
Abstract
We have employed first-principles calculations based on density functional theory (DFT) to investigate the underlying physics of unusual magnetism in Mn-doped MgO surface. We have studied two distinct scenarios. In the first one, two Mn atoms are substitutionally added to the surface, occupying the Mg sites. Both are stabilized in the Mn valence state carrying a local moment of 4.3
having a high-spin configuration. The magnetic interaction between the local moments display a very short-ranged characteristic, decaying very quickly with distance, and having antiferromagnetic ordering lower in energy. The energetics analysis also indicates that the Mn ions prefer to stay close to each other with an oxygen atom bridging the local interaction. In the second scenario, we started exploring the effect of native defects on the magnetism by crafting both Mg and O vacancies, which are p- and n-type defects, respectively. It is found that the electrons and holes affect the magnetic interaction between Mn ions in a totally different manner. The n-type defect leads to very similar magnetism, with the AFM configuration being energetically preferred. However, in the presence of Mg vacancy, the situation is quite different. The Mn atoms are further oxidized, giving rise to mixed Mn(d) ionic states. As a consequence, the Mn atoms couple ferromagnetically, when placed in the close configuration, and the obtained electronic structure is coherent with the double-exchange type of magnetic interaction. To guarantee the robustness of our results, we have benchmarked our calculations with three distinct theory levels, namely DFT-GGA, DFT-GGA+U and DFT-hybrid functionals. On the surface, the Mg vacancy displays lower formation energy occurring at higher concentrations. Therefore, our model systems can be the basis to explain a number of controversial results regarding transition metal doped oxides.
Introduction
The idea behind the spin transistor, proposed by Dutta and Das [Citation1], is to combine the functionalities of an s and p electron-based semiconductor with the d electron-based ferromagnetism. The proposed device would solve the purpose of information processing, as well as data storage. The idea behind a dilute magnetic semiconductor (DMS) is to dope a small concentration of magnetic ion, such as a transition metal, to a suitable host semiconductor while retaining its semiconducting properties along with its magnetism at room temperature. Following to this idea, the search for such ferromagnetic semiconducting materials has been stimulated by many experimental findings and theoretical models. One such study described the low-temperature ferromagnetism in (In,Mn)As and (Ga,Mn)As DMS [Citation2, Citation3]. The theoretical studies by Sato et al have revealed the electronic structure and the underlying mechanism ensuing the ferromagnetism in several DMSs, such as (Ga, Mn)N and (Ga, Mn)As [Citation4, Citation5], which are found to have low Curie temperatures (). However, despite the many investigations, the proposed spintronic device has not been realized to date due to unavailability of a stable DMS with
higher than 300 K. A significant magnetic percolation in these wide-bandgap semiconductors, where magnetic exchange interactions are short ranged, also gives a wrong estimation of
. For such systems, a spinodal nano-decomposition is predicted to estimate the
more precisely [Citation6–Citation11].
Nevertheless, much effort has been made to investigate the ferromagnetism in DMSs based on group II-VI, [Citation12] III-V [Citation13] and III-IV [Citation14] elements while doping with transition metal impurities like Mn, Co, and Cr. Several experimental groups have reported ferromagnetism in wide-bandgap semiconductors, such as TiO and ZnO, while doping with transition metal dopants like Co and V and even with Li [Citation15–Citation18]. Many theoretical works have also reported ferromagnetism in C-doped CaO [Citation19], which opens their possible applications in electro-optical devices. Similar to TiO
, CaO and ZnO systems, MgO is also a wide-bandgap semiconductor, which could be a promising host material for a high Curie temperature dilute magnetic oxide (DMO), but has not yet been fully explored. There are few studies reporting the magnetism in MgO at different configurations when doped with metals like Mn, Co, Ni, and V [Citation20–Citation23] and even with non-metals like C [Citation22, Citation24, Citation25]. A few first-principles calculations [Citation26, Citation27] and experiments [Citation28] have even reported
magnetism in the pristine MgO system just by crafting Mg vacancies.
In general, the ferromagnetic ordering in a DMS doped with a transition metal, such as Mn mostly comes from a large density of holes () arising from the Mn dopants. At the same time, such a large hole density can induce a semiconducting-to-metallic phase transition in the DMSs, making them undesirable for their practical applications in spin transport. In addition, the p-type (holes) and n-type (electrons) intrinsic defects are crafted in the DMSs during sample preparation and play a crucial role in modulating the magnetic properties of the DMSs. To understand the ferromagnetism of the Mn-doped GaAs having
as high as 110 K, the first-principles calculation by Dietl et al have explained that the ferromagnetic coupling between the Mn, which is mostly mediated by the holes. Comes from the shallow acceptors of the doped system [Citation29]. Thus, possibilities are always there to manipulate the defects by artificially crafting holes and electrons inside the DMS materials, which would further improve the ferromagnetic and semiconducting properties of a DMS in a controlled fashion. Very recently, Mahadeva et al have reported the room temperature ferromagnetism in Mn-doped MgO thin films of various thicknesses. They saw that by increasing film thickness, the ferromagnetism is suppressed, and beyond certain thickness the film shows diamagnetism as expected in a bulk structure. They predicted the cation defects to be one possible answer to the origin of such ferromagnetism in the Mn-doped MgO films [Citation30]. However, this aspect of ferromagnetism has so far not been investigated carefully.
In this work, we report defect-induced ferromagnetism of a ∼0.4 nm thick MgO slab doped with two Mn atoms on the surface maintained with a dilute concentration of 2.7 %. The two Mn atoms energetically prefer to be at the shortest-possible distance apart (4.21 Å) [referred as ‘near’ configuration] than at a distance of 6.65 Å [referred as ‘far’ configuration]. We have introduced defects to the Mn-doped MgO slab by artificially creating Mg and O vacancy separately. We find, for the ‘far’ configuration the exchange coupling between the two Mn atoms always maintains the degeneracy between the ferromagnetic (FM) and antiferromagnetic (AFM) coupling, even in the presence of both defects. For the defect-free Mn-doped MgO slab in its ‘near’ configuration, the exchange coupling between the Mn atoms also favors the AFM coupling over the FM coupling. However, for the same Mn-doped MgO slab, the Mg vacancy generates a strong FM exchange coupling and the O vacancy leads to an AFM coupling between the Mn atoms. Interestingly, we find that the strength of FM exchange coupling decreases while bringing the Mg vacancy close to Mn atoms. In the presence of a Mg vacancy, the magnetic moment of the Mn atoms get reduced compared with that in the defect-free Mn-doped MgO slab.
Now the question is how the defects, like one Mg vacancy, could mediate a strong ferromagnetic exchange coupling between the Mn ions? Is it only a surface effect? Despite of many efforts, it is still difficult to determine the origin of ferromagnetism in a Mn-doped MgO system, which might be due to the spin-polarized holes localized at the cation vacancy or coming all total from the d-orbital magnetic moment of the doped impurities. So far, the hole-induced ferromagnetism, which is definitely a defect-induced phenomenon for the Mn-doped MgO surface, is not well addressed in the literature. To address the defect-induced unusual ferromagnetic properties, we have done systematic spin-polarized calculations for the Mn-doped MgO slab with density functional theory (DFT), GGA+U and with a very accurate hybrid functional approach. The Mg vacancy is found to initiate a double exchange type of interaction, which is responsible for the strong ferromagnetic exchange coupling between the Mn atoms doped at the surface of the MgO slab.
Theoretical approach
In this work, the spin-polarized DFT calculations are performed by using a plane wave pseudopotential method as prescribed within Vienna ab initio package (VASP) [Citation31, Citation32]. The exchange correlation interaction is considered within GGA approximation by using the projected augmented wave (PAW) potential with Perdew–Bruke–Ernzerhof (PBE) functional. Very often, the conventional GGA calculation fails to describe the correct electronic structure for the strongly correlated systems having d and f electrons due to inadequate description of exchange and correlation energy. GGA even underestimates the band gap of such systems. Several groups have carried out self-interaction-corrected local-density approximation (LDA-SIC) to overcome shortcomings of conventional DFT calculations [Citation33–Citation35]. However, for the Mn-doped MgO system, we have used the rotationally invariant DFT+U approach as explained by Dudarev et al and implemented within the VASP code [Citation36] where the strong correlation is captured via an additional local Hubbard repulsion of magnitude U. Within GGA+U approach, the on-site effective Hubbard parameter, = U − J, where U is the on-site Coulomb energy and J is the on-site Hundʼs rule coupling constant, is taken to be a converged value of 6 eV. We have also used the hybrid functional calculation by using the approach of Wu et al as executed within the VASP code to see the effect of an improved version of exchange functional in our calculation [Citation37]. Here, the exchange correlation functional
includes a portion of nonlocal Hartree–Fock exchange to the local or semi-local DFT exchange and takes the form
1 where E
is the exact exchange and
and
are the GGA exchange and correlation energy, respectively. The slab geometry was optimized using a spin-polarized GGA approach. Otherwise, all calculations throughout this work were performed with spin-polarized GGA, GGA+U and with hybrid functional approaches. For the plane wave basis set, we have taken a very high cut-off value of 860 eV. During structure optimization the criterion for the Hellman–Feynman force component on each atom in the supercell is chosen to be 1 meV/Å. The criterion for the energy convergence was taken to be
. A fine Monkhorst–Pack k-point grid (4 × 2 × 2) is used throughout the calculation.
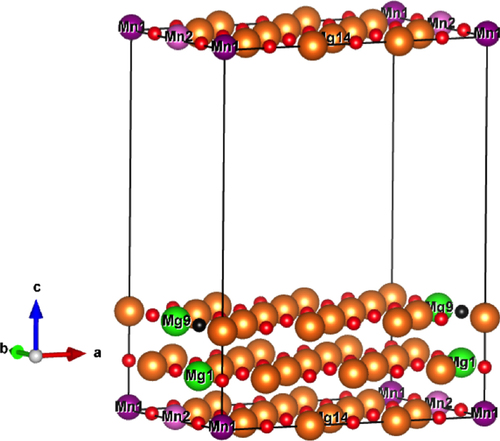
For this work, we model a [3a × 2a] two-layer slab having (1 0 0) surface orientation (with a = 4.21 Å) and 98 atoms in the supercell. We have removed the topmost layer with 24 atoms to create the required symmetric structure for the slab as shown in figure . A vacuum space of 15 Å along the conventional z-direction is maintained to minimize the interaction between the periodic images of the slab. Further, we have doped the slab with Mn atoms at the Mg site (Mn1 and Mn2 in figure ) and introduced Mg (Mg9 in figure ) and O (black ball in figure ) vacancy to the Mn-doped MgO slab.
Figure 1 A 72-atom MgO slab doped with two Mn atoms. Mg: golden yellow, Mn1 and Mn2 at ‘near’ configuration: purple. For ‘far’ configuration the Mn1 is replaced with Mg14. First Mg vacancy (): light green, O vacancy: black. Second Mg vacancy (
): light green.
Since only the p-type defects initiate ferromagnetism in the Mn-doped MgO slab, to know the p-type defect limits for the MgO slab, we have calculated the formation energy for a Mg vacancy in the pristine MgO system by following the routine described by Arhammar et al [Citation38] as,2
is the Mg defect formation energy.
and
are the DFT energies of the MgO slab having one Mg vacancy and that of the perfect MgO slab, respectively.
is the chemical potential of the reservoir containing Mg.
The thermodynamic equilibrium between Mg and O, and MgO is maintained when the chemical potential of Mg and O satisfy the relation, where,
(MgO) is the calculated formation energy of MgO.
Depending on the growth condition, there can be one O-rich condition where (Mg poor),
3 else there will be an O-poor condition where
(Mg rich),
4 Here, the atomic chemical potentials are the variables that are controlled by experimental growth conditions.
The exchange energy for the Mn-doped MgO slab is estimated as5 where
with negative sign indicates the FM being energetically favored over the AFM.
Result and discussion
We present our results and discussions in three parts. In the first part, we describe the electronic structure of a Mn-doped MgO slab with and without the presence of Mg and O vacancies. In the second part, we discuss the reduced magnetic moment of the Mn atoms in the presence of a Mg vacancy in the slab. The third part elaborates the hole-mediated exchange interaction between the two Mn atoms doped on the MgO surface. At the end, we have summarized our results.
Electronic structure
At the beginning, we have studied the electronic structure of the pristine MgO slab (figure ) and continued further by introducing a Mg vacancy and an O vacancy to the slab separately. Both the pristine and the slab with an O vacancy are found to be wide-bandgap () non-magnetic semiconducting systems, whereas the experimentally measured band gap of a MgO system is
[Citation40]. For the MgO slab, the formation energy of Mg-vacancy is calculated to be 3.89 eV and 5.76 eV for O-rich (equation (Equation3
3 )) and for O-poor (equation (Equation4
4 )) conditions, respectively. In another context, the formation energy of Mg-vacancy in a MgO bulk structure at O-poor condition is calculated to be 7.42 eV [Citation23]. Thus, the formation of a Mg vacancy on MgO is predicted to be energetically more favorable in slab structure than in bulk structure.
However, the formation energy exactly reflects the concentration of defects introduced to the system. The ferromagnetism reported in the case of a MgO thin film is due to the formation of magnetic point defects that percolate in the crystal structure and results in a long-range ferromagnetic ordering [Citation41]. With a minimal defect concentration, the ferromagnetism is established only when the crystal structure is grown under non-equilibrium conditions, whereas lowering the defect concentration results in a reduced magnetic moment for the MgO thin film. Thus, the magnitude of the estimated p-type defect formation energy of Mn-doped MgO surface is not of concern. Instead, the ferromagnetism due to the defect concentration is our main concern.
The modeled MgO slab with one Mg vacancy is found to be spin polarized and has a net magnetic moment of 1.6 (table ). Here, the magnetism comes from the spin-polarized oxygen atoms lying in the vicinity of the Mg vacancy. Similar spin-polarization of O atoms has also been predicted in a Zn vacancy-containing ZnO thin film [Citation18, Citation39]. For the modeled MgO slab, the Mn as the extrinsic impurity prefers to be doped at the surface than at the subsurface with an energy difference of nearly 2.3 eV. In the next step, the two Mn atoms are introduced at the Mg site of the MgO slab surface while maintaining a dilute concentration of 2.71%. We have one ‘near’ configuration where the two Mn atoms are placed at a distance of 4.2 Å (Mn1 and Mn2 in figure ) and one ‘far’ configuration where both the Mn atoms are at a distance of 6.65 Å (Mn1 and Mn14 is replaced to be Mn2 in figure ). The two Mn atoms prefer to be in ‘near’ configuration than in ‘far’ configuration by an energy difference of 58 meV. For the ‘far’ configuration, the FM and AFM coupling between the Mn atoms are energetically degenerate even in the presence of Mg and O vacancies (table ). Whereas in the ‘near’ configuration, the AFM coupling between the two Mn atoms is found to be energetically favored over the FM coupling. However, by introducing one Mg vacancy (Mn9 in figure ), the FM exchange interaction becomes energetically more favored, and with one O vacancy (black ball in figure ) the coupling becomes AFM.
Table 1 The calculated total magnetic moment μ, band gap and the exchange coupling
, which is the energy difference between FM and AFM couplings.
and
are the pristine and Mg-vacancy-containing MgO, respectively, whereas
,
, and
correspond to MgO with no defects, with Mg vacancy and with O vacancy, respectively.
Now, one needs to understand the unusual ferromagnetism in the hole-induced MgO slab when the two Mn atoms are doped on the slab surface and are at the closest possible distance apart. Here, we try to compare the electronic structures of the Mn-doped perfect MgO slab, and when one Mg vacancy is created in the slab while keeping both Mn atoms in FM configuration.
For the electronic structures, we have plotted the spin-polarized density of states (DOS) for the Mn-doped MgO slab and that for the slab with one Mg vacancy. From the DOS plots in figure (a), one can see that following the substitution of Mg by Mn, some impurity bands are formed deeply in the band gap region. The conventional DFT-GGA level theory might not be able to describe the width and position of such impurity bands correctly due to the presence of artificial self-interaction in the GGA functional. The consequence is an unphysical delocalization of impurity states, which in turn can lead to an artificial long-range interaction in the lattice. From the partial density of states (PDOS) plots in figure (a), those impurity states are found to be Mn-d states which are very difficult to describe accurately in crystal lattices and particularly in a wide-bandgap oxide such as MgO. To address such issues, we went beyond the framework of GGA and used standard alternatives like GGA+U and hybrid functional approaches. The GGA+U takes the orbital dependence of the Coulomb and exchange interactions into account and explicitly introduces the on-site Coulomb interaction. In contrast, for hybrid functionals, a percentage of exchange energy is obtained from the exact Hartree–Fock based calculations.
Figure 2 Dark maroon shows the majority spin density, and dark blue gives the minority spin density. Panels a, b and c present the total spin-polarized density of states of Mn-doped MgO slab calculated with the DFT, DFT+U and hybrid functional methods. Panels a’, b’ and c’ present the corresponding results for the Mn-doped MgO slab containing Mg vacancy.
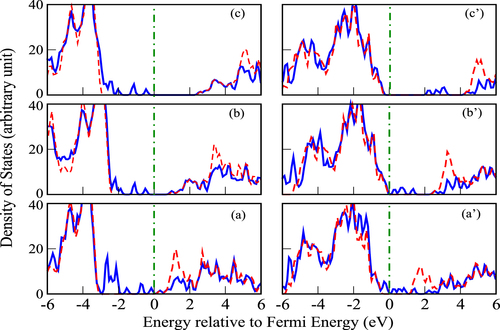
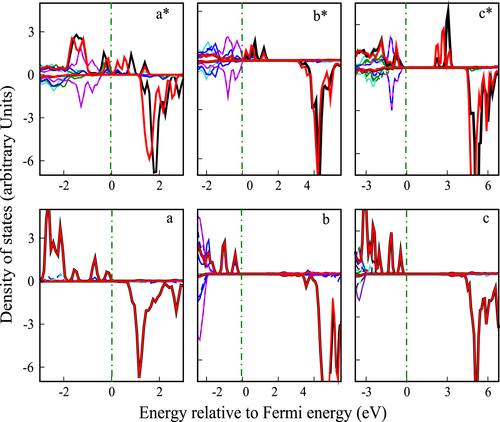
Figure 3 Solid black and red lines present the spin-polarized PDOS of the two Mn(d) ions. Color lines other than red and black show the O(p) PDOS in the vicinity of Mn ions. Panels a, b and c present the PDOS for the ‘perfect’ Mn-doped slab calculated with the DFT, DFT+U and hybrid functional approaches, whereas panels and a*, b* and c* show the corresponding results for the Mn-doped MgO containing Mg vacancy.
The hybrid functionals and GGA+U used to remedy the unphysical self-interaction effect introduced by local-density approximations, leads to delocalization of electrons in the system and gives a better description for the excited-state properties, such as energy band gap, and also the ground-state properties such as magnetic moments and interatomic exchange parameters. However, the effect of these functionals depends upon the degree of unphysical delocalization obtained in the standard GGA calculation, which is very much system dependent. Thus, these self-interaction correction approaches are expected to predict similar results. Comparing the DOS plots calculated with GGA+U and hybrid functional (figure (b) and (c)) with those calculated with GGA (figure (a)), one can observe that both the impurity Mn(d) states are more localized with GGA+U and hybrid functional calculations otherwise, both the alternative approaches leads to similar magnetic properties.
Afterwards, by removing one Mg atom from the Mn-doped MgO slab, the hole states result in a p-type electronic structure. The O atoms coordinating the Mg vacancy become more reactive, which further increases the oxidative power of the crystal environment. As a consequence the Mn atoms get further oxidized, i.e. the electron transfer from Mn to oxygen sub-lattice takes place. However, this process is quite heterogeneous, and the two Mn atoms stabilize at difference between oxidation states. Such a different magnetic states depends on the geometrical distribution of Mn atoms and Mg vacancy.
Henceforth, a significant charge transfer happens from Mn d-bands to the crystal states, which we can see in figures (a)–(c), where the Fermi energy is shifted to the top of the valence band and the Mn-d bands become empty lying in the conduction band region. The single Mg vacancy in the Mn-doped MgO system gives two different oxidation states for the two Mn atoms, which is also apparent from the PDOS plots for the two Mn atoms as shown in figures (a*)–(c*). The Bader analysis further confirms the formation of a mixed oxidation state for the Mn ions in the hole-induced system. We have estimated the energy gap between the valence band and the conduction band with the impurity levels staying close the Fermi level for the perfect Mn-doped MgO slab and subsequently for the slab with p-type and n-type defects, which are summarized in table .
Quenching in magnetic moment
In table , we show the net magnetic moment associated with the perfect Mn-doped MgO slab and that of the slab containing one Mg vacancy and one O vacancy, separately. One can see that the net magnetic moment of the slab decreases as the holes are created by crafting one Mg vacancy in the slab. The magnetization–density–distribution plot associated with the Mn and O atoms for the perfect slab and the slab with one Mg vacancy are shown in figures (a)–(c) and (aʼ)–(cʼ), respectively. We see, for the slab with a Mg vacancy, the O atom positioned in-between the Mn atoms is strongly spin polarized (figures (aʼ)–(cʼ) and has a net magnetic moment of 1.6. The magnetic moment of the Mn atoms in the hole-doped slab is ∼0.8
smaller when compared to that of the perfect slab. Compared to perfect slab, the Mn-O interatomic bond distance also gets reduced in the hole-doped slab.
Figure 4 Panels a, b and c show the magnetization density of Mn and O atoms in ‘perfect’ Mn-doped MgO slab calculated with DFT, DFT+U and hybrid functional approaches. Panels a’, b’ and c’ present that of the hole-doped slab calculated in similar fashion. The dark maroon color presents the positive magnetization density, and dark blue color around the O presents the negative magnetization density. Green: Mn, golden yellow: oxygen, cyan: Mg.
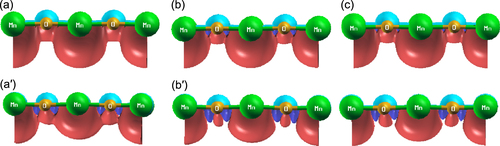
For the hole-doped slab, from the PDOS plot in figure , the valence band region is found to be influenced by the O(p), and mixed Mn(d) ionic states with similar weight. As a consequence, in the hole-doped slab the Mn(d) electrons get strongly hybridized with the neighboring O(p) states. The corresponding PDOS plots calculated with DFT+U and hybrid functionals also produce similar results, as shown in figures (b), (b*) and (c), (c*). The hybridization results delocalization of the valence electrons, which explains the quenching in Mn magnetic moment in the hole-doped slab (table ). However, with an n-type defect (O-vacancy), we find neither quenching in Mn magnetic moment nor spin polarization of neighboring O atoms. A similar effect has also been reported in an O vacancy-containing Mn-doped MgO bulk structure [Citation22].
Table 2 The magnetic moment and charge associated with the individual Mn atoms in the unit of and e, respectively. The symbol Mn12 reflects that both the Mn atoms in the perfect slab have the same magnetic moment and charge distribution, whereas symbols Mn1 and Mn2 correspond to different Mn atoms in the Mg-vacancy containing slab.
Hole mediated double exchange interaction
In the Mn-doped MgO slab, when the Mn atoms are kept in their ‘far’ configuration, the calculated exchange energies predict degeneracy for FM and AFM states even in the presence of p-type and n-type defects. Being at far, there is a very little overlap between the exponential decaying Mn wave functions, which leads to a lack of preferential orientation for their spin moments. For Mn-doped ZnO thin films, in similar situation, a paramagnetic behavior has been reported by Cheng and Chiang et al [Citation42] and also has been predicted by several other theoretical calculations [Citation43, Citation44].
On the other hand, when the two Mn atoms are in their ‘near’ configuration the changes its sign depending upon the nature of the carrier within the system. In the defect-free slab, the exchange interaction between the two Mn
states, which is mediated via the oxygen atom lying in-between, is of the super-exchange type, giving rise to an AFM state. However, having holes as the carriers, the two Mn ions get reformed into two different oxidation states. The two distinct, unequal peaks of the Mn-d density of states in figures (a*)–(c*), compared to the degenerate Mn-d states in figures (a)–(c) confirms mixed oxidation states for the Mn ions. The O atoms lying in the vicinity of Mn ions also get strongly spin polarized, as evident from the magnetization density plot in figures (aʼ)–(cʼ).
The unequal charge distribution allows the hopping of 3d electrons from the partially occupied one Mn ionic states to the second Mn ion via the ion when both the Mn ions have parallel spin configuration. An interaction of this type is termed as a ferromagnetic (FM) double-exchange mechanism. By using mean-field approximations, for certain hole concentrations, a similar FM exchange interaction is also predicted in a Mn-doped DMS, which is mediated by the delocalized holes [Citation47]. The double-exchange interaction that is qualitatively described in both the d-p Zener model and the Anderson impurity model was originally proposed to explain the ferromagnetism in transition metals [Citation45, Citation46]. Later, the mechanism was successfully used to explain the room temperature ferromagnetism in Mn-doped in GaAs and ZnTe type of DMSs [Citation29].
Further, by bringing the Mg vacancy closer to the Mn ions (replacing Mg9 vacancy by Mg1), we find that the strength of the FM exchange interaction decreases from to
. Having the vacancy at Mg1 site (figure ), we find the two Mn ions are with equal change distribution and have equal magnetic moments, and the double-exchange mechanism is no longer valid to explain the small ferromagnetism in the Mn-doped MgO slab. The position of the vacancies, which play a role in interfering the valence states of the Mn atoms and also in Mn-O charge transfer, might affect the strength of exchange interaction, and thus needs further attention.
Schematic model
Based on the individual magnetic moment and charge distribution on Mn and O atoms, we have drawn a schematic model to visualize the super-exchange and the double-exchange types of interactions between the Mn ions in the perfect slab and the one having p-type defects. For the perfect slab, the two Mn ions have an equal magnetic moment of 4.38 and have equal charge distribution of 5.4 e on each (table ). We draw a Mn
oxidation state for both the Mn ions (figure (a)), and we see a super-exchange type of interaction between both the Mn ions, which results in an AFM coupling (figure (a)). For the hole-doped slab, we find the Mn atoms to have an unequal magnetic moment of 3.7 and 3.5
, respectively, and showing a charge distribution of 5.5 and 5.6 e, respectively (table ). We draw two asymmetric oxidation states for both the Mn ions as shown in figure (b). The O atom lying in between the Mn ions is spin polarized with a net spin-down density as evident from the magnetization density plot in figures (aʼ)–(cʼ). We suggest a simultaneous electron hop from the first Mn-3d ionic state to the second Mn-3d ionic state via the spin-down states of O ensuing a double-exchange type of interaction between the Mn ions, resulting in FM states (figure (b)). It is worthy to mention that, though the numbers estimated for the magnetic moment and charge are different for the Mn and O ions when calculated within DFT, GGA+U and hybrid functional approaches (table ), the trend remains the same in all the three cases, which justifies our proposed model.
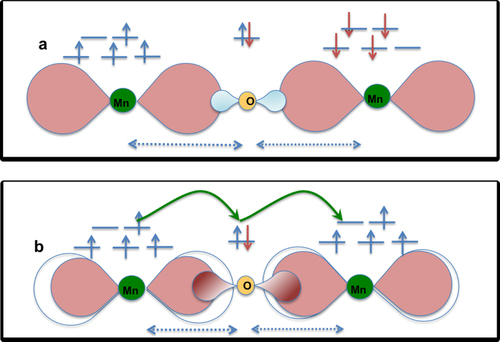
Figure 5 A schematic picture showing (a) super exchange interaction favoring AFM coupling (b) and double exchange interaction favoring FM coupling between the Mn ions, which is mediated via the O ion lying in between. The blue dotted line shows the Mn-O interatomic distance in both cases. The green connecting curved line shows the simultaneous charge transfer between Mn and O atoms. Pink lobes around Mn atoms in both panel (a) and (b) and around O in panel b present the spin-polarized charge density, whereas the blue lobes around O in panel (a) present spin-unpolarized charge density. The blue and red arrows present the positive and negative spin densities, respectively.
Summary
By using density functional theory, we have studied the magnetic properties of a MgO surface while doped with two Mn atoms at the Mg sites. On the slab surface, the magnetic interaction between the two Mn atoms is short ranged and decays very fast with distance. The two Mn atoms energetically prefer to stay in a close-configuration with one O atom bridging their local interaction. In close configuration, the Mn atoms stabilize at a Mn ionic state, having a local magnetic moment of 4.3
on each. The AFM interaction between the Mn3+ ionic states is favored over the FM interaction. Further, crafting the slab with p-type defects, the two Mn atoms resulted in mixed oxidation states, while the O atom bridging in between gets strongly spin polarized. The simultaneous charge transfer between the Mn mixed ionic states via the O ion results in a ferromagnetic coupling between the Mn ions which can be explained by the double-exchange type of magnetic interaction. On the other hand, crafting the slab with n-type defects results in an AFM state similar to the perfect slab. To get a complete insight to the electronic structures of such complex systems, we attempt to overcome the self-interaction errors of the conventional GGA approach by using GGA+U and hybrid functional calculations. Both GGA+U and hybrid functional calculations follow a similar trend with the GGA results for the electronic structure and magnetic properties of the Mn-doped MgO slab, which supports for the accuracy of our results.
Acknowledgments
We would like to thank Professor K V Rao for valuable discussions in this project. We are grateful to the VR, STINT, and Energimyndigheten for financial support. Puspamitra Panigrahi would like to thank the Swedish Institute for a postdoctoral fellowship, and Tanveer Husen is thankful to the Higher Education Commission of Pakistan for a PhD fellowship. We are also thankful to NSC, HPC2N and UPPMAX for providing us with computing resources.
References
- DattaSDasB 1990 Appl. Phys. Lett. 56 665 10.1063/1.102730
- OhnoHShenAMatsukuraFOiwaAEndoA 1996 Appl. Phys. Lett. 69 363 10.1063/1.118061
- OhnoHMunekataHPenneyTvon MolnarSChangL L 1992 Phys Rev. Lett. 68 2664 10.1103/PhysRevLett.68.2664
- SatoKSchweikaWDederichsP H 2004 Phys. Rev. B 70 201202 10.1103/PhysRevB.70.201202
- SatoK 2010 Rev. Mod. Phys. 82 1633 10.1103/RevModPhys.82.1633
- SatoKFukushimaTKatayama-YoshidaH 2005 Jpn. J. Appl. Phys. Lett. 2. Exps Lett. 44 L948 10.1143/JJAP.44.L948
- FukushimaTSatoKKatayama-YoshidaHDederichsP H 2006 Jpn. J. Appl. Phys. 45 L416 10.1143/JJAP.45.L416
- Katayama-YoshidaHSatoKFukushimaTToyodaMKizakiHDinhV ADederichsP HMagJ 2007 Mag. Materials 310 2070 10.1016/j.jmmm.2006.10.1120
- SatoKFukushimaTKatayama-YoshidaH 2007 Jpn. J. Appl. Phys. Lett. 2. Exps Lett. 46 L682 10.1143/JJAP.46.L682
- Katayama-YoshidaHSatoKFukushimaTToyodaMKizakiHDinhV ADederichsP H 2007 Phys. Stat. Sol. A 204 15 10.1002/pssa.v204:1
- SeikeMDinhV AFukushimaTSatoKKatayama-YoshidaH 2012 Jpn. J. Appl. Phys. 51 050201 10.7567/JJAP.51.050201
- HauryAWasielaAArnoultACibertJTatarenkoSDietlTMerleY 1997 Phys. Rev. Lett. 79 511 10.1103/PhysRevLett.79.511
- HansenLFerrandDRichterGThierleyMHockV 2001 Appl. Phys. Lett. 79 3125 10.1063/1.1416160
- OhnoHMunekataHVon MolnrSChangL L 1991 J. Appl. Phys. 69 6103 10.1063/1.347780
- MatsumotoYMurakamiMShonoTHasegawaTFukumuraTKawasakiMAhmetPChikyowTKoshiharaSKoinumaH 2001 Science 291 854 10.1126/science.1056186
- UedaKTabataHKawaiT 2001 Appl. Phys. Lett. 79 988 10.1063/1.1384478
- SaekiHTabataHKawaiT 2001 Solid State Commun. 120 439 10.1016/S0038-1098(01)00400-8
- YiJ 2010 Phys. Rev. Lett. 104 137201 10.1103/PhysRevLett.104.137201
- KenmochiKSeikeMSatoKYanaseAYoshidaH K 2005 J. Superconduc. 18 37 10.1007/s10948-005-2147-7
- IbrahimM MFengZDeanJ CSeehraM S 1992 J. Phys.: Condens. Matter 4 7127 10.1088/0953-8984/4/34/012
- LiuGJiSYinLFeiGYeC 2010 J. Phys.: Condens. Matter 22 046002
- SharmaVPilaniaGLowtherJ E 2011 AIP Adv. 1 032129 10.1063/1.3625411
- RhammarCMoyses AraujoCRaoK VNorgrenSJohanssonBAhujaR 2010 Phys. Rev. B 82 134406 10.1103/PhysRevB.82.134406
- DroghettiASanvitoS 2009 Appl. Phys. Lett. 94 252505 10.1063/1.3152781
- KenmochiKDinhVSatoKYanaseAYoshidaH K 2004 J. Phys. Soc. Jpn. 73 2952 10.1143/JPSJ.73.2952
- GaoaFHuaJYangbCZhengaYQinaHSunaLKongaXJiangaM 2009 Solid State Commun. 149 855 10.1016/j.ssc.2009.03.010
- DroghettiAPemmarajuCSanvitoS 2010 Phys. Rev. B 81 092403 10.1103/PhysRevB.81.092403
- LiJJiangYLiYYangDXuYYanM 2013 Appl. Phys. Lett. 102 072406
- DietlTOhnoHMatsukuraFCibertJFerrandD 2000 Science 287 5455 10.1126/science.287.5455.1019
- MahadevaaS KQuanaZ YFanaJ CSreelathaaK SBelovaaLPuzniakRRaoK V 2013 MRS Proc. 1552 83 88 83–88 10.1557/opl.2013.611
- KresseGHafnerJ 1993 Phys. Rev. B 47 558 10.1103/PhysRevB.47.558
- KresseGJoubertJ 1999 Phys. Rev. B 59 1758 10.1103/PhysRevB.59.1758
- ToyodaMAkaiHSatoKKatayama-YoshidaH 2006 Physica B Condens. Matter 376 647 10.1016/j.physb.2005.12.163
- ToyodaMAkaiHSatoKKatayama-YoshidaH 2006 Phys. Stat. Sol. C 3 4155 10.1002/pssc.200672846
- An DinhVToyodaMSatoKKatayama-YoshidaH 2006 J. Phys. Soc. Jpn. 75 093705 10.1143/JPSJ.75.093705
- DudarevS LBottonG ASavrasovS YHumphreysC JSuttonA P 1998 Phys. Rev. B 57 1505 10.1103/PhysRevB.57.1505
- WuXSelloniACarR 2009 Phys. Rev. B 79 85102 10.1103/PhysRevB.79.184208
- ArhammarCArajoC MAhujaR 2009 Phys. Rev. B 80 115208 10.1103/PhysRevB.80.115208
- WangQSunQChenGKawazoeYJenaP 2008 Phys. Rev. B. 77 205411 10.1103/PhysRevA.77.033834
- RoesslerD MWalkerW C 1967 Phys. Rev. 159 733 10.1103/PhysRev.159.733
- AraujoC 2010 Appl. Phys. Lett. 96 232505 10.1063/1.3447376
- ChengXChienC 2003 J. Appl. Phys. 93 7876 10.1063/1.1556125
- SatoKKatayamaH 2002 Semicond. Sci. Technol. 17 367 10.1088/0268-1242/17/4/309
- WangQSunQRaoB KJenaP 2004 Phys. Rev. B 69 233310 10.1103/PhysRevB.69.233310
- ZenerC 1950 Phys. Rev. 83 299 10.1103/PhysRev.83.299
- AndersonP WHasegawaH 1955 Phys. Rev. 100 675 10.1103/PhysRev.100.675
- DietlTOhnoHMatsukuraF 2001 Phys. Rev. B 63 195205 10.1103/PhysRevB.63.195205