUA from the PBNPs. Indeed, it was seen that the MUA–PBNPs prepared with 50 mM MUA showed a significant absorption decrease (i.e. desorption of MUA) after 12 and 24 h immersion in PBS. In contrast, the MUA–PBNPs prepared with 5 mM MUA exhibited relatively steady absorbance characteristics after long-term immersion in PBS. This suggests that 5 mM MUA is better than 50 mM MUA for obtaining a robust decorating layer of MUA on PBNPs.
Figure 3. FTIR spectra of the as-prepared MUA-PBNPs from (a) 50 mM and (b) 5 mM MUA and those immersed in PBS after 12 and 24 h.
3.2. Cytotoxicity assessment of the MUA–PBNPs
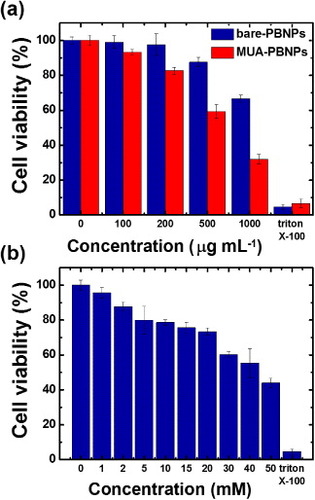
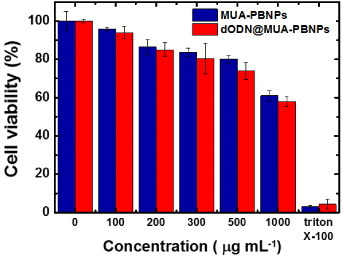
In this study, the 22rv1 human prostate carcinoma epithelial cell was chosen as the target for demonstration of PBNPs-assisted DNA drug delivery. To assess the cytotoxicity of the DNA drug carrier MUA–PBNPs, the 22rv1 carcinoma cells were incubated with the MUA–PBNPs (modified with 20 mM MUA) for 16 h. Then the standard MTT assay was used to evaluate the cell viability. Figure (a) compares the cell viability of bare PBNPs with that of MUA–PBNPs with different incubation concentrations. The cytotoxicity of the bare PBNPs observed here is similar to that reported in a previous work, which used mesoporous PBNPs as the drug carriers [Citation13]. The 22rv1 carcinoma cell remained its 90% of viability after a treatment with 500 μg ml−1 MUA–PBNPs, and exhibited 80% of viability when increased the dosage to 750 μg ml−1 (figure S1 (supplementary data available from stacks.iop.org/STAM/14/044405/mmedia)). The result proves that the synthesized PBNPs are of high intrinsic biocompatibility. In comparison, MUA–PBNPs showed some degree of the cytotoxicity effect. The 22rv1 carcinoma cell viability decreased to 60% after a treatment with 500 μg ml−1 MUA–PBNPs, which was a 30% drop when compared with bare-PBNPs. The reason needs further investigation, since such cytotoxicity effect is not common in the MUA-modified NPs reports for intracellular drug delivery. For example, the MUA-modified gold nanoparticles (MUA–AuNPs) showed an high biocompatibility where the MUA–AuNPs treated cell remained its viability at the initial level after treating with a high concentration of MUA–AuNPs [Citation35]. We suspect that the cytotoxicity of MUA–PBNPs is attributed to the desorption of MUA, since the desorbed MUA might cause the genetic damages of a 22rv1 carcinoma cell [Citation36]. Therefore, the higher concentration of desorbed MUA is, the higher possibility of genetic damage in the cell will occur. This is supported by figure (b). The cytotoxicity seems to be a disadvantage of the MUA–PBNPs for DDS applications, but we can minimize this unwanted effect by tuning the MUA modification on the PBNPs. As shown in figure (b), the viability of the 22rv1 carcinoma cell was able to remain 80% of its initial level after treating with 500 μg ml−1 MUA–PBNPs (a 20% increase), when 5 mM MUA was used for PBNPs functionalization. Hence, the MUA–PBNPs used in the following in vitro DDS application were prepared with 5 mM MUA.
Figure 4. (a) Cell viabilities of 22rv1 carcinoma cells with bare-PBNPs and MUA-PBNPs. (b) Effect of MUA concentration for PBNPs modification on the cell viability. In this experiment, the MUA-PBNPs concentration is fixed at 300 μg ml−1.
3.3. Characteristics of the dODN-grafted MUA–PBNPs
The covalent grafting of dODN on MUA-PBNPs was achieved with amide bond or disulfide bond formation (see the experimental sections 3 and 4 for the details), and the evidence of successful covalent conjugation between the dODN and the MUA–PBNPs is provided here. The UV–visible spectroscopy and FTIR spectroscopy were used to confirm the presence of the dODN on the MUA–PBNPs surface, as shown in figure . The characteristic absorption bands of the dODN and the FAM reporter, which imply the successful conjugation between dODN and MUA–PBNPs, can be observed. The absorption maxima of dODN and FAM in the UV–Vis spectrum were 260 and 490 nm, respectively, and the absorption maxima at 980, 1129 and 1205 cm−1 in the FTIR spectrum indicate the presence of dODN. The observed absorption maxima are consistent with those reported in literature [Citation24]. In addition to qualitative aspect, the spectral absorption maxima was used to estimate how many dODN molecules were grafted onto PBNPs. Accordingly, we estimated that 100 pmol dODN was grafted to each gram of MUA–PBNPs, corresponding to a conjugation efficiency of 17% (a ratio of the amount of conjugated dODN to the initial amount of dODN used for conjugation). Also, fluorescence microscopy images confirmed the presence of FAM-labeled dODN covalently grafted on the MUA–PBNPs surface (see figure S2 (supplementary data available from stacks.iop.org/STAM/14/044405/mmedia)). It is seen that dODN@MUA–PBNPs display intense fluorescence, while the MUA–PBNPs do not.
Figure 5. Characterization of dODN@MUA–PBNPs using UV–Vis and FTIR spectra.
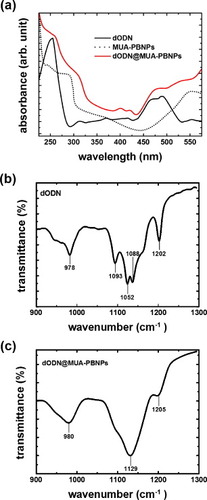
The as-synthesized dODN@MUA–PBNPs were dispersed in PBS (10 mM phosphate, pH 7.4) as a stock solution and stored in a light-resistant container at 4 °C. The average hydrodynamic size and zeta-potential of the dODN@MUA–PBNPs in PBS are about 100 nm and about −20 mV, respectively. Because of the small size and negative charged surface, the dODN@MUA–PBNPs were homogenously dispersed in the solution.
3.4. Cellular uptake and subcellular localization of dODN@MUA–PBNPs
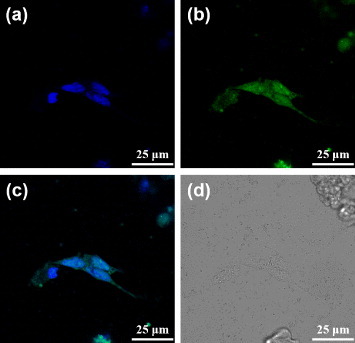
The dODN used in this study inhibits STAT3 gene expression. To demonstrate that the DNA drugs were successfully sent to the cancer cells by PBNPs, the dODN@MUA–PBNPs were dispersed in a culture of 22rv1 carcinoma cell and co-incubated for 16 h. To visualize the internalization of dODN@MUA–PBNPs, the dODN molecules were labeled with a FAM reporter at the counter end against the linker end, and the confocal fluorescence microscopy was applied to track the subcellular localization of the internalized dODN@MUA–PBNPs. The nuclei of cells were stained with DAPI before imaging. With the aid of fluorescent molecules, the nuclei emit blue fluorescence due to DAPI, while dODN@MUA–PBNPs emit green one due to the FAM reporter. The results are given in figure . It is seen that the 22rv1 carcinoma cell treated with dODN@MUA–PBNPs not only exhibits the blue fluorescence emitted from the stained nuclei (figure (a) but also displays uniform green fluorescence emission from the FAM-labeled dODN all over the cell (figure (b)). The merged image (figure (c)) suggests that dODN@MUA–PBNPs were successfully delivered into the cancer cell and were homogeneously distributed throughout the cell including both the cytoplasmic and the nucleic regions. The cross-views of the confocal images taken at different depths of focus (figure S3 (supplementary data available from stacks.iop.org/STAM/14/044405/mmedia)) unequivocally indicate that the DNA-nano drugs were not merely adsorbed on the cell membrane. The overlapping of green and blue emission further confirms that the dODN@MUA–PBNPs were internalized indeed.
Figure 6. Confocal fluorescence microscopy images of the dODN@MUA–PBNPs treated 22rv1 cells: (a) blue fluorescence from the nuclei stained with DAPI, (b) green fluorescence from the FAM-labeled dODN, (c) the merged image of (a) and (b), and (d) the bright-field image of the cells.
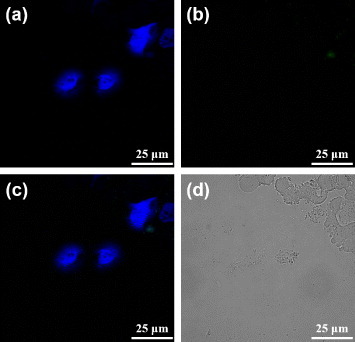
In an attempt of verifying the hypothesis that the MUA-PBNPs act like the nanocargoes for escorting dODNs to the cancer cells, the 22rv1 carcinoma cells were incubated with 1 μM free FAM-labeled dODN under the same condition as that for figure . The confocal fluorescence images of dODN incubated cells are shown in figure . It is clear that the dODN cannot enter the cell without MUA–PBNPs, since the cell emitted blue emission only. Another test indicates that the surface functionalization by an adaptor molecule like MUA is indispensable to render PBNPs as nanocarriers for transporting the DNA drugs. Figure tells that without MUA, the dODN also cannot enter the cell even when bare PBNPs were present and co-incubated with the 22rv1 carcinoma cell. To sum up, the dODN drug molecules with amino or thiol linkers can be readily delivered into the cancer cells and their nuclei with the nanocargoes of MUA–PBNPs.
Figure 7. Confocal fluorescence microscopy images of the free dODN treated 22rv1 cells: (a) the channel for observing the nuclei stained with DAPI, (b) the channel for observing the FAM-labeled dODN, (c) the merged image of (a) and (b), and (d) the bright-field image of the cells.
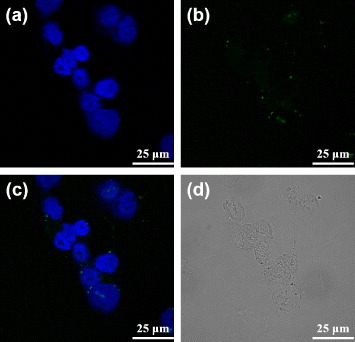
Figure 8. Confocal fluorescence microscopy images of the mixture (dODN and bare PBNPs) treated 22rv1 cells: (a) the channel for observing the nuclei stained with DAPI, (b) the channel for observing the FAM-labeled dODN, (c) the merged image of (a) and (b), and (d) the bright-field image of the cells.
3.5. Dose response of dODN@MUA–PBNPs on 22rv1 cell's viability
In an attempt to understand the dosage response of dODN@MUA–PBNPs on 22rv1 carcinoma cell's viability, the cells were incubated with the various dosage (0–1000 μg ml−1) of dODN@MUA–PBNPs for 4 h and then evaluated by using MTT assays. The concentration of the dODN corresponding to each dosage was estimated to be in the range of 0–0.1 nM on the basis of the dODN grafting efficiency mentioned before. After incubating with the dODN@MUA–PBNPs, the 22rv1 carcinoma cell was expected to exhibit a decrease in cell viability with an increasing dosage of the dODN@MUA–PBNPs. Figure shows that the LC50 value (the dosage required to kill a half of the test population in a specified duration) was achieved until the concentration of the dODN@MUA–PBNPs was higher than 1000 μg ml−1 (i.e. a dODN concentration over 0.1 nM). The measured LC50 value here is much higher than the value estimated from literature [Citation37]. This implies that even though the dODN molecules were successfully delivered into the 22rv1 carcinoma cell with the aid of MUA–PBNPs, the STAT3 inhibition might not work efficiently. The anticipated apoptosis-inducing mechanism of the dODN@MUA–PBNPs is that the dODN takes effect on the signal transducer and activator of transcription 3 (abbreviated as STAT3), which is the key regulator of the cell survival and proliferation. Without STAT3, 22rv1 carcinoma cell ends up with the apoptosis because of the blocked gene expression [Citation38]. The possible reasons why dODN inside the 22rv1 carcinoma cell does not work as expected are considered below. Firstly, it could be the result of inappropriate folding of the dODN molecule on MUA–PBNPs, since the dODN is like an conformation-dependent aptamer drug [Citation39]. Secondly, the poor STAT3 inhibition ability might come from the spatial obstacle of MUA–PBNPs, which hinders the dODN from interacting with STAT3. Third, the dODN might bind to the MUA–PBNPs tightly and difficult to release. It may lead to the reducing of dODN release rate. The proofs of the reasons require further investigation, but significant decreases in cell viability after treating with the DNA-nano drug are still observed (when compared with the controlled experiments with MUA–PBNPs), which imply potential cancer cell-killing capability.
Figure 9. Cell viability test for the dODN@MUA–PBNPs with different incubation concentrations.
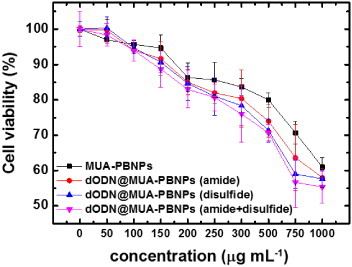
3.6. Effect of the dODN-grafting method
The MUA on the PBNPs provides both carboxyl and thiol groups for dODN grafting. Therefore, the dODN molecules can be grafted onto MUA–PBNPs via both amide bond and disulfide bond formation simultaneously to increase the efficiency of conjugation. That is, MUA–PBNPs can carry more dODNs if both amide and disulfide grafting mechanisms are used in the conjugation process. Once the amount of the dODN conjugation increases, the dODN@MUA–PBNPs will be able to deliver more dODNs into the 22rv1 carcinoma cell. To evaluate the effect of the dODN grafting method, the 22rv1 cells were incubated with three types of dODN@MUA–PBNPs, which were prepared via amide grafting, disulfide grafting, and both grafting, respectively. Figure shows the dose effects of different types of dODN@MUA–PBNPs on the cancer cell viability, and the significant differences in cell viability resulted from different dODN grafting methods are observed. It is seen that the cancer cells have similar viability trends for the amide and disulfide grafting methods, respectively, while the disulfide grafting method seems better. By contrast, the dODN@MUA–PBNPs prepared with a co-grafting process provides a remarkable enhancement in the 22rv1 carcinoma cell-killing capability. Hence, the co-grafting method that anchors both dODNs with an amino and thiol linkers on the MUA–PBNPs seems to be a good strategy to increase the loading of the DNA drugs on the MUA–PBNPs. In addition to the co-grafting strategy, the porous and hollow structures are also possible for enhancing the molecule loading capacity [Citation40, Citation41]. Therefore, the combination of multi-functionalization and nanostructure may contribute to the higher efficacy of DDS.
Figure 10. Cell viability tests for the dODN@MUA–PBNPs prepared with different grafting procedures and different incubation concentrations.
4. Conclusions
In this work, the MUA functionalization of cubic PBNPs, dODN–MUA–PBNPs conjugation methods, potential killing effects of dODN@MUA–PBNPs on the 22rv1 prostate carcinoma cell, and the subcellular drug localization tracking have been investigated. This proof-of-concept study demonstrates the feasibility of a method for covalently grafting decoy oligodeoxynucleotide (dODN) drugs on PBNPs with the aid of an adaptor layer of bifunctional MUA molecules. After simple adsorption of MUA on the surfaces, PBNPs can individually graft or simultaneously co-graft the dODN drug with an amino or a thiol linker. It is discovered that such surface functionalization of PBNPs for DNA grafting is necessary to deliver the dODN drug into cancer cells. It is also found that the novel PBNPs nanocargoes indeed help achieving internalization and homogeneous distribution of the dODN drug in the tested cells. The cancer cell-killing ability is found to be enhanced when the amount of dODN conjugated onto PBNPs and the dosage of DNA–PBNPs drug increase. In conclusion, the present work demonstrates the use of PBNPs as novel nanocarriers for sending DNA drugs into cancer cells.
Acknowledgments
We thank the joint center for instruments and researches in the College of Bioresources and Agriculture of National Taiwan University and the instrument center of National Taiwan University for the technical aids. This work was supported by the National Science Council of ROC. (Taiwan) under a grant number of 101-2221-E-002-174-MY3.
References
- MedinaO PPillarsettyNGlekasAPunzalanBLongoVGonenMZanzonicoPSmith-JonesPLarsonS M 2011 J. Control Release 149 292 10.1016/j.jconrel.2010.10.024
- HuC M JZhangL F 2012 Biochem. Pharmacol. 83 1104 10.1016/j.bcp.2012.01.008
- FangCZhangM Q 2010 J. Control Release 146 2 10.1016/j.jconrel.2010.05.013
- BallyFGargD KSerraC AHoarauYAntonNBrochonCParidaDVandammeTHadziioannouG 2012 Polymer 53 5045 10.1016/j.polymer.2012.08.039
- KimuraTKatoK 2007 Studies in Surface Science and Catalysis vol 165 pp 579–82
- ShokouhimehrMSoehnlenE SKhitrinABasuSHuangS D 2010 Inorg. Chem. Commun. 13 58 10.1016/j.inoche.2009.10.015
- LiTSiZHuLQiHYangM 2012 Sensors Actuators B 172–2 1060 10.1016/j.snb.2012.06.034
- ChenZLiXHeHRenZLiuYWangJLiZShenGHanG 2012 Colloids Surf. B 95 274 10.1016/j.colsurfb.2012.03.012
- UemuraTOhbaMKitagawaS 2004 Inorg. Chem. 43 7339 10.1021/ic0488435
- ChiuJ-YYuC-MYenM-JChenL-C 2009 Biosens. Bioelectron. 24 2015 10.1016/j.bios.2008.10.010
- HongS-FChenL-C 2012 Sol. Energy Mater. Sol. Cells 104 64 10.1016/j.solmat.2012.04.032
- MiaoY QLiuJ W 2009 Sci. Technol. Adv Mater. 10 025001 10.1088/1468-6996/10/2/025001
- LianH YHuMLiuC HYamauchiYWuK C 2012 Chem. Commun. 48 5151 10.1039/c2cc31708g
- BrewerMZhangTDongWRutherfordMTianZ R 2007 Med. Clin. North Am. 91 963 10.1016/j.mcna.2007.05.006
- MingHToradN L KChiangY-DWuK C WYamauchiY 2012 Cryst. Eng. Commun. 14 3387 10.1039/c2ce25040c
- SraK KTorresGRadyPHughesT KPayneD ATyringS K 2005 J. Am. Acad. Dermatol. 53 749 10.1016/j.jaad.2004.08.052
- ZhangXLiuPZhangBWangAYangM 2010 Cancer Genet. Cytogenet. 197 46 10.1016/j.cancergencyto.2009.10.004
- PalmerL JChappleI LWrightH JRobertsACooperP R 2012 J. Periodontal Res. 47 439 10.1111/j.1600-0765.2011.01451.x
- SramkoskiR MPretlowT IIGiaconiaJPretlowTSchwartzSSyM-SMarengoSRhimJZhangDJacobbergerJ 1999 In Vitro Cell Dev. Biol. 35 403 10.1007/s11626-999-0115-4
- OoyaTYamashitaAKurisawaMSugayaYMaruyamaAYuiN 2004 Sci. Technol. Adv. Mater. 5 363 10.1016/j.stam.2003.12.014
- PeerDLiebermanJ 2011 Gene Ther. 18 1127 10.1038/gt.2011.56
- KanedaM MSasakiYLanzaG MMilbrandtJWicklineS A 2010 Biomaterials 31 3079 10.1016/j.biomaterials.2010.01.006
- KermanKMatsubaraYMoritaYTakamuraYTamiyaE 2004 Sci. Technol. Adv. Mater. 5 351 10.1016/j.stam.2004.01.009
- GeinguenaudFSouissiIFagardRMotteLLalatonneY 2012 Nanomedicine 8 1106 10.1016/j.nano.2011.12.010
- YanJLeiMZhuLAnjumM NZouJTangH 2011 J. Hazard. Mater. 186 1398 10.1016/j.jhazmat.2010.12.017
- ChuMShaoYPengJDaiXLiHWuQShiD 2013 Biomaterials 34 4078 10.1016/j.biomaterials.2013.01.086
- ChenJWangFHuangKLiuYLiuS 2009 J. Alloys Compounds 475 898 10.1016/j.jallcom.2008.08.064
- HuMFurukawaSOhtaniRSukegawaHNemotoYReboulJKitagawaSYamauchiY 2012 Angew. Chem. Int. Edn Engl. 51 984 10.1002/anie.201105190
- HartelADidierAPfafflM WMeyerH H D 2003 J. Steroid Biochem. Mol. Biol. 84 231 10.1016/S0960-0760(03)00033-5
- StockertJ CBlazquez-CastroACaneteMHorobinR WVillanuevaA 2012 Acta Histochem. 114 785 10.1016/j.acthis.2012.01.006
- WinK YFengS S 2005 Biomaterials 26 2713 10.1016/j.biomaterials.2004.07.050
- MoraisTSoaresM EDuarteJ ASoaresLMaiaSGomesPPereiraEFragaSCarmoHBastosMde L 2012 Eur. J. Pharm. Biopharm. 80 185 10.1016/j.ejpb.2011.09.005
- TimkoMKopcanskýPAntalikMSimsikovaMValusovaEMolcanMKovácJ 2012 Acta Phys. Pol. A 121 1321
- AndersonM RBaltzersenR 2003 J. Colloid Interface Sci. 263 516 10.1016/S0021-9797(03)00291-1
- ZhangZZhangPGuoKLiangGChenHLiuBKongJ 2011 Talanta 85 2695 10.1016/j.talanta.2011.06.032
- FragaSFariaHSoaresM EDuarteJ ASoaresLPereiraECosta-PereiraCTeixeiraJ Pde Lourdes BastosMCarmoH 2013 J. Appl. Toxicol. 33 1111 10.1002/jat.2865
- Shakeri-ZadehAMansooriG AHashemianA REshghiHSazgarniaAMontazerabadiA R 2010 Cancerous cells targeting and destruction using folate conjugated gold nanoparticles DBPBMB vol 4 Global Science Books 7 11 pp 7–11
- Håkansson-OvesjöM-LCollinMMeisterA R 2000 Endocrinology 141 3946 10.1210/en.141.11.3946
- KeefeA DPaiSEllingtonA 2010 Nature Rev. Drug Discov. 9 537 10.1038/nrd3141
- HuMBelikA AImuraMYamauchiY 2013 J. Am. Chem. Soc. 135 384 10.1021/ja3096703
- HuMToradN LYamauchiY 2012 Eur. J. Inorg. Chem. 2012 4795 10.1002/ejic.201200654