
Abstract
Carbon nanotubes (CNTs) have been recognized as a promising material in a wide range of applications from biotechnology to energy-related devices. However, the poor solubility in aqueous and organic solvents hindered the applications of CNTs. As studies have progressed, the methodology for CNT dispersion was established. In this methodology, the key issue is to covalently or non-covalently functionalize the surfaces of the CNTs with a dispersant. Among the various types of dispersions, polymer wrapping through non-covalent interactions is attractive in terms of the stability and homogeneity of the functionalization. Recently, by taking advantage of their stability, the wrapped-polymers have been utilized to support and/or reinforce the unique functionality of the CNTs, leading to the development of high-performance devices. In this review, various polymer wrapping approaches, together with the applications of the polymer-wrapped CNTs, are summarized.
1. Introduction
CNTs are cylindrical graphene tubes with a one-dimensional π-conjugated structure [Citation1]. Due to the outstanding electrical, mechanical, thermal and optical properties, CNTs are promising candidates for various applications such as electronics, sensors, energy conversion and storage devices [Citation2]. During the early stage of CNT research, the limited solubility of CNTs, due to their high aspect ratios and strong van der Waals interactions, hindered the development of CNT applications [Citation3]. Only a limited number of organic solvents, such as o-dichlorobenzene (ODCB), N-methylpyrrolidinone (NMP), N,N-dimethylformamide (DMF) and N,N-dimethylacetamide (DMAc), are known to disperse CNTs to some extent, but the degree of isolation and the stability are not sufficient for many of the applications [Citation4–Citation12].
Based on the results of the significant amount of research, the methodology to exfoliate the bundled structures of the CNTs and disperse them in solvents has been established in which the engineering of the surface of the CNTs using small molecules or polymers in a covalent or non-covalent way is the major strategy (figure ) [Citation13–Citation15].
Figure 1. Main CNT functionalization methods.
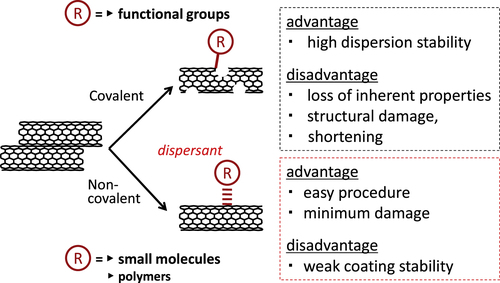
For the covalent modifications, the oxidation of the surface to introduce the carboxyl groups [Citation16–Citation24] and the chemical reactions to connect the functional moieties are typical approaches. Generally speaking, the covalent modifications are superior to the non-covalent modification in terms of the stability of the functionalization. In particular, the covalent approach easily leads to the effective reinforcement of the polymer films due to the effective load transfer from the polymer matrix to the CNTs through the covalent bonding [Citation25–Citation30]. However, the covalent modification changes the intrinsic properties of the CNTs, such as conductivity and mechanical toughness, and often cuts the CNTs into shorter tubes [Citation31]; thus, the non-covalent modification is superior in most cases in order to utilize the inherent properties of the CNTs [Citation14, Citation15, Citation30, Citation32–Citation39]. In addition, non-covalent modifications are characterized as their simple procedure, typically just by the mixing of CNTs with molecules under a shear force treatment such as sonication. In this review, the molecules used for the non-covalent modification of CNTs are called ‘dispersants’.
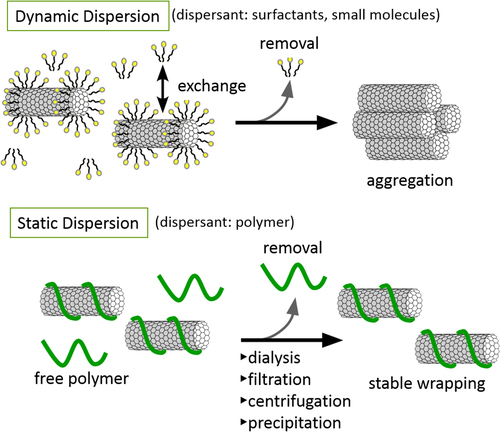
Non-covalent functionalization is realized via 1) enthalpy-driven interactions, such as π−π, CH−π, NH−π, etc, between the CNT surface and the dispersants and/or 2) entropy-driven interaction; i.e. hydrophobic interaction using surfactants (figure ) [Citation40]. In the case of the surfactant dispersion, sodium dodecyl sulfate (SDS) [Citation41–Citation43], sodium dodecylbenzene sulfonate (SDBS) [Citation44–Citation48], sodium cholate (SC) [Citation49–Citation51], cethyltrimethylammonium bromide (CTAB) [Citation46, Citation52], Brij [Citation46, Citation51], Tween [Citation46, Citation51] and Triton X [Citation44, Citation46, Citation51, Citation53] have typically been used due to their availability and cost [Citation50]. It is important to understand that the surfactant molecules on the surface of the CNTs are in a dynamic equilibrium between the surfactants in the bulk solution (figure , upper) [Citation54]. Therefore, the dispersants are easily removed by filtration or dialysis, resulting in the aggregation of the CNTs (figure ) [Citation55]. Similar dynamic dispersions also take place in the case of the enthalpy-driven functionalization using small molecules as the dispersant. On the other hand, when the polymer is used in the enthalpy-driven functionalization, static dispersion due to the multi-point interaction between polymers and the surface of the CNTs is often realized [Citation55]. In this case, the non-covalently surrounded polymers remained even after the washing process, such as filtration, to provide the ‘polymer-wrapped CNTs’. In some applications, such wrapped dispersants act as a contaminate, but in some cases, the wrapped CNTs synergistically improve the performance of the CNTs if the polymers are strategically designed. In this review, such polymers that offer additional functions to the CNTs are categorized as ‘functional dispersants’. Due to the tailorable design of the polymers, the concept of the functional dispersant has recently been widely recognized and utilized.
Figure 2. Dispersant-CNT interactions.
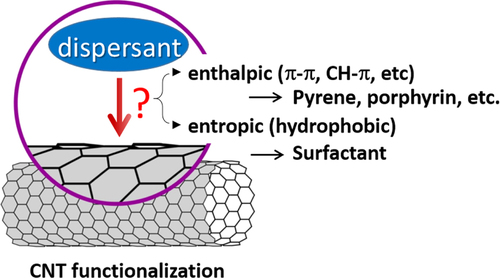
Figure 3. Schematic illustrations for the dynamic (upper) and static (lower) dispersion of CNTs.
In this review, some examples of the non-covalent functionalization of CNTs based on the polymer wrapping approach and the roles of the functional dispersants are summarized.
2. Type of polymer for wrapping
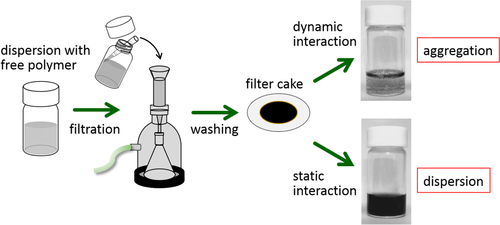
For the polymer wrapping of CNTs, various interactions, including π−π, CH−π and cation−π, function to adsorb on the surfaces of the CNTs. In this chapter, the types of polymers used for the wrapping are summarized based on their structural features. One of the advantages of the polymer wrapping is the thermodynamically stable coating on the surface of the CNTs, and it is possible to remove the unbound (free) polymer while leaving the wrapping polymer on the CNT surface (figure , lower). Removal of the unbound polymer in the bulk solution can be carried out by (1) dialysis [Citation56, Citation57], (2) a precipitation/decantation cycle [Citation58], (3) a filtration/washing process [Citation59–Citation61], (4) a ultracentrifugation/decantation process [Citation58, Citation62] and (5) a chromatographic separation [Citation57]. In the case of the filtration process, it is quite simple to distinguish the stable dispersion since the nice dispersion is achieved again from the washed materials when the stable wrapping has taken place (figure ). Note that not all the papers detail the presence of the unbound polymer in the solution, and it is highly recommended to recognize the effect of the unbound polymer even when the authors did not describe it since such polymers sometimes affect displayed data.
Figure 4. Schematic procedure of the filtration process.
2.1. π-conjugated polymers
It is reasonable to use π-conjugated polymers for the wrapping of the π-conjugated surfaces of the CNTs in which effective interactions, such as π−π and/or CH−π, are expected. Pioneering studies of dispersing CNTs by a π-conjugated polymer into solvents were carried out using poly(p-phenylenevinylene) derivatives (PPVs) as the polymer dispersant [Citation63–Citation65]. The CNTs formed a stable dispersion in the organic solution of PPVs, suggesting the formation of the polymer-wrapped CNTs. In addition to the dispersion of CNTs [Citation66–Citation82], the PPV wrapping was also utilized for the extraction of single-walled carbon nanotubes (SWCNTs) with specific chiral indices. Keogh et al and Coleman et al used the poly(m-phenylene-co-2,5-dioctoxy-p-phenylenevinylene) (PmPV, figure ) to preferentially disperse SWCNTs with specific chiral indices, leaving the others in the precipitate [Citation83–Citation86]. Although a detailed mechanism of the selective dispersion is still unknown, it is assumed that the π-conjugated polymers with a rigid backbone exhibit the selectivity for the specific chiral indices by aligning their backbones along the SWCNTs’ surfaces with a preferential angle in order to maximize the interaction on the π-surface [Citation87]. Selective extraction of semiconducting SWCNTs (s-SWCNTs) by π-conjugated polyfluorene (PFO) and their derivatives has been reported by Nish et al in which poly(9,9-dioctylfluorenyl-2,7-diyl) (PFO) and poly[(9,9-dioctylfluorenyl-2,7-diyl)-alt-co-(1,4-benzo-2,1,3-thiadiazole)] (PFO-BT or P8BT) (figure ) were used [Citation88].
Figure 5. Chemical structure of PmPV.
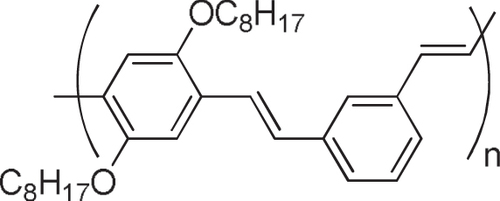
Figure 6. PFO and its derivatives that dissolve s-SWCNTs reported from other groups.
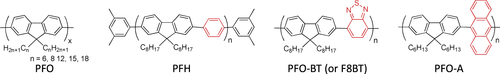
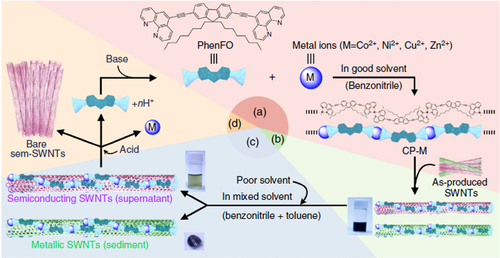
Compared to the other separation techniques, such as gel chromatography [Citation89–Citation93] and density gradient ultracentrifugion (DGU) [Citation94, Citation95], PFO-based extraction of s-SWCNTs is quite attractive due to its simplicity as well as its high purity [Citation96]. Very importantly, the tailorable design of the PFO allowed researchers to develop a wide variety of PFO-based copolymers for the separation of the SWCNTs [Citation97]. Indeed, we demonstrated that many monomers are incorporated into the PFO for the s-SWCNT extraction (figure ) [Citation98–Citation105]. Thanks to the high purity of the obtained s-SWCNTs, the extracted s-SWCNTs are considered as a promising material for next-generation SWCNT-based semiconducting devices. Izard et al reported an on/off ratio greater than 105 in a field effect transistor (FET) fabricated from the s-SWCNT network extracted by PFO wrapping, while the unsorted SWCNTs gave only 102 ∼ 103 [Citation61]. In 2010, Bindl et al reported light harvesting in the near-IR (NIR) region using PFO-sorted s-SWCNTs as the chromophore, where C60 was incorporated as the acceptor (figure ) [Citation106–Citation108]. In these examples, due to the high-quality separation of the s-SWCNTs by PFO, a significant enhancement of the efficiency was obtained compared to the device fabricated with unsorted SWCNTs. However, in these applications, the wrapped PFOs were used only for the separation but remained wrapped in the devices. It was pointed out that the organic residual remaining on the surface of CNTs often diminishes the performance of the devices, especially for semiconducting applications [Citation94], and in such cases the complete removal of the wrapped polymers is required. For instance, Bisri et al developed a two-step ultracentrifugation method to remove the polymer from the polymer-dispersed CNT solution [Citation62]. In this method, they obtained a polymer less than 7 mg mL−1, but wrapping of the polymer was so tight that the perfect removal of the polymer was not possible. Recently, we have designed the fluorene monomer carrying two ligand units (PhenFO), which polymerize in the presence of metal ions to give a PFO derivative (CP-M) by coordination (figure ). In this system, the wrapped PFO was easily removed by the addition of an acid to decompose the coordination after the extraction of the s-SWCNTs (figure ) [Citation109]. As the result, complete removal of the polymer was realized.
Figure 7. PFO derivatives and the analogs selectively disperse s-SWCNTs reported from our group.
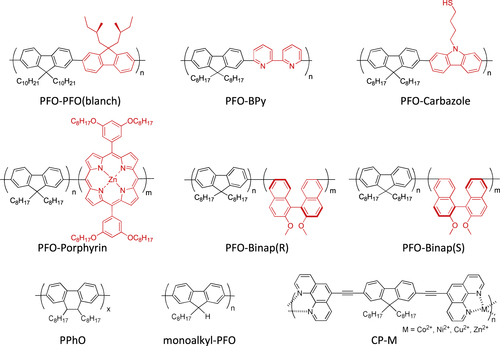
Figure 8. (left) Schematic depicting charge transfer at the s-SWCNT/C60 interface under NIR irradiation. (right) Energy diagram of s-SWCNTs and C60, where LUMO stands for lowest unoccupied molecular orbital. Reproduced with permission from D J Bindl and M S Arnold 2013 J. Phys. Chem. C 117 2390. Copyright 2013 American Chemical Society.
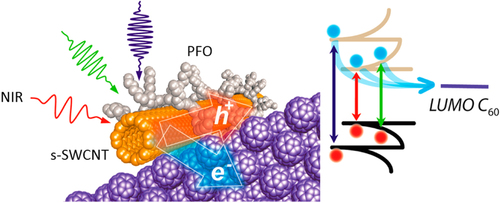
Figure 9. A method for s-SWCNT sorting using removable dispersant. The purification method starts from (a) the preparation of CP-M (M = Co, Ni, Cu and Zn), (b) dispersion of as-produced SWCNTs, (c) separation of s- and m-SWCNTs and (d) removal and recovery of the adsorbents. Chemical structure of PhenFO is shown in step (a). Reprinted by permission from Macmillan Publishers Ltd: F Toshimitsu and N Nakashima 2014 Nat. Commun. 5 5041, copyright 2014.
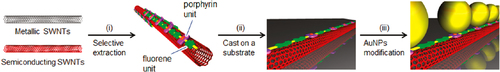
In contrast to such an approach, it is not necessary to remove the wrapped polymer in the concept of ‘functional dispersant’. In order to utilize the wrapping PFO as the functional dispersant, we developed a PFO-based dispersant by introducing a carbazole-based co-monomer bearing a thiol group (PFO-carbazole, figure ). Since the thiol group binds to the Ag surface, the PFO-wrapped s-SWCNTs were decorated with the Ag nanoparticles after the separation [Citation103]. A similar decoration with Au nanoparticles was also achieved using the s-SWCNTs wrapped with a porphyrin-containing PFO copolymer (PFO-porphyrin, figures and ) [Citation102]. In these examples, the wrapped PFO were functioned for the anchoring of metal nanoparticles.
Figure 10. Schematic illustration of the separation of s-SWCNTs using PFO-porphyrin and an attachment of gold nanoparticles (AuNPs). Reprinted with permission from H Ozawa et al 2011 J. Am. Chem. Soc. 133 14771. Copyright 2011 American Chemical Society.
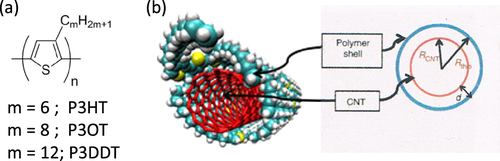
Poly(3-alkylthiophenes), figure (a), such as poly(3-hexylthiophene) (P3HT), are also known to wrap CNTs and are used mostly for organic photovoltaic applications [Citation110]. It was pointed out that not only the π−π interaction but also the sulfur atom in the backbone plays an important role for the adhesion based on MD calculations [Citation111]. Goutam et al found that P3HT rapidly degrades in organic solvents containing dissolved molecular oxygen when irradiated with an UV light, but that was not the case for the P3HT-wrapped CNTs [Citation112]. It was suggested that the π–π interaction between the P3HT-CNT composite improves the stability of the π-conjugation system, thereby preventing photosensitization and a reduced opportunity for the reaction of singlet oxygen with the P3HT. This enhanced stability explained the higher stability and efficiency of the P3HT/CNT devices compared with the devices composed from P3HT [Citation113]. In this example, the function of the polymer was reinforced by the incorporation of CNTs. In 2011, Lee et al found that regioregular poly(3-alkylthiophenes) selectively disperse s-SWCNTs by a polymer wrapping mechanism (figure (b)) [Citation114]. Using this technique, they fabricated high-performance transistors formed with a s-SWCNT network and observed a charge-carrier mobility as high as 12 cm2 V−1 s−1 and an on/off ratio of >106.
Figure 11. (a) Chemical structure of poly(3-alkylthiophenes). (b) Schematics of a cross-sectional geometrical view of the polymer–SWCNT supramolecular structure. Reprinted by permission from Macmillan Publishers Ltd: H W Lee et al 2011 Nat. Commun. 2 541, copyright 2011.
Other π-conjugated polymers, such as polypyrrole (PPy) and polyaniline (PANI) [Citation115], as well as their derivatives also wrap the CNTs, and many such studies are summarized in an excellent review paper [Citation115]. In particular, for these polymers, electropolymerization using CNTs as the electrode enables homogeneous wrapping by the polymers. It is quite unique since, in this approach, stable wrapping was achieved even in the absence of the strong interaction between the wrapping polymer and CNT surfaces.
2.2. Aromatic polymers
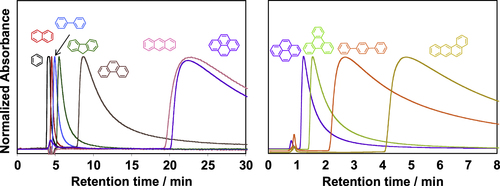
Aromatic condensation polymers having an aromatic system in the main-chain, such as polyimides (PIs), are the polymers that can wrap the CNTs [Citation116]. Our chromatography studies using CNTs as the stationary phase revealed that the one-dimensional aromatic compounds, such as tetraphene, exhibited a stronger affinity with the CNT surfaces than the other analogs with the same number of the aromatic rings, such as pyrene or triphenylene, due to the effective overwrapping with the one-dimensional CNT surfaces (figure ) [Citation117, Citation118].
Figure 12. Chromatogram of the series of aromatic compounds analyzed using CNT as the stationary phase. Chromatograms of (black) benzene, (red) naphthalene, (light blue) biphenyl, (green) fluorene, (brown) phenanthrene, (pink) anthracene, (purple) pyrene, (light green) triphenylene, (orange) p-terphenyl and (yellow) tetraphene obtained from the SWCNT-column; eluent: tetrahydrofuran, flow rate 0.1 mL min−1 (left) and 0.5 mL min−1 (right).
We considered that the results well explained the effective interaction of the aromatic condensation polymers, such as PIs, with the CNTs. By taking advantage of the remarkable thermal stability of these polymers and CNTs, the composites are fascinating for the use under harsh conditions such as aerospace applications. We have reported that a PI having a sulfonic acid salt (PI-SO3Na) effectively exfoliated and dispersed SWCNTs in organic solvents for long periods of time [Citation116]. It is interesting to note that the concentration of SWCNTs in a dimethyl sulfoxide (DMSO) solution of the PI-SO3Na reached as high as 2–3 mg mL−1, and the mixtures formed physical gels above 1.8 mg mL−1. Polybenzimidazoles (PBIs, figure ) also individually and effectively dispersed both SWCNTs [Citation119] and multi-walled carbon nanotubes (MWCNTs) [Citation59] in organic solvents, such as DMAc and NMP, via a polymer wrapping mechanism. PBI-wrapped CNTs were obtained after filtering the dispersion and successive washing with DMAc to remove the unbound PBIs in the solution.
Figure 13. Chemical structure of PBI.
Wrapping of CNTs by PBIs was so effective that the re-dispersion of the wrapped CNTs was possible (figure ). When using PBI, the wrapping thickness was estimated to be ∼1 nm by dividing the total volume of the PBI by the surface area of the CNTs [Citation59, Citation60]. Similar stable wrapping was confirmed in polyamide [Citation120].
2.3. Non-aromatic polymers
Non-aromatic polymers, such as commercially available poly(vinyl alcohol) [Citation121] and poly(vinylpyrrolidone) [Citation122], have been reported to disperse CNTs via the wrapping mechanism. Baskaran et al successfully prepared a stable dispersion of MWCNTs in organic solvents with the aid of polybutadiene, polyisoprene, poly(methyl methacrylate) (PMMA) or poly(ethylene oxide) [Citation123]. They pointed out the importance of the CH–π interaction for the dispersion of these non-aromatic polymers. Biological molecules, including peptides [Citation124–Citation129] and proteins [Citation130–Citation132], are also important CNT dispersants in which the hydrophobic domains play a significant role in the interaction [Citation132]. Other biopolymers, such as chitosan [Citation133] and gelatin [Citation134], also wrap the CNTs and assist their dispersion. Significant examples of the wrapping by biological polymers are the helical wrapping of CNTs by amylose [Citation135, Citation136] and β-1,3-glucans [Citation137, Citation138] to render the hybrid water dispersible in which the multi-point OH−π interaction is considered to contribute to the wrapping. Among the biopolymer dispersants, carboxymethyl cellulose sodium salt (CMC) is known to be one of the most effective dispersants for CNTs, and stable individual isolation of SWCNTs was achieved in high concentrations [Citation139]. We utilized the stable isolation of SWCNTs by CMC for in situ spectroelectrochemical studies of the SWCNTs in which a stable photoluminescence (PL) of SWCNTs is necessary upon potential cycling [Citation140–Citation143].
In addition, these biopolymer-wrapped CNTs have often been used for the application of cell or tissue culturing since the fibrous 2D or 3D network structure formed by the CNTs resembles native extracellular matrices. In these applications, polymer wrapping using chitosan, gelatin, collagen and CMC served to weaken the potential toxicity and to reinforce the mechanical toughness of the scaffold [Citation144–Citation147].
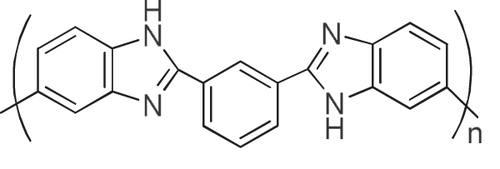
Another interesting example was reported by Naito et al [Citation148]. They developed poly(dialkylsilane)-wrapped SWCNTs through a strong CH−π interaction between the alkyl side chains and the SWCNT surfaces. In another example, the CH–π interaction between polyethylene (PE) and highly ordered graphitic surfaces of the CNTs induced the crystallization PE [Citation149]. As a result, formation of the ordered shish kebab structure was observed (figure ) [Citation150, Citation151].
Figure 14. (a) SEM image of MWCNTs decorated by disc-shaped PE single crystals. (b) TEM image of enlarged PE/MWCNT shish kebab structure. (c) Schematic representation of the PE/CNT shish kebab structure. The PE forms folded lamellar single crystals on the CNT surface with polymer chains perpendicular to the lamellae. Reproduced with permission from C Y Li et al Adv. Mater. 17 1198. Copyright 2005 John Wiley and Sons.
2.4. Cationic polymers
Cationic polymers, such as poly(diallyldimethylammonium chloride) (PDDA) [Citation152, Citation153], poly(allylamine hydrochloride) [Citation154] and PANI, were often used for the non-covalent CNT wrapping. For PDDA, Yang et al pointed out the role of the π−π interaction [Citation152]. They excluded a possible electrostatic interaction between the positively charged PDDA and COO− groups of the CNTs by using unoxidized CNTs (oxygen concentration <3%). Generally speaking, we need to recognize that pristine CNTs are not always free from the oxygen functional groups, and it is rather hard to deny the possibility of the ionic interaction between the positively charged polymer and the pristine CNTs. Some commercially available CNTs are covered with amorphous carbon that contains groups. Therefore, characterizing and reporting the purity of CNTs is crucial for many experiments.
2.5. Block polymers
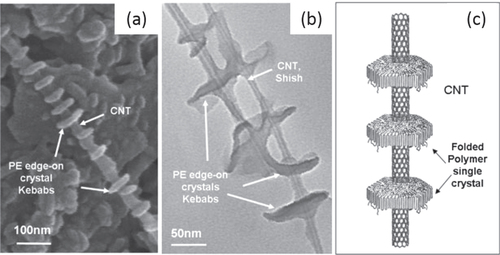
It is also believed that amphiphilicity of the polymers may contribute to the dispersion of the CNTs through a micelle-encapsulation mechanism. Kang and Taton found that the micelle formation in a DMF solution of polystyrene-b-poly(acrylic acid) (PS-PAA, figure ) [Citation155] induced by water addition enabled the dispersion of SWCNTs. A wide range of block copolymers were reported to disperse CNTs through the micelle encapsulation mechanism in which either polystyrene (PS) [Citation155–Citation163] or polyethylene oxide (PEO) [Citation158, Citation159, Citation164, Citation165] units were introduced as the blocks in most of the cases (figure (a)). Shin et al found by TEM analysis that in the case of a PS-b-poly(4-vinyl pyridine) (P4VP) block copolymer as the dispersant, PS domains were exposed to the outer surfaces in a non-polar toluene solution, while a P4VP domain was located to an outer surface in the polar ethanol solution (figures (b)–(d)) [Citation161]. This observation provided a strong indication of the mechanism of the micelle encapsulation of CNTs using the block copolymers.
Figure 15. (a) Examples of the chemical structures of the block copolymer-based dispersants. (b) TEM bright-field image of SWCNTs dispersed with PS-P4VP. Micelles are located between two nanotubes, by indicated arrows, implying a possible de-bundling of SWCNTs by micelles. (c), (d) Schematic model of the nanostructure of SWCNTs and block copolymer. PS and P4VP are selectively adsorbed on the surface of nanotubes in (c) toluene and (d) ethanol, respectively. Parts (b)–(d) reproduced with permission from H-i Shin et al 2005 Macromol. Rapid Commun. 26 1451. Copyright 2005 John Wiley and Sons.
2.6. Pendant polymers
Even if the backbone of the polymers only possess a weak interaction on the surfaces of the CNTs, the tailorable design of the polymers enables stable wrapping by incorporating the pendant moieties having a strong affinity to the CNT surfaces in the polymer side chains. This concept was pioneered by Petrov et al using pyrene as the pendant moiety [Citation166]. In this approach, polycyclic aromatic moieties, such as pyrene [Citation167–Citation191] and porphyrin [Citation192], were often introduced [Citation166, Citation193–Citation197] since they have been proved to show an effective non-covalent interaction with the CNT surfaces [Citation167–Citation191, Citation198]. As a matter of fact, we revealed that the pyrene moiety acted as a more efficient functional group compared to the naphthyl and phenyl groups for the dispersion [Citation167]. While the monomeric dispersants having a pyrene moiety precipitated CNTs upon heating at around 50 °C, we found that the polymers containing pyrenes in the side chain showed a higher stability and no precipitation up to 95 °C [Citation194]. This result clearly indicates the advantage of a polymer-based dispersant when compared to the monomeric one.
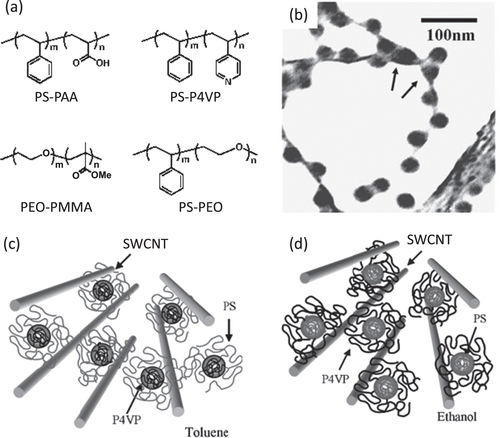
As for the effective design of the pendant-type dispersants, in 2006, Bahun et al reported the effect of the pendant sequence between the random versus block and found a limited solubility for the randomly labeled polymers, while a higher solubility was observed for the block copolymer with one pyrene block (figure (a)) [Citation196]. The result was reasonable because for the block copolymer, the other domain can be free to extend into the solution to achieve high solvation. They also revealed that a much longer pyrene block resulted in a decrease in solubility (figure (b)) [Citation196]. In 2008, it was reported that only one pyrene unit in the end of the polymer chain was long enough to disperse CNTs [Citation199–Citation201]. Essentially, a multipoint interaction is required for the formation of a stable polymer wrapping, and a more detailed study in terms of the wrapping stability is required in this approach.
Figure 16. Schematic diagram of nanotube interactions with (a) a pyrene-containing random copolymer (left) versus a block copolymer (right) and (b) short pyrene-functionalized blocks (left) and long pyrene-functionalized blocks (right) in block copolymers. Reproduced with permission from G J Bahun et al 2006 J. Polym. Sci., A: Polym. Chem 44 1941. Copyright 2006 John Wiley and Sons.
Polypeptides with aromatic side chains were also used to disperse CNTs through the π−π interactions. In this regard, polypeptides containing tryptophan [Citation202] and phenylalanine [Citation203] are known to provide favorable stabilizations.
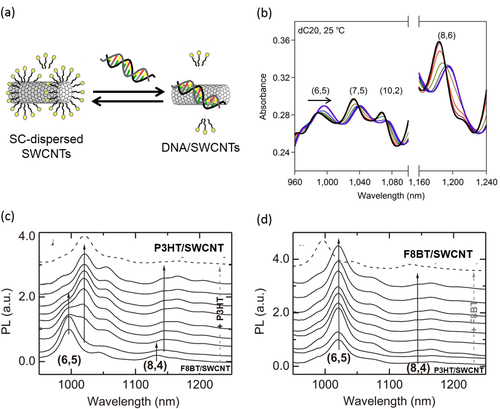
Individual wrapping of SWCNTs using double-strand DNA (dsDNA) and single-strand DNA (ssDNA) in an aqueous system was reported by our group [Citation204] and Zheng et al [Citation205], respectively. In these cases, the aromatic nucleic-acid base in DNA are also regarded as the pendant moiety used for the wrapping of CNTs [Citation206–Citation209] in which the stacking of the nucleic-acid base on the SWCNT surfaces leaving highly charged phosphate backbones exposed to water has been proposed both experimentally [Citation206–Citation208, Citation210] and theoretically (figure (a)) [Citation209, Citation211]. As a matter of fact, dissolution of the CNTs is highly sequence-dependent, and poly-d(T) and d(GT)10-45 provides the highest concentration of individual SWCNT aqueous solutions [Citation205, Citation212]. It was reported that the dispersion efficiency is so high that it exhibits a lyotropic LC phase in the high concentration region [Citation213]. The thermodynamic stability of the DNA wrapping on the CNTs was proved by our group using the gel permeation chromatography (GPC) technique. After removing the unbound DNA in a solution by the GPC technique, no peak attributed to the free DNA was detected even after 1 month, obviously indicating the absence of the detachment of the DNA from the CNT surfaces (figure (b)) [Citation55]. Due to the unique combination between biological materials and nano-carbon materials, together with the stable wrapping, a wide range of studies have been carried out for the DNA-wrapped CNTs, such as the conformation transition monitoring of DNA [Citation214], redox sensing of glucose and hydrogen peroxide [Citation215], hybridization detection between ssDNA and their complimentary DNA [Citation56] and uptake estimation of DNA/SWCNTs into a cell [Citation216, Citation217].
Figure 17. (a) A schematic model of DNA wrapped on SWCNTs. Color coding: orange, thymine; green, adenine; yellow ribbons, backbones. (b) Size exclusion chromatograms of SWCNTs dispersed by dsDNA (left) and the fraction re-injected after 1 month of the separation (right). The chromatograms were measured at 260 nm [Citation55]. Part (a) reprinted by permission from Macmillan Publishers Ltd: X Tu et al 2009 Nature 460 250, copyright 2009.
![Figure 17. (a) A schematic model of DNA wrapped on SWCNTs. Color coding: orange, thymine; green, adenine; yellow ribbons, backbones. (b) Size exclusion chromatograms of SWCNTs dispersed by dsDNA (left) and the fraction re-injected after 1 month of the separation (right). The chromatograms were measured at 260 nm [Citation55]. Part (a) reprinted by permission from Macmillan Publishers Ltd: X Tu et al 2009 Nature 460 250, copyright 2009.](/cms/asset/3ba7e671-13cc-4e9b-a973-940825d6df2d/tsta_a_11661258_f0017_oc.jpg)
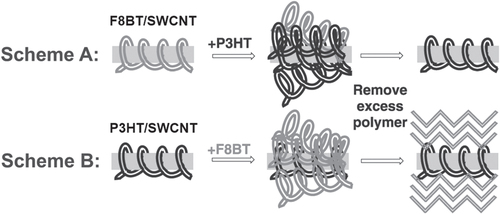
It is important to note that the degree of the stability depends on the polymer structure; thereby, in some cases, exchange of the wrapped polymer with the other polymer dispersant is possible. Sprafke et al demonstrated the correlation between the length of the porphyrin oligomers and the binding strength and found that the longer oligomers are able to replace the shorter ones and that the replacement was attributed to an increasing binding affinity with the oligomer length [Citation219]. Chen et al demonstrated competitive binding between PFO and PFO-BT and found that the SWCNTs were preferentially functionalized by PFO-BT [Citation220]. In 2013, Stranks et al found that PFO-BT was replaced by P3HT in the solution (figure ) [Citation58]. This fact also suggests that the polymer wrapping can be realized also by the exchange of the surfactant-dispersed CNTs. Indeed, Jeng et al reported the wrapping of DNA by adding DNA to the solution of SC dispersed SWCNTs [Citation56]. Such an exchanging procedure is advantageous to avoid damage of the polymers by the dispersion process, such as sonication.
Figure 18. Scheme A shows excess P3HT being added to F8BT/SWCNT. Three days after the addition of excess polymer, any unbound polymer is removed to give P3HT/SWCNT. In Scheme B, the addition of F8BT in P3HT/SWCNT gave P3HT/SWCNT. Reproduced with permission from S D Stranks et al 2013 Adv. Mater. 25 4365. Copyright 2013 John Wiley and Sons.
3. Characterizations of polymer wrapping
The homogeneous dispersion of the CNTs using polymers as the dispersants is clear evidence of the wrapping of the polymer onto the surfaces of CNTs. However, analysis of further information, such as wrapping geometry, composition ratio and degree of interaction requires detailed characterizations.
The wrapping geometry can be visualized by microscopy techniques such as transmission electron microscopy (TEM), scanning electron microscopy (SEM) and atomic force microscopy (AFM). For instance, helical wrapping of the DNA on CNTs has been extensively studied, especially by TEM [Citation221, Citation222] and AFM [Citation223–Citation226]. In the case of the PBI wrapping of the CNTs, we successfully visualized the coating structures by SEM based on the difference in the efficiency of the electron scattering from the polymer and CNTs in which non-coated bare islands were also observed [Citation227]. However, since only a limited area is observed in these techniques, and the sample structure sometimes depends on the preparation conditions of the specimen, these microscopic observations need careful interpretation. Generally speaking, it is highly recommended that some spectroscopic evidence, such as absorption or fluorescent spectroscopy, is required to support the microscopic information.
Absorption spectroscopy, especially in the NIR region, provides useful information, including the dispersion degree of the SWCNTs [Citation228], the degree of wrapping [Citation56] and the replacement of the dispersants [Citation58, Citation229], since SWCNTs act as the pigment that is sensitive to the surrounding environment. We found that the addition of ssDNA to a SC-dispersed SWCNT solution led to the clear shifts in the absorption spectra in the NIR region with an isosbestic point due to the thermodynamic exchange from SC to ssDNA (figure ). This finding allowed us to estimate the ΔT and ΔS values involved in the exchange reaction [Citation229].
Figure 19. (a) A schematic drawing of the exchange reaction between SC-dissolved SWCNTs (left) and ssDNA/SWCNTs (right). (b) Absorption spectra of the SWCNT in the mixed solution of SC containing ssDNA of 0 (black), 0.0625 (red), 0.156 (orange), 0.313 (yellow), 0.469 (green), 0.938 (blue) and 15.6 μM (purple) at 25 °C. Isosbestic points were observed in the spectral changes. (c), (d) PL spectra of (c) F8BT/SWCNT and (d) P3HT/SWCNT in a chloroform solution excited at 580 nm with an increasing amount of excess (c) P3HT and (d) F8BT and measured 10 days after the addition of excess polymer. Part (b) reprinted by permission from Macmillan Publishers Ltd: Y Kato et al 2012 Sci. Rep. 2 733, copyright 2012. Parts (c) and (d) reproduced with permission from S D Stranks et al 2013 Adv. Mater. 25 4365. Copyright 2013 John Wiley and Sons.
In Raman spectroscopy, the wrapped polymers often lead to a shift in the D∗ band, the second-order overtone of the D band that is sensitive to the strain or stress applied to the CNTs from the surrounding media [Citation230–Citation233]. Indeed, the wrapping of PBI on the SWCNTs resulted in the upshifting of the spectrum by 16 cm−1 [Citation119]. When the electronic interaction was present, the G-band shift and/or the RBM shift were often observed due to the softening or hardening of the tubes [Citation234].
Fluorescence measurement of the polymer provides strong evidence for the wrapping since an effective energy or electron transfer from the wrapped polymer to the CNTs leads to fluorescence quenching [Citation235–Citation237] when the unbound fluorophore in the bulk solution is successfully removed. In the case of the electron transfer, not only metallic CNTs but also s-SWCNTs can serve as the fluorescent quencher when the LUMO of the s-SWCNTs is lower than the LUMO of the fluorophore [Citation238]. Tezuka et al prepared P3HT-wrapped SWCNTs without containing the unbound P3HT and revealed that the short-lived singlet excited state relaxes to yield the exciplex state with the SWCNTs and then rapidly decays to the ground state [Citation239]. On the other hand, not only the excited-state interaction but also the ground-state interaction provides useful information. When SWCNTs are individually dispersed, the peak shifts of the SWCNT fluorescence are observed due to the energy stabilization through the interaction between the polymer and SWCNTs [Citation240].
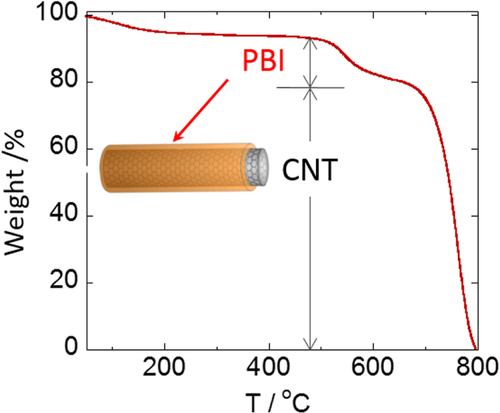
Characterizations of the polymer-wrapped CNTs after vigorous washing to remove unbound polymer by filtration support information on the average structures. Based on the thermogravimetric analysis (TGA), it is possible to evaluate the composition ratios between the wrapped-polymers and CNTs [Citation59]. For instance, the PBI-wrapped CNTs exhibit a two-step weight reduction at around 520 °C and 700 °C, corresponding to the thermal degradation of the wrapped PBI and CNTs, respectively, and the composition ratio was successfully estimated (figure ) [Citation59].
Figure 20. TGA curve of the PBI/CNT.
4. Functions of polymer-wrapped CNTs
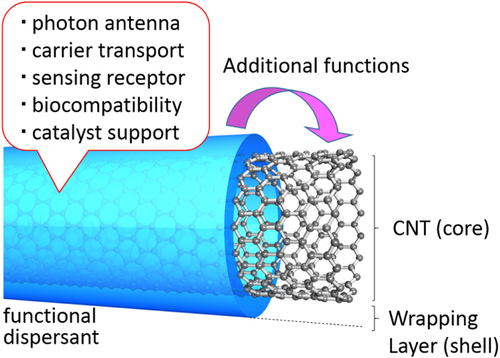
One of the advantages of polymer wrapping is the synergetic functionalization of CNTs by combining the functions of the polymers. For this target, the strategic design of polymers for wrapping is required. When the wrapped polymer possesses a sufficient affinity to the surfaces of the CNTs, the polymers can remain wrapped even after the vigorous removal of the unbound polymers. As a result, the CNT surfaces are subjected to decoration by the polymer to provide a core–shell structure with an extremely thin polymer layer (figure ).
Figure 21. Concept and the role of the functional dispersant.
4.1. Polymer composites
One of the most promising applications of the CNTs is polymer composites in which the CNTs are embedded in the polymer matrices as a filler material to improve the electron conductivity or mechanical strength of the matrices. In order to realize the effective reinforcement using CNTs, a homogeneous dispersion is always the primary requirement [Citation25–Citation30]. To prepare the composite films with a homogeneous dispersion of the CNTs, several approaches were proposed, including (1) melt mixing, (2) in situ polymerization and (3) solution blending using either pristine or chemically-modified (oxidized or grafted) CNTs [Citation25–Citation30]. For this purpose, CNTs wrapped by polymers may also serve to increase the miscibility of the matrix polymers. Yan et al reported the preparation of a PS-based composite using SWCNTs wrapped by a pyrene-terminated PS in a chloroform solution and found an improvement in the dispersion efficiency compared to the composites prepared in the absence of the pyrene-terminated PS [Citation241]. In this approach, unbound dispersant also remained in the solution, and such a residual possibly contaminates the matrix, depending on the amount of the unbound polymers. On the other hand, if the unbound polymer is removed prior to the composition, well-defined composites can be prepared with a minimum side effect.
In our group, MWCNTs wrapped by a newly developed polybenzoxazole (PBO) precursor (polyhydroxyphenyl amide) was used to blend with the PBO precursor [Citation120]. This two-step blending (figure ) showed a better homogeneity than the direct blending in the solution since the pristine MWCNTs were hard to disperse, especially in the highly concentrated solution used for film preparation, which is typically over 10 wt% due to their high viscosity. On the other hand, we found that the polymer-wrapped MWCNTs can disperse even in a highly viscous polymer solution without applying any significant shear force, such as sonication, due to the good miscibility of the matrix with the wrapping polymer. Since this approach required a milder condition compared to the reinforcement with the oxidized CNTs involving the severe cutting of the CNTs, effective reinforcements are expected.
Figure 22. Schematic illustration showing the advantage of the polymer-wrapped CNTs for the preparation of a PBO/CNT composite [Citation120].
![Figure 22. Schematic illustration showing the advantage of the polymer-wrapped CNTs for the preparation of a PBO/CNT composite [Citation120].](/cms/asset/66b3a6d6-fbf9-4d4f-810f-6a8727945e93/tsta_a_11661258_f0022_oc.jpg)
4.2. Photovoltaic and optoelectronic applications
When the wrapping polymers act as the pigment, light-harvesting systems using the CNTs as an acceptor can be fabricated [Citation242]. The unique charge transport features of the CNTs provide an efficient percolation network with a highly efficient exciton dissociation in polymeric bulk heterojunctions with the polymer acting as the donor and the CNT as the acceptor. Based on this concept, poly(3-alkylthiophenes) have been widely used [Citation110, Citation239]. In 2005, poly(3-octylthiophene) (P3OT) was reported to wrap the SWCNTs and improved the photovoltaic behavior by the photo-induced electron transfer at the P3OT/SWCNT interfaces [Citation243]. The combination of poly(3-alkylthiophenes) and s-SWCNTs forms a heterojunction with a type-II band alignment in which the HOMO and LUMO of the donor are higher than those of the acceptor (figure ) [Citation244]. This band structure leads to exciton dissociation at the interface with an ultrafast charge transfer from the P3HT to the s-SWCNTs [Citation244–Citation247], and the s-SWCNTs can act as efficient acceptors at the interface [Citation244]. The time-resolved microwave conductivity measurements revealed that the photoexcitation of P3HT results in long-lived carriers due to the efficient spatial separation at the SWCNT–P3HT interface, which readily move electrons away to avoid recombination [Citation248]. Another advantage is that organization of P3HT induced by SWCNTs [Citation249] leads to the improvement of the exciton diffusion as well as the charge mobility in a P3HT/SWCNT composite [Citation250]. It is important to note that the key progress in this field is the developments of the extraction technology of the s-SWCNTs, such as DGU [Citation251] and PFO wrapping [Citation88], since the contamination of m-SWCNTs in the active layer have a detrimental impact on the photocurrent [Citation245], and theoretical studies have predicted that the band alignment of the P3HT/m-SWCNT is unfavorable for charge separation [Citation244]. By utilizing s-SWCNTs, Dabera et al revealed the ground-state electron transfer from s-SWCNT to P3HT in the P3HT-wrapped s-SWCNTs, which facilitated the hole transportation property of the s-SWCNTs. The hybrid was used as hole transport layers for bulk heterojunction organic photovoltaics, and the device showed the highest power conversion efficiency (PCE = 7.6%) [Citation252].
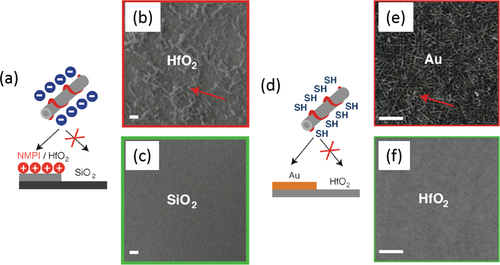
Figure 23. (a) Selective deposition of CNTs based on electrostatic directed assembly. (b), (c) SEM images of (b) HfO2 and the (c) SiO2 area of the patterned substrate. Scale bars = 1 μm. (d) Selective deposition of CNT based on the metal-ligand directed assembly. (e), (f) SEM images of (e) Au and the (f) HfO2 area of the patterned substrate. Scale bars = 100 nm. Reprinted with permission from J M Lobez and A Afzali 2013 Chem. Mater. 25 3662. Copyright 2013 American Chemical Society.
Nicholas et al developed a unique polymer-wrapping system; namely two different semiconducting polymers, P3HT and PFO-BT, are sequentially wrapped SWCNTs [Citation247]. Due to the difference in the band levels, when the PFO-BT is the inner layer, it acts as a barrier layer, preventing holes created in the P3HT from recombining with electrons in the SWCNTs (figure (b)). By incorporating an excess P3HT in order to allow the hole to move away from the interface [Citation58, Citation247], such structures could be integrated into OPVs, enabling the SWCNTs to be used as electron transporters [Citation253].
4.3. Sensors
The high mobility of charge carriers in s-SWCNTs coupled with their high surface area makes them ideal candidates for sensor applications [Citation254]. When the wrapping polymer possesses a stronger affinity for the target analytes, highly selective amperometric sensors can be fabricated [Citation255]. Staii et al used s-SWCNTs wrapped by ssDNA in which the DNA sequences were chosen to have a specific binding affinity for a series of analytes, including methanol, propionic acid, trimethylamine (TMA), dinitrotoluene (DNT) and dimethyl methylphosphonate (DMMP) [Citation256]. The introduction of ssDNA significantly enhanced the current signal to as high as 20–30% versus 0.1% for the bare control CNT device with the same exposure dose [Citation257]. Pang et al used a cationic polythiophene as the wrapping polymer, providing the binding sites for glucose oxidase and achieving a highly sensitive amperometric biosensor for glucose [Citation258]. Polymer-wrapped CNTs can provide a better biocompatibility compared to the CNTs having a bare surface. For example, higher cell viability was reported for the amylose-wrapped SWCNTs than for the non-wrapped SWCNTs [Citation259], which is advantageous for biosensing applications. In addition, wrapped polymers were also used to incorporate the receptors for sensing. One of the examples is the incorporation of a trinitrotoluene (TNT)-binding peptide into the wrapping layer for the TNT sensing [Citation260]. Tam and Hieu proposed an incorporation of an antibody via electropolymerization of pyrrole in the presence of the antibody, resulting in the formation of PPy-wrapped CNTs containing an antibody [Citation261]. The hybrid functioned as an immune sensor to explore the interactions between the antibody and antigen based on conductivity measurements.
Not only biosensors but also gas sensing are promising applications of polymer-wrapped CNTs. Wang et al wrapped CNTs by a hexafluoroisopropanol functionalized thiophene (HFIP-PT). In this hybrid, the HFIP unit was chosen due to the strong hydrogen binding with phosphate esters, which are common structures in many chemical warfare agents, including sarin gas [Citation262]. A FET-based sensor employing polyethyleneimine (PEI)-wrapped CNTs was demonstrated by Qi et al and was highly sensitive to NO2, while the Nafion-coated FET was selective to NH3 [Citation263]. Lobez and Afzali proposed the use of side-chain functionalized polymers for the selective deposition on the patterned substrate in which the selectivity was obtained based on the electrostatic interactions or metal-ligand interaction between the surfaces of CNTs and the substrates (figure ) [Citation264]. Thiophene-based polymers having phosphonic acid and thiol groups were used to deposit onto the HfO2 and Au surface selectively, respectively.
4.4. Biotechnology
CNTs also attract much attention in the field of biotechnology [Citation265], including biomedicine [Citation266], biomedical imaging [Citation267], biomedical engineering [Citation268], tissue engineering [Citation269], neurobiology [Citation270], drug discovery [Citation271], drug delivery [Citation272], cancer therapy [Citation273], gene therapy [Citation274] and cell therapy [Citation275] due to their characteristic nano-size as well as their unique optical properties showing a strong light absorption and emission in the NIR region [Citation276]. NIR light is quite useful since most of the components in the body are transparent in the NIR region (figure (a)); thus, it is possible to monitor and irradiate CNTs from the outside of the body [Citation276].
Figure 24. (a) SWCNT fluoresce (blue) in the NIR region. Blood (red) and water (black) absorbance occurs in the visible and NIR region, respectively. The gap in tissue absorbance, which occurs in the NIR region, ensures minimal tissue interference with SWCNT PL emission. (b) A schematic drawing showing various approaches for CNT-based drug delivery and cancer therapies based on PEG-PL as the platform. Part (a) reproduced with permission from A A Boghossian et al 2011 Chem. Sus. Chem. 4 848. Copyright 2011 John Wiley and Sons. Part (b) reproduced based on [Citation300].
![Figure 24. (a) SWCNT fluoresce (blue) in the NIR region. Blood (red) and water (black) absorbance occurs in the visible and NIR region, respectively. The gap in tissue absorbance, which occurs in the NIR region, ensures minimal tissue interference with SWCNT PL emission. (b) A schematic drawing showing various approaches for CNT-based drug delivery and cancer therapies based on PEG-PL as the platform. Part (a) reproduced with permission from A A Boghossian et al 2011 Chem. Sus. Chem. 4 848. Copyright 2011 John Wiley and Sons. Part (b) reproduced based on [Citation300].](/cms/asset/296fb71f-c77b-4331-b105-74fb754b09e1/tsta_a_11661258_f0024_oc.jpg)
However, many toxicological screenings for CNTs revealed the potential risks of the toxicity of CNTs mainly due to the asbestos-like shape of the CNTs [Citation277–Citation279]. Although the degree of toxicity varies from report to report, probably originating from the difference in the length, diameter, metal impurity, dose, type of CNTs, etc [Citation280–Citation283], it is widely recognized that well-functionalized CNTs by biocompatible wrapping exhibit a remarkably reduced toxicity in vivo [Citation284–Citation286]. Along this line, one of the most studied molecules for improving long blood circulation of CNTs through a covalent or non-covalent fashion is polyethylene glycol (PEG) [Citation287–Citation293]. The surface coverage with PEG lowers the immunogenicity of the CNTs and prevents their non-specific phagocytosis by the reticuloendothelial system (RES); thereby, their half-life in blood circulation is prolonged [Citation285, Citation286]. Once the CNTs are stably wrapped with biocompatible materials, they are extremely attractive for biomedicine due to their incredible ability for passing biological barriers across the cytoplasmic and nuclear membrane without generating an immunogenic response [Citation294, Citation295]. Many researchers have focused on the potential of CNTs for drug delivery, which might be attributed to their exclusive physicochemical features [Citation296, Citation297].
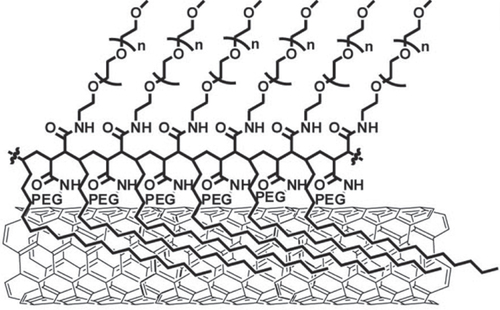
One of the most successful examples in this field is the series of excellent works explored by Liu et al [Citation298, Citation299]. They wrapped SWCNTs with a PEG-functionalized phospholipid (PEG-PL) and successfully achieved a long blood circulation [Citation285, Citation300]. By taking advantage of the unique optical properties of non-oxidized SWCNTs, they successfully realized NIR- [Citation301–Citation303] and Raman [Citation304] imaging of the tumor [Citation302, Citation305] and vessels [Citation303, Citation306, Citation307] in vivo using PEG-PL/SWCNT. Furthermore, PEG-PL/SWCNT was used as the platform for 1) ligand functionalization for targeting [Citation308], 2) a photo-thermal molecular heater to treat cancer cells and 3) labeling for radio-active imaging (figure (b)) [Citation309]. Importantly, SWCNTs wrapped by polymeric PEG synthesized based on the PEGylation of poly(maleic anhydride-alt-1-octadecene) (figure ) showed a much longer blood circulation half-life than that of PEG-PL/SWCNT due to the pronounced PEG loading [Citation300, Citation310]. While the packing density of PEG coatings immobilized on PEG-PL/SWCNT with single anchoring points is limited by steric hindrance, polymeric PEG allows continuous binding of the polymer onto the SWCNT surface, yielding a highly dense PEG coating.
Figure 25. Structure of SWCNTs modified by polymeric PEG. Reprinted with permission from A J Andersen et al 2013 ACS Nano 7 1108. Copyright 2013 American Chemical Society.
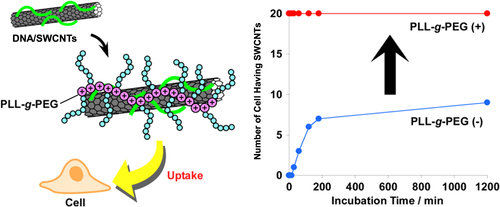
In our group, negatively charged ssDNA-wrapped SWCNTs were further hybridized by positively charged poly(L-lysine) grafted by polyethylene glycol (PLL-g-PEG) (figure ). We found that the obtained ternary hybrid exhibited a dramatic enhancement in the cell uptake efficiency compared to that of the SWCNT wrapped by ssDNA without PLL-g-PEG [Citation57]. In this system, since unbound DNA can be removed prior to the hybridization due to the stable wrapping of ssDNA on SWCNTs, the contamination of the composite without containing SWCNTs was avoided.
Figure 26. (left) Schematic drawing of PEGylation of DNA/SWCNTs with PLL-g-PEG based on the electrostatic interaction. (right) Plots of the number of cells containing SWCNTs as a function of the incubation time. Dramatic enhancement of the cell uptake efficiency is achieved after the PEGylation. Reproduced from T Fujigaya et al 2011 Nanoscale 3 4365. Copyright 2011 Royal Society of Chemistry.
4.5. Conducting support for the electrocatalyst
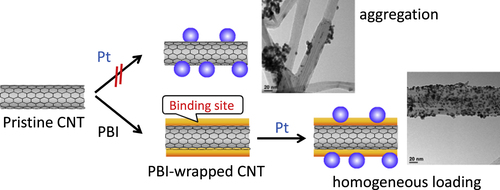
CNTs are recognized as an ideal supporting material for catalysts used in electrocatalysts, especially for fuel cells due to their higher electrical conductivities [Citation311, Citation312], lower impurities [Citation313] and higher electrochemical durability [Citation314–Citation318] compared to conventional supporting materials such as carbon blacks (CBs). However, due to the lack of binding sites, such as –COOH and –OH groups, loading of the metal catalyst onto the surfaces of pristine CNTs has been rather difficult. Therefore, the strong oxidation of the CNTs was carried out to introduce the hydrophilic groups [Citation311, Citation313, Citation316, Citation317, Citation319–Citation325]. However, since the oxidation is known to severely damage the graphitic structure of CNTs and damage the excellent electrochemical stability, a novel methodology to load the catalyst onto the non-oxidized CNTs has been required to utilize the intrinsic electrochemical stability of the pristine CNTs. In this issue, the introduction of binding sites by polymer wrapping of the pristine CNTs offers a promising solution. Until now, CNTs wrapped by poly(allylamine hydrochloride) [Citation326, Citation327], chitosan [Citation328], PANI [Citation329–Citation331], PDDA [Citation332, Citation333] and PPy [Citation331, Citation334, Citation335] were successfully used to anchor metal nanoparticles onto the surfaces of the pristine CNTs. In our group, PBI-wrapped pristine CNTs were used for the loading of Pt-nanoparticles (figure ) [Citation59, Citation60] in which imidazole units in PBI acted as the binding sites for Pt ions through a coordination mechanism, and quantitative loading of the Pt was achieved by a conventional polyol method using an ethylene glycol aqueous solution, H2PtCl6, as the reducing agent and Pt salt, respectively. The thus-obtained composite (CNT/PBI/Pt) was employed as the electrocatalyst in a polymer electrolyte fuel cell (PEFC) for the first time. As a result of the durability tests using the cells, it was revealed that the fuel cell using PBI-wrapped CNTs showed a remarkable durability compared to the conventional cell using the CB as the supporting material. In this strategy, since PBIs are known to have an excellent proton conductivity after acid doping [Citation336], the wrapping layer of PBI also functioned to fabricate the proton conduction pathway along with CNT networks in the electrocatalyst.
Figure 27. Schematic drawing of the two different approaches for Pt growth, namely the direct growth (upper) and PBI-assisted growth (lower) method.
We developed a novel function in the wapping polymer for the purpose of the electrocatalyst application. In this approach, the transformation of the wrapped PBI into a nitrogen-containing (N-doped) graphitic structure by heating in the presence of a metal ion as a graphitization catalyst produced a catalyst layer for the oxygen reduction reaction (ORR) around the CNTs. Recently, N-doped graphitic nanostructures, such as N-doped carbon nanoshells [Citation337], N-doped CNTs [Citation338], N-doped graphite [Citation339, Citation340] and N-doped graphene [Citation341], have emerged as candidates for the cathode catalyst due to their potential ORR activity. In this application, incorporation of the electron conductivity is one of the key issues for the practical applications. Conversion of the wrapping polymers into the N-doped graphene may facilitate the effective electron delivery into all of the active sites on the surface of CNTs; therefore, a highly active ORR can be expected.
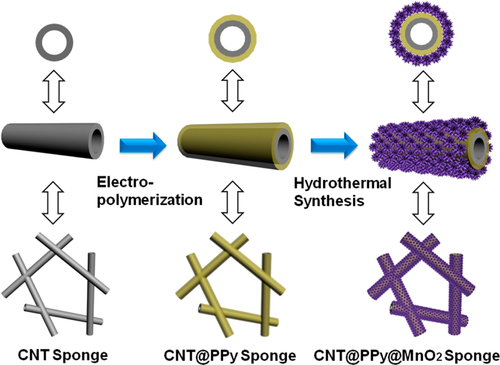
For supercapacitor applications, conducting polymers wrapped on CNTs serve to increase the specific capacitance [Citation342–Citation344]. In these examples, PPy and PANI were often used owing to the ease of wrapping via either chemical or electrochemical polymerization on the surfaces of the CNTs. The recent trend is the additional functionalization with a metal oxide, such as MnO2, on the polymer-wrapped CNTs to further increase the specific capacitance [Citation345]. A representative example demonstrated by Li et al is the coating of a CNT 3D network (CNT sponge) via electropolymerization of PPy, followed by the MnO2 loading by hydrothermal synthesis (figure ) [Citation346]. Such a ternary system showed the synergetic effect of the components and long cycling life stability [Citation346, Citation347].
Figure 28. Schematic for the coating of the CNT sponge by PPy and the successive loading of MnO2. Reprinted with permission from P Li et al 2014 ACS Appl. Mater. Interfaces 6 5228. Copyright 2014 American Chemical Society.
5. Summary
We have reviewed polymers that can help to disperse pristine CNTs in solvents via the polymer wrapping mechanism. A wide variety of polymers were found to non-covalently wrap CNTs. Wrapping of CNTs by dispersants based on π–π, CH–π and cation–π interactions is a typical mechanism of CNT wrapping. Recently, many polymer dispersants have been developed not only for dispersion of CNTs but also for adding new functions to CNTs. The concept of the functional dispersant is now widely recognized and utilized in many fields, including biotechnology, energy application, etc. The tailorable design of the polymers will further expand the functionality of the polymer-wrapped CNTs, and novel applications using the hybrid are expected.
Acknowledgments
The authors would like to acknowledge the funding support from Low-Carbon Research Network (LCnet); the Nanotechnology Platform Project (Molecules and Materials Synthesis) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan; the Center of Innovation Science and Technology based Radical Innovation and Entrepreneurship Program (COI Program); and the Advanced Low Carbon Technology Research and Development Program (ALCA) from the Japan Science and Technology Agency (JST).
References
- IijimaS 1991 Nature 354 56 10.1038/354056a0
- JorioADresselhausGDresselhausM S 2008 Carbon Nanotubes: Advanced Topics in the Synthesis, Structure, Properties and Applications Heidelberg Springer
- DykeC ATourJ M 2004 J. Phys. Chem. A 108 11151 10.1021/jp046274g
- BoulP J 1999 Chem. Phys. Lett. 310 367 10.1016/S0009-2614(99)00713-7
- LiuJ 1999 Chem. Phys. Lett. 303 125 10.1016/S0009-2614(99)00209-2
- AusmanK DPinerRLourieORuoffR SKorobovM 2000 J. Phys. Chem. B 104 8911 10.1021/jp002555m
- BahrJ LMickelsonE TBronikowskiM JSmalleyR ETourJ M 2001 Chem. Commun. 193 10.1039/b008042j
- FurtadoC AKimU JGutierrezH RPanLDickeyE CEklundP C 2004 J. Am. Chem. Soc. 126 6095 10.1021/ja039588a
- LandiB JRufH JWormanJ JRaffaelleR P 2004 J. Phys. Chem. B 108 17089 10.1021/jp047521j
- KimD SNepalDGeckelerK E 2005 Small 1 1117 10.1002/smll.200500167
- LiQKinlochI AWindleA H 2005 Chem. Commun. 3283 10.1039/b419039d
- GiordaniSBerginSNicolosiVLebedkinSBlauW JColemanJ N 2006 Phys. Status Solidi B 243 3058 10.1002/pssb.200669217
- NakashimaNFujigayaT 2007 Chem. Lett. 36 692 10.1246/cl.2007.692
- MurakamiHNakashimaN 2006 J. Nanosci. Nanotechnol. 6 16
- NakashimaN 2005 Int. J. Nanosci. 4 119 10.1142/S0219581X05002985
- RameshS 2004 J. Phys. Chem. B 108 8794 10.1021/jp036971t
- ShafferM S PFanXWindleA H 1998 Carbon 36 1603 10.1016/S0008-6223(98)00130-4
- DujardinEEbbesenT WKrishnanATreacyM M J 1998 Adv. Mater. 10 611 10.1002/(SICI)1521-4095(199805)10:8<611::AID-ADMA611>3.0.CO;2-8
- RaiP KPinnickR AParra-VasquezA N GDavisV ASchmidtH KHaugeR HSmalleyR EPasqualiM 2006 J. Am. Chem. Soc. 128 591 10.1021/ja055847f
- LiuJ 1998 Science 280 1253 10.1126/science.280.5367.1253
- KovtyukhovaN IMalloukT EPanLDickeyE C 2003 J. Am. Chem. Soc. 125 9761 10.1021/ja0344516
- ZhaoWSongCPehrssonP E 2002 J. Am. Chem. Soc. 124 12418 10.1021/ja027861n
- ChenJ 2001 J. Phys. Chem. B 105 2525 10.1021/jp002596i
- McKayS F 1964 J. Appl. Phys. 35 1992 10.1063/1.1713794
- ThostensonE TLiCChouT-W 2005 Compos. Sci. Technol. 65 491 10.1016/j.compscitech.2004.11.003
- ColemanJ NKhanUBlauW JGun’koY K 2006 Carbon 44 1624 10.1016/j.carbon.2006.02.038
- ColemanJ NKhanUGun’koY K 2006 Adv. Mater. 18 689 10.1002/adma.200501851
- Yerushalmi-RozenRSzleiferI 2006 Soft Matter 2 24 10.1039/b513344k
- MoniruzzamanMWineyK I 2006 Macromolecules 39 5194 10.1021/ma060733p
- TasisDTagmatarchisNBiancoAPratoM 2006 Chem. Rev. 106 1105 10.1021/cr050569o
- SkakalovaVKaiserA BDettlaff-WeglikowskaUHrncarikovaKRothS 2005 J. Phys. Chem. B 109 7174 10.1021/jp044741o
- SunY-PFuKLinYHuangW 2002 Acc. Chem. Res. 35 1096 10.1021/ar010160v
- NiyogiSHamonM AHuHZhaoBBhowmikPSenRItkisM EHaddonR C 2002 Acc. Chem. Res. 35 1105 10.1021/ar010155r
- HirschA 2002 Angew. Chem. Int. Edn 41 1853 10.1002/1521-3773(20020603)41:11<1853::AID-ANIE1853>3.0.CO;2-N
- BanerjeeSKahnM G CWongS S 2003 Chem. Eur. J. 9 1898 10.1002/chem.200204618
- BanerjeeSHemraj-BennyTWongS S 2005 Adv. Mater. 17 17 10.1002/adma.200401340
- BanerjeeSWongS S 2002 J. Phys. Chem. B 106 12144 10.1021/jp026304k
- HirschAVostrowskyO 2005 Top. Curr. Chem. 245 193
- BalasubramanianKBurghardM 2005 Small 1 180 10.1002/smll.200400118
- StranoM SMooreV CMillerM KAllenM JHarozE HKittrellCHaugeR HSmalleyR E 2003 J. Nanosci. Nanotechnol. 3 81 10.1166/jnn.2003.194
- RichardCBalavoineFSchultzPEbbesenT WMioskowskiC 2003 Science 300 775 10.1126/science.1080848
- DuesbergG SBurghardMMusterJPhilippGRothS 1998 Chem. Commun. 435 10.1039/a707465d
- BurghardMDuesbergGPhilippGMusterJRothS 1998 Adv. Mater. 10 584 10.1002/(SICI)1521-4095(199805)10:8<584::AID-ADMA584>3.0.CO;2-9
- IslamM FRojasEBergeyD MJohnsonA TYodhA G 2003 Nano Lett. 3 269 10.1021/nl025924u
- ParedesJ IBurghardM 2004 Langmuir 20 5149 10.1021/la049831z
- MooreV CStranoM SHarozE HHaugeR HSmalleyR ESchmidtJTalmonY 2003 Nano Lett. 3 1379 10.1021/nl034524j
- HoughL AIslamM FHammoudaBYodhA GHeineyP A 2006 Nano Lett. 6 313 10.1021/nl051871f
- IslamM FNobiliMYeFLubenskyT CYodhA G 2005 Phys. Rev. Lett. 95 148301 /1 10.1103/PhysRevLett.95.148301
- IshibashiANakashimaN 2006 Chem. Eur. J. 12 7595 10.1002/chem.200600326
- IshibashiANakashimaN 2006 Bull. Chem. Soc. Jpn. 79 357 10.1246/bcsj.79.357
- WenseleersWVlasov I I GoovaertsEObraztsovaE DLobachA SBouwenA 2004 Adv. Funct. Mater. 14 1105 10.1002/adfm.200400130
- KimYHongSJungSStranoM SChoiJBaikS 2006 J. Phys. Chem. B 110 1541 10.1021/jp055110c
- LisunovaM OLebovkaN IMelezhykO VBoikoY P 2006 J. Colloid Interface Sci. 299 740 10.1016/j.jcis.2006.03.012
- KeP C 2007 Phys. Chem. Chem. Phys. 9 439 10.1039/b611142d
- NoguchiYFujigayaTNiidomeYNakashimaN 2008 Chem. Phys. Lett. 455 249 10.1016/j.cplett.2008.02.089
- JengE SMollA ERoyA CGastalaJ BStranoM S 2006 Nano Lett. 6 371 10.1021/nl051829k
- FujigayaTYamamotoYKanoAMaruyamaANakashimaN 2011 Nanoscale 3 4352 10.1039/c1nr10635j
- StranksS DHabisreutingerS NDirksBNicholasR J 2013 Adv. Mater. 25 4365 10.1002/adma.201205250
- OkamotoMFujigayaTNakashimaN 2009 Small 5 735 10.1002/smll.200801742
- FujigayaTOkamotoMNakashimaN 2009 Carbon 47 3227 10.1016/j.carbon.2009.07.038
- IzardNKazaouiSHataKOkazakiTSaitoTIijimaSMinamiN 2008 Appl. Phys. Lett. 92 243112 10.1063/1.2939560
- BisriS ZGaoJDerenskyiVGomulyaWIezhokinIGordiichukPHerrmannALoiM A 2012 Adv. Mater. 24 6147 10.1002/adma.201202699
- RomeroD BCarrardMDe HeerWZuppiroliL 1996 Adv. Mater. 8 899 10.1002/adma.19960081105
- CurranS A 1998 Adv. Mater. 10 1091
- ColemanJ NCurranSDaltonA BDaveyA PMcCarthyBBlauWBarklieR C 1998 Phys. Rev. B 58 R7492 10.1103/PhysRevB.58.R7492
- AgoHPetritschKShafferM S PWindleA HFriendR H 1999 Adv. Mater. 11 1281 10.1002/(SICI)1521-4095(199910)11:15<1281::AID-ADMA1281>3.0.CO;2-6
- ColemanJ NCurranSDaltonA BDaveyA PMcCarthyBBlauWBarklieR C 1999 Synth. Metals 102 1174 10.1016/S0379-6779(98)01065-0
- DaltonA BByrneH JColemanJ NCurranSDaveyA PMcCarthyBBlauW 1999 Synth. Metals 102 1176 10.1016/S0379-6779(98)01067-4
- CurranS 1999 Synth. Metals 103 2559 10.1016/S0379-6779(98)00279-3
- AgoHShafferM S PGingerD SWindleA HFriendR H 2000 Phys. Rev. B 61 2286 10.1103/PhysRevB.61.2286
- McCarthyBDaltonA BColemanJ NByrneH JBernierPBlauW J 2001 Chem. Phys. Lett. 350 27 10.1016/S0009-2614(01)01184-8
- McCarthyB 2001 Nanotechnology 12 187 10.1088/0957-4484/12/3/301
- McCarthyBColemanJ NCzerwRDaltonA BCarrollD LBlauW J 2001 Synth. Metals 121 1225 10.1016/S0379-6779(00)00906-1
- McCarthyB 2002 J. Phys. Chem. B 106 2210 10.1021/jp013745f
- SteuermanD WStarANarizzanoRChoiHRiesR SNicoliniCStoddartJ FHeathJ R 2002 J. Phys. Chem. B 106 3124 10.1021/jp014326l
- StarAStoddartJ F 2002 Macromolecules 35 7516 10.1021/ma0204150
- StarALiuYGrantKRidvanLStoddartJ FSteuermanD WDiehlM RBoukaiAHeathJ R 2003 Macromolecules 36 553 10.1021/ma021417n
- DruryAMaierSRuetherMBlauW J 2003 J. Mater. Chem. 13 485 10.1039/b208439b
- YangCWohlgenanntMVardenyZ VBlauW JDaltonA BBaughmanRZakhidovA A 2003 Physica B: Condensed Matter 338 366 10.1016/j.physb.2003.08.022
- MulazziEPeregoRAarabHMihutLLefrantSFaulquesEWeryJ 2004 Phys. Rev. B 70 155206 /1 10.1103/PhysRevB.70.155206
- KeoghS MHeddermanT GRuetherM GLyngF MGreganEFarrellG FChambersGByrneH J 2005 J. Phys. Chem. B 109 5600 10.1021/jp044755u
- KeoghS MHeddermanT GLynchPFarrellG FByrneH J 2006 J. Phys. Chem. B 110 19369 10.1021/jp056321k
- ColemanJ NO’BrienD FMcCarthyBLahrBDruryABarklieR CBlauW JDaltonA B 2000 Chem. Commun. 2001 10.1039/b006164f
- ColemanJ N 2000 Adv. Mater. 12 213
- DaltonA BStephanCColemanJ NMcCarthyBAjayanP MLefrantSBernierPBlauW JByrneH J 2000 J. Phys. Chem. B 104 10012 10.1021/jp002857o
- in het PanhuisMMaitiADaltonABvan den NoortAColemanJ NMcCarthyBBlauW J 2003 J. Phys. Chem. B 107 478 10.1021/jp026470s
- ChenJLiuHWeimerW AHallsM DWaldeckD HWalkerG C 2002 J. Am. Chem. Soc. 124 9034 10.1021/ja026104m
- NishAHwangJ-YDoigJNicholasR J 2007 Nat. Nanotech. 2 640 10.1038/nnano.2007.290
- TanakaTLiuHFujiiSKatauraH 2011 Phys. Status Solidi PRL 5 301
- LiuHNishideDTanakaTKatauraH 2011 Nat. Commun. 2 309 10.1038/ncomms1313
- HiranoATanakaTKatauraH 2011 J. Phys. Chem. C 115 21723 10.1021/jp207786g
- TanakaTJinHMiyataYKatauraH 2008 Appl. Phys. Express 1 114001 10.1143/apex.1.114001
- TanakaT 2009 Nano Lett. 9 1497 10.1021/nl8034866
- ArnoldM SGreenA AHulvatJ FStuppS IHersamM C 2006 Nat. Nanotech. 1 60 10.1038/nnano.2006.52
- EngelMSmallJ PSteinerMFreitagMGreenA AHersamM CAvourisP 2008 ACS Nano 2 2445 10.1021/nn800708w
- HersamM C 2008 Nat. Nanotech. 3 387 10.1038/nnano.2008.135
- MistryK SLarsenB ABlackburnJ L 2013 ACS Nano 7 2231 10.1021/nn305336x
- OzawaHFujigayaTNiidomeYHottaNFujikiMNakashimaN 2011 J. Am. Chem. Soc. 133 2651 10.1021/ja109399f
- FukumaruTToshimitsuFFujigayaTNakashimaN 2014 Nanoscale 6 5879 10.1039/c4nr00809j
- AkazakiKToshimitsuFOzawaHFujigayaTNakashimaN 2012 J. Am. Chem. Soc. 134 12700 10.1021/ja304244g
- YiXOzawaHNakagawaGFujigayaTNakashimaNAsanoT 2011 Jpn. J. Appl. Phys. 50 098003 10.7567/jjap.50.098003
- OzawaHYiXFujigayaTNiidomeYAsanoTNakashimaN 2011 J. Am. Chem. Soc. 133 14771 10.1021/ja2055885
- OzawaHIdeNFujigayaTNiidomeYNakashimaN 2011 Chem. Eur. J. 17 13438 10.1002/chem.201101669
- OzawaHFujigayaTSongSSuhHNakashimaN 2011 Chem. Lett. 40 470 10.1246/cl.2011.470
- OzawaHFujigayaTNiidomeYNakashimaN 2011 Chem. Asian J. 6 3281 10.1002/asia.201100362
- BindlD JWuM-YPrehnF CArnoldM S 2010 Nano Lett. 11 455 10.1021/nl1031343
- BindlD JArnoldM S 2013 J. Phys. Chem. C 117 2390 10.1021/jp310983y
- BindlD JSheaM JArnoldM S 2013 Chem. Phys. 413 29 10.1016/j.chemphys.2012.08.001
- ToshimitsuFNakashimaN 2014 Nat. Commun. 5 5041 10.1038/ncomms6041
- CataldoSSalicePMennaEPignataroB 2012 Energy Environ. Sci. 5 5919 10.1039/c1ee02276h
- ZaminpaymaEMirabbaszadehK 2012 Polymer Composites 33 548 10.1002/pc.22182
- GoutamP JSinghD KGiriP KIyerP K 2011 J. Phys. Chem. B 115 919 10.1021/jp109900m
- PradhanBBatabyalS KPalA J 2006 J. Phys. Chem. B 110 8274 10.1021/jp060122z
- LeeH W 2011 Nat. Commun. 2 541 10.1038/ncomms1545
- OueinyCBerliozSPerrinF-X 2014 Prog. Polym. Sci. 39 707 10.1016/j.progpolymsci.2013.08.009
- ShigetaMKomatsuMNakashimaN 2006 Chem. Phys. Lett. 418 115 10.1016/j.cplett.2005.10.088
- YooJFujigayaTNakashimaN 2013 Nanoscale 5 7419 10.1039/c3nr01828h
- YooJOzawaHFujigayaTNakashimaN 2011 Nanoscale 3 2517 10.1039/c1nr10079c
- OkamotoMFujigayaTNakashimaN 2008 Adv. Funct. Mater. 18 1776 10.1002/adfm.200701257
- FukumaruTFujigayaTNakashimaN 2013 Macromolecules 46 4034 10.1021/ma4004117
- ZhangXLiuTSreekumarT VKumarSMooreV CHaugeR HSmalleyR E 2003 Nano Lett. 3 1285 10.1021/nl034336t
- O’ConnellM J 2001 Chem. Phys. Lett. 342 265
- BaskaranDMaysJ WBratcherM S 2005 Chem. Mater. 17 3389 10.1021/cm047866e
- DieckmannG R 2003 J. Am. Chem. Soc. 125 1770 10.1021/ja029084x
- ZorbasVOrtiz-AcevedoADaltonA BYoshidaM MDieckmannG RDraperR KBaughmanR HJose-YacamanMMusselmanI H 2004 J. Am. Chem. Soc. 126 7222 10.1021/ja049202b
- ZorbasVSmithA LXieHOrtiz-AcevedoADaltonA BDieckmannG RDraperR KBaughmanR HMusselmanI H 2005 J. Am. Chem. Soc. 127 12323 10.1021/ja050747v
- Ortiz-AcevedoAXieHZorbasVSampsonW MDaltonA BBaughmanR HDraperR KMusselmanI HDieckmannG R 2005 J. Am. Chem. Soc. 127 9512 10.1021/ja050507f
- XieHOrtiz-AcevedoAZorbasVBaughmanR HDraperR KMusselmanI HDaltonA BDieckmannG R 2005 J. Mater. Chem. 15 1734 10.1039/b413262a
- DaltonA B 2004 Adv. Funct. Mater. 14 1147 10.1002/adfm.200400190
- ErlangerB FChenB-XZhuMBrusL 2001 Nano Lett. 1 465 10.1021/nl015570r
- KarajanagiS SYangHAsuriPSellittoEDordickJ SKaneRS 2006 Langmuir 22 1392 10.1021/la0528201
- KarajanagiS SVertegelA AKaneR SDordickJ S 2004 Langmuir 20 11594 10.1021/la047994h
- TakahashiTLuculescuC RUchidaKIshiiTYajimaH 2005 Chem. Lett. 34 1516 10.1246/cl.2005.1516
- TakahashiTTsunodaKYajimaHIshiiT 2002 Chem. Lett. 31 690 10.1246/cl.2002.690
- StarASteuermanD WHeathJ RStoddartJ F 2002 Angew. Chem. Int. Edn 41 2508 10.1002/1521-3773(20020715)41:14<2508::AID-ANIE2508>3.0.CO;2-A
- KimO-KJeJBaldwinJ WKooiSPehrssonP EBuckleyL J 2003 J. Am. Chem. Soc. 125 4426 10.1021/ja029233b
- NumataMAsaiMKanekoKHasegawaTFujitaNKitadaYSakuraiKShinkaiS 2004 Chem. Lett. 33 232 10.1246/cl.2004.232
- NumataMAsaiMKanekoKBaeA-HHasegawaTSakuraiKShinkaiS 2005 J. Am. Chem. Soc. 127 5875 10.1021/ja044168m
- MinamiNKimYMiyashitaKKazaouiSNaliniB 2006 Appl. Phys. Lett. 88 093123 10.1063/1.2180870
- TanakaYHiranaYNiidomeYKatoKSaitoSNakashimaN 2009 Angew. Chem. Int. Edn 48 7655 10.1002/anie.200902468
- TanakaYNiidomeYNakashimaN 2009 Chem. Lett. 38 864 10.1246/cl.2009.864
- HiranaYTanakaYNiidomeYNakashimaN 2010 J. Am. Chem. Soc. 132 13072 10.1021/ja105980a
- ParkJ SHiranaYMouriSMiyauchiYNakashimaNMatsudaK 2012 J. Am. Chem. Soc. 134 14461 10.1021/ja304282j
- LiXFanYWatariF 2010 Biomed. Mater. 5 022001 10.1088/1748-6041/5/2/022001
- ChaoT-IXiangSChenC-SChinW-CNelsonA JWangCLuJ 2009 Biochem. Biophys. Res. Commun. 384 426 10.1016/j.bbrc.2009.04.157
- LovatVPantarottoDLagostenaLCacciariBGrandolfoMRighiMSpallutoGPratoMBalleriniL 2005 Nano Lett. 5 1107 10.1021/nl050637m
- SadaTFujigayaTNiidomeYNakazawaKNakashimaN 2011 ACS Nano 5 4414 10.1021/nn2012767
- NaitoMNobusawaKOnouchiHNakamuraMYasuiK-IIkedaAFujikiM 2008 J. Am. Chem. Soc. 130 16697 10.1021/ja806109z
- FukushimaTKosakaAIshimuraYYamamotoTTakigawaTIshiiNAidaT 2003 Science 300 2072 10.1126/science.1082289
- LiC YLiLCaiWKodjieS LTennetiK K 2005 Adv. Mater. 17 1198 10.1002/adma.200401977
- ZhengXXuQ 2010 J. Phys. Chem. B 114 9435 10.1021/jp103932b
- YangD QRochelteJ FSacherE 2005 J. Phys. Chem. B 109 4481 10.1021/jp044511+
- LeubnerSKatsukisGGuldiD M 2012 Faraday Discuss. 155 253 10.1039/c1fd00102g
- YanX BHanZ JYangYTayB K 2007 J. Phys. Chem. C 111 4125 10.1021/jp0651844
- KangYTatonT A 2003 J. Am. Chem. Soc. 125 5650 10.1021/ja034082d
- LuLZhouZZhangYWangSZhangY 2007 Carbon 45 2621 10.1016/j.carbon.2007.08.025
- GilmoreK JMoultonS EWallaceG G 2007 Carbon 45 402 10.1016/j.carbon.2006.09.015
- CotiugaIPicchioniFAgarwalU SWoutersDLoosJLemstraP J 2006 Macromol. Rapid Commun. 27 1073 10.1002/marc.200600163
- Shvartzman-CohenRLevi-KalismanYNativ-RothEYerushalmi-RozenR 2004 Langmuir 20 6085 10.1021/la049344j
- SlusarenkoN NHeurtefeuBMaugeyMZakriCPoulinPLecommandouxS 2007 Carbon 45 903 10.1016/j.carbon.2006.12.003
- Shin H-iMin B GJeongWParkC 2005 Macromol. Rapid Commun. 26 1451 10.1002/marc.200500290
- MountrichasGPispasSTagmatarchisN 2007 Small 3 404 10.1002/smll.200600476
- MountrichasGTagmatarchisNPispasS 2007 J. Phys. Chem. B 111 8369 10.1021/jp067500k
- Nativ-RothEShvartzman-CohenRBouniouxCFlorentMZhangDSzleiferIYerushalmi-RozenR 2007 Macromolecules 40 3676 10.1021/ma0705366
- WangZLiuQZhuHLiuHChenYYangM 2007 Carbon 45 285 10.1016/j.carbon.2006.09.025
- PetrovPStassinFPagnoulleCJeromeR 2003 Chem. Commun. 2904 10.1039/b307751a
- TomonariYMurakamiHNakashimaN 2006 Chem. Eur. J. 12 4027 10.1002/chem.200501176
- NakashimaNTomonariYMurakamiH 2002 Chem. Lett. 638 10.1246/cl.2002.638
- GomezF JChenR JWangDWaymouthR MDaiH 2003 Chem. Commun. 190 10.1039/b211194b
- FifieldL SDaltonL RAddlemanR SGalhotraR AEngelhardM HFryxellG EAardahlC L 2004 J. Phys. Chem. B 108 8737 10.1021/jp037977l
- GuldiD M 2006 Chem. Eur. J. 12 3975 10.1002/chem.200600114
- KavakkaJ SHeikkinenSKilpelaeinenIMattilaMLipsanenHHelajaJ 2007 Chem. Commun. 519 10.1039/b611461j
- NakashimaNTanakaYTomonariYMurakamiHKatauraHSakaueTYoshikawaK 2005 J. Phys. Chem. B 109 13076 10.1021/jp050958m
- ChenR JZhangYWangDDaiH 2001 J. Am. Chem. Soc. 123 3838 10.1021/ja010172b
- LiuLWangTLiJGuoZ-XDaiLZhangDZhuD 2002 Chem. Phys. Lett. 367 747 10.1016/S0009-2614(02)01789-X
- ArtyukhinA BBakajinOStroevePNoyA 2004 Langmuir 20 1442 10.1021/la035699b
- SgobbaVRahmanG M AGuldiD MJuxNCampidelliSPratoM 2006 Adv. Mater. 18 2264 10.1002/adma.200501493
- EhliC 2006 J. Am. Chem. Soc. 128 11222 10.1021/ja0624974
- GuldiD MRahmanG M APratoMJuxNQinSFordW 2005 Angew. Chem. Int. Edn 44 2015 10.1002/anie.200462416
- GuldiD MRahmanG M AJuxNBalbinotDTagmatarchisNPratoM 2005 Chem. Commun. 2038 10.1039/b418406h
- GuldiD MRahmanG M ASgobbaVKotovN ABonifaziDPratoM 2006 J. Am. Chem. Soc. 128 2315 10.1021/ja0550733
- RahmanG M AGuldiD MCagnoliRMucciASchenettiLVaccariLPratoM 2005 J. Am. Chem. Soc. 127 10051 10.1021/ja050396k
- GuldiD MRahmanG M AJuxNBalbinotDHartnagelUTagmatarchisNPratoM 2005 J. Am. Chem. Soc. 127 9830 10.1021/ja050930o
- GeorgakilasVTzitziosVGournisDPetridisD 2005 Chem. Mater. 17 1613 10.1021/cm0483590
- LiXLiuYFuLCaoLWeiDWangY 2006 Adv. Funct. Mater. 16 2431 10.1002/adfm.200600339
- GranotEBasnarBCheglakovZKatzEWillnerI 2006 Electroanalysis 18 26 10.1002/elan.200503403
- OuY-YHuangM H 2006 J. Phys. Chem. B 110 2031 10.1021/jp055920o
- PaloniemiHLukkarinenMAeaeritaloTArevaSLeiroJHeinonenMHaapakkaKLukkariJ 2006 Langmuir 22 74 10.1021/la051736i
- HolderP GFrancisM B 2007 Angew. Chem. Int. Edn 46 4370 10.1002/anie.200700333
- ChittaRSandanayakaA S DSchumacherA LD’SouzaLArakiYItoOD’SouzaF 2007 J. Phys. Chem. C 111 6947 10.1021/jp0704416
- OgoshiTTakashimaYYamaguchiHHaradaA 2007 J. Am. Chem. Soc. 129 4878 10.1021/ja070457+
- GuldiD MTaiebHRahmanG M ATagmatarchisNPratoM 2005 Adv. Mater. 17 871 10.1002/adma.200400641
- LouXDaussinRCuenotSDuwezA-SPagnoulleCDetrembleurCBaillyCJeromeR 2004 Chem. Mater. 16 4005 10.1021/cm0492585
- NakashimaNOkuzonoSTomonariYMurakamiH 2004 Trans. Mater. Res. Soc. Jpn. 29 525
- YuanW Z 2006 Macromolecules 39 8011 10.1021/ma061856c
- BahunG JWangCAdronovA 2006 J. Polym. Sci., A. Polym. Chem 44 1941 10.1002/pola.21308
- WangDJiW-XLiZ-CChenL 2006 J. Am. Chem. Soc. 128 6556 10.1021/ja060907i
- MurakamiHNakamuraGNomuraTMiyamotoTNakashimaN 2007 J. Porphyrins Phthalocyanines 11 418 10.1142/S1088424607000473
- MeuerSBraunLZentelR 2008 Chem. Commun. 3166 10.1039/b803099e
- MeuerSBraunLSchillingTZentelR 2009 Polymer 50 154 10.1016/j.polymer.2008.10.039
- SchopfEBroyerRTaoLChenYMaynardH D 2009 Chem. Commun. 4818 10.1039/b908282d
- SalzmannC GWardM A HJacobsR M JTobiasGGreenM L H 2007 J. Phys. Chem. C 111 18520 10.1021/jp076013h
- LovellC SWiseK EKimJ WLilleheiP THarrisonJ SParkC 2009 Polymer 50 1925 10.1016/j.polymer.2009.02.016
- NakashimaNOkuzonoSMurakamiHNakaiTYoshikawaK 2003 Chem. Lett. 32 456 10.1246/cl.2003.456
- ZhengMJagotaASemkeE DDinerB AMcLeanR SLustigS RRichardsonR ETassiN G 2003 Nat. Mater. 2 338 10.1038/nmat877
- GuoZSadlerP JTsangS C 1998 Adv. Mater. 10 701 10.1002/(SICI)1521-4095(199806)10:9<701::AID-ADMA701>3.0.CO;2-4
- TsangS CGuoZChenY KGreenM L HHillH A OHambleyT WSadlerP J 1997 Angew. Chem. Int. Edn 36 2198 10.1002/anie.199721981
- BuzanevaE 2002 Mater. Sci. Eng. C 19 41 10.1016/S0928-4931(01)00425-8
- WangYBuY 2007 J. Phys. Chem. B 111 6520 10.1021/jp0700433
- MengSMaragakisPPapaloukasCKaxirasE 2007 Nano Lett. 7 45 10.1021/nl0619103
- JohnsonR RJohnsonA T CKleinM L 2008 Nano Lett. 8 69 10.1021/nl071909j
- ZhengM 2003 Science 302 1545 10.1126/science.1091911
- BadaireSZakriCMaugeyMDerreABarisciJ NWallaceGPoulinP 2005 Adv. Mater. 17 1673 10.1002/adma.200401741
- HellerD AJengE SYeungT-KMartinezB MMollA EGastalaJ BStranoM S 2006 Science 311 508 10.1126/science.1120792
- XuYPehrssonP EChenLZhangRZhaoW 2007 J. Phys. Chem. C 111 8638 10.1021/jp0709611
- KamNWongSLiuZDaiH 2006 Angew. Chem. Int. Edn 45 577 10.1002/anie.200503389
- BeckerM LFaganJ AGallantN DBauerB JBajpaiVHobbieE KLacerdaS HMiglerK BJakupciakJ P 2007 Adv. Mater. 19 939 10.1002/adma.200602667
- TuXManoharSJagotaAZhengM 2009 Nature 460 250 10.1038/nature08116
- SprafkeJ KStranksS DWarnerJ HNicholasR JAndersonH L 2011 Angew. Chem. Int. Edn 50 2313 10.1002/anie.201007295
- ChenF 2009 J. Phys. Chem. C 113 14946 10.1021/jp904431u
- CathcartHNicolosiVHughesJ MBlauW JKellyJ MQuinnS JColemanJ N 2008 J. Am. Chem. Soc. 130 12734 10.1021/ja803273s
- MalikSVogelSRoesnerHArnoldKHennrichFKoehlerA-KRichertCKappesM M 2007 Compos. Sci. Technol. 67 916 10.1016/j.compscitech.2005.11.041
- GigliottiBSakizzieBBethuneD SShelbyR MChaJ N 2006 Nano Lett. 6 159 10.1021/nl0518775
- CampbellJ FTessmerIThorpH HErieD A 2008 J. Am. Chem. Soc. 130 10648 10.1021/ja801720c
- ChoiJ HNguyenF TBaroneP WHellerD AMollA EPatelDBoppartS AStranoM S 2007 Nano Lett. 7 861 10.1021/nl062306v
- JengE SBaroneP WNelsonJ DStranoM S 2007 Small 3 1602 10.1002/smll.200700141
- HafezI HBerberM RFujigayaTNakashimaN 2014 Sci. Rep 4 6295 10.1038/srep06295
- O’ConnellM J 2002 Science 297 593 10.1126/science.1072631
- KatoYInoueANiidomeYNakashimaN 2012 Sci. Rep 2 733
- ZhaoQFrogleyM DWagnerH D 2002 Polym. Adv. Technol. 13 759 10.1002/pat.246
- ZhaoQWoodJ RWagnerH D 2001 Appl. Phys. Lett. 78 1748 10.1063/1.1357209
- AjayanP MSchadlerLSGiannarisCRubioA 2000 Adv. Mater. 12 750 10.1002/(SICI)1521-4095(200005)12:10<750::AID-ADMA750>3.0.CO;2-6
- AmerM SEl-AshryM MMaguireJ F 2004 J. Chem. Phys. 121 2752 10.1063/1.1768157
- RaoA MEklundP CBandowSThessASmalleyR E 1997 Nature 388 257 10.1038/40827
- LiMXuPYangJYingHHaubnerKDunschLYangS 2011 J. Phys. Chem. C 115 4584 10.1021/jp112330n
- CollisonC JSpencerSPreskeAPalumboCHelenicABaileyRPellizzeriS 2010 J. Phys. Chem. B 114 11002 10.1021/jp104705y
- ZouJLiuLChenHKhondakerS IMcCulloughR DHuoQZhaiL 2008 Adv. Mater. 20 2055 10.1002/adma.200701995
- GengJKongB-SYangS BYounS CParkSJooTJungH-T 2008 Adv. Funct. Mater. 18 2659 10.1002/adfm.200800496
- TezukaN 2011 Energy Environ. Sci. 4 741
- CherukuriPGannonC JLeeuwT KSchmidtH KSmalleyR ECurleyS AWeismanR B 2006 Proc. Natl Acad. Sci. USA 103 18882 10.1073/pnas.0609265103
- YanYCuiJPötschkePVoitB 2010 Carbon 48 2603 10.1016/j.carbon.2010.03.065
- ArnoldM SBlackburnJ LCrochetJ JDoornS KDuqueJ GMohiteATelgH 2013 Phys. Chem. Chem. Phys. 15 14896 10.1039/c3cp52752b
- ValentiniLKennyJ M 2005 Polymer 46 6715 10.1016/j.polymer.2005.05.025
- KanaiYGrossmanJ C 2008 Nano Lett. 8 908 10.1021/nl0732777
- HoltJ MFergusonA JKopidakisNLarsenB ABultJRumblesGBlackburnJ L 2010 Nano Lett. 10 4627 10.1021/nl102753z
- BindlD JSafronN SArnoldM S 2010 ACS Nano 4 5657 10.1021/nn1012397
- StranksS DWeisspfennigCParkinsonPJohnstonM BHerzL MNicholasR J 2011 Nano Lett. 11 66 10.1021/nl1036484
- FergusonA JBlackburnJ LHoltJ MKopidakisNTenentR CBarnesT MHebenM JRumblesG 2010 J. Phys. Chem. Lett. 1 2406 10.1021/jz100768f
- SchuettfortTSnaithH JNishANicholasR J 2010 Nanotechnology 21 025201 10.1088/0957-4484/21/2/025201
- KymakisEServatiPTzanetakisPKoudoumasEKorniliosNRompogiannakisIFranghiadakisYAmaratungaG A J 2007 Nanotechnology 18 435702 10.1088/0957-4484/18/43/435702
- ArnoldM SStuppS IHersamM C 2005 Nano Lett. 5 713 10.1021/nl050133o
- DaberaG D M R 2013 ACS Nano 7 556 10.1021/nn304705t
- RenSBernardiMLuntR RBulovicVGrossmanJ CGradečakS 2011 Nano Lett. 11 5316 10.1021/nl202796u
- BestemanKLeeJ-OWiertzF G MHeeringH ADekkerC 2003 Nano Lett. 3 727 10.1021/nl034139u
- Santiago-RodríguezLSánchez-PomalesGCabreraC R 2010 Isr. J. Chem. 50 277 10.1002/ijch.201000034
- StaiiCJohnsonA TChenMGelperinA 2005 Nano Lett. 5 1774 10.1021/nl051261f
- JohnsonA T CStaiiCChenMKhamisSJohnsonRKleinM LGelperinA 2006 Semicond. Sci. Technol. 21 S17 10.1088/0268-1242/21/11/S03
- PangXIminPZhitomirskyIAdronovA 2010 Macromolecules 43 10376 10.1021/ma101862b
- ZhangXMengLLuQ 2009 ACS Nano 3 3200 10.1021/nn9006362
- KimT HLeeB YJaworskiJYokoyamaKChungW JWangEHongSMajumdarALeeS W 2011 ACS Nano 5 2824 10.1021/nn103324p
- TamP DHieuN V 2011 Appl. Surf. Sci. 257 9817 10.1016/j.apsusc.2011.06.028
- WangFGuHSwagerT M 2008 J. Am. Chem. Soc. 130 5392 10.1021/ja710795k
- QiPVermeshOGrecuMJaveyAWangQDaiHPengSChoK J 2003 Nano Lett. 3 347 10.1021/nl034010k
- LobezJ MAfzaliA 2013 Chem. Mater. 25 3662 10.1021/cm401881w
- BiancoAKostarelosKPratoM 2005 Curr. Opin. Chem. Biol. 9 674 10.1016/j.cbpa.2005.10.005
- BandaruN MVoelckerN H 2012 J. Mater. Chem. 22 8748 10.1039/c2jm16636d
- LiuZTabakmanS MChenZDaiH 2009 Nat. Protoc. 4 1372 10.1038/nprot.2009.146
- BiancoAKostarelosKPartidosC DPratoM 2005 Chem. Commun. 571 10.1039/b410943k
- HarrisonB SAtalaA 2007 Biomaterials 28 344 10.1016/j.biomaterials.2006.07.044
- MalarkeyE BParpuraV 2007 Neurodegener. Dis. 4 292 10.1159/000101885
- PratoMKostarelosKBiancoA 2008 Acc. Chem. Res. 41 60 10.1021/ar700089b
- MadaniS YNaderiNDissanayakeOTanASeifalianA M 2011 Int. J. Nanomed. 6 2963
- PrakashSMalhotraMShaoWTomaro-DuchesneauCAbbasiS 2011 Adv. Drug Delivery Rev. 63 1340 10.1016/j.addr.2011.06.013
- YangR 2006 Gene Ther. 13 1714 10.1038/sj.gt.3302808
- CiofaniGRaffaV 2009 Mini-Rev. Med. Chem. 9 1251 10.2174/138955709789878132
- BoghossianA A 2011 Chem. Sus. Chem. 4 848 10.1002/cssc.201100070
- CuiH-FVashistS KAl-RubeaanKLuongJ H TSheuF-S 2010 Chem. Res. Toxicol. 23 1131 10.1021/tx100050h
- SharifiSBehzadiSLaurentSLaird ForrestMStroevePMahmoudiM 2012 Chem. Soc. Rev. 41 2323 10.1039/c1cs15188f
- LacerdaLBiancoAPratoMKostarelosK 2006 Adv. Drug Delivery Rev. 58 1460 10.1016/j.addr.2006.09.015
- SerranoM CGutiérrezM Cdel MonteF 2014 Prog. Polym. Sci. 39 1448 10.1016/j.progpolymsci.2014.02.004
- DingLStilwellJZhangTElboudwarejOJiangHSelegueJ PCookeP AGrayJ WChenF F 2005 Nano Lett. 5 2448 10.1021/nl051748o
- Manna SunilKSarkarSBarrJWiseKBarrera EnriqueVJejelowoORice-Ficht AllisonCRamesh GovindarajanT 2005 Nano Lett. 5 1676 10.1021/nl0507966
- TianFCuiDSchwarzHEstradaG GKobayashiH 2006 Toxicol. in Vitro 20 1202 10.1016/j.tiv.2006.03.008
- AdeliMSoleymanRBeiranvandZMadaniF 2013 Chem. Soc. Rev. 42 5231 10.1039/c3cs35431h
- LiuZDavisCCauWHeLChenXDaiH 2008 Proc. Natl Acad. Sci. USA 105 1410 10.1073/pnas.0707654105
- SchipperM LNakayama-RatchfordNDavisC RKamN W SChuPLiuZSunXDaiHGambhirS S 2008 Nat. Nanotech. 3 216 10.1038/nnano.2008.68
- KojimaCReginoCUmedaYKobayashiHKonoK 2010 Int. J. Pharm. 383 293 10.1016/j.ijpharm.2009.09.015
- WelsherKLiuZSherlockS PRobinsonJ TChenZDaranciangDDaiH 2009 Nat. Nanotech. 4 773 10.1038/nnano.2009.294
- NamaziHAdeliM 2003 Eur. Polym. J. 39 1491 10.1016/S0014-3057(02)00385-3
- NamaziHAdeliMZarnegarZJafariSDadkhahAShuklaA 2007 Colloid Polym. Sci. 285 1527 10.1007/s00396-007-1717-6
- D’EsteMDe NardiMMennaE 2006 Eur. J. Org. Chem. 11 2517 10.1002/ejoc.200600196
- ZhaoBHuHYuAPereaDHaddonR C 2005 J. Am. Chem. Soc. 127 8197 10.1021/ja042924i
- CordellaFDe NardiMMennaEHébertCLoiM A 2009 Carbon 47 1264 10.1016/j.carbon.2009.01.004
- PantarottoDBriandJ-PPratoMBiancoA 2004 Chem. Commun. 16 10.1039/b311254c
- LeeYGeckelerK E 2010 Adv. Mater. 22 4076 10.1002/adma.201000746
- KrajcikRJungAHirschANeuhuberWZolkO 2008 Biochem. Biophys. Res. Commun. 369 595 10.1016/j.bbrc.2008.02.072
- ThakareV SDasMJainA KPatilSJainS 2010 Nanomedicine 5 1277 10.2217/nnm.10.95
- LiuZTabakmanSWelsherKDaiH 2009 Nano Res 2 85 10.1007/s12274-009-9009-8
- LiuZRobinsonJ TTabakmanS MYangKDaiH 2011 Mater. Today 14 316 10.1016/S1369-7021(11)70161-4
- AndersenA JRobinsonJ TDaiHHunterA CAndresenT LMoghimiS M 2013 ACS Nano 7 1108 10.1021/nn3055175
- WelsherKLiuZDaranciangDDaiH 2008 Nano Lett. 8 586 10.1021/nl072949q
- AntarisA LRobinsonJ TYaghiO KHongGDiaoSLuongRDaiH 2013 ACS Nano 7 3644 10.1021/nn4006472
- HongGLeeJ CRobinsonJ TRaazUXieLHuangN FCookeJ PDaiH 2012 Nat. Med. 18 1841 10.1038/nm.2995
- LiuZLiXTabakmanS MJiangKFanSDaiH 2008 J. Am. Chem. Soc. 130 13540 10.1021/ja806242t
- RobinsonJ THongGLiangYZhangBYaghiO KDaiH 2012 J. Am. Chem. Soc. 134 10664 10.1021/ja303737a
- HongG 2014 Nat. Photon. 8 723 10.1038/nphoton.2014.166
- DiaoSHongGRobinsonJ TJiaoLAntarisA LWuJ ZChoiC LDaiH 2012 J. Am. Chem. Soc. 134 16971 10.1021/ja307966u
- KamN W SO’ConnellMWisdomJ ADaiH 2005 Proc. Natl Acad. Sci. USA 102 11600 10.1073/pnas.0502680102
- LiuZCaiWHeLNakayamaNChenKSunXChenXDaiH 2007 Nat. Nanotech. 2 47 10.1038/nnano.2006.170
- PrencipeGTabakmanS MWelsherKLiuZGoodwinA PZhangLHenryJDaiH 2009 J. Am. Chem. Soc. 131 4783 10.1021/ja809086q
- LiWLiangCZhouWQiuJZhouZSunGXinQ 2003 J. Phys. Chem. B 107 6292 10.1021/jp022505c
- TianZ QJiangS PLiangY MShenP K 2006 J. Phys. Chem. B 110 5343 10.1021/jp056401o
- LiWLiangCQiuJZhouWHanHWeiZSunGXinQ 2002 Carbon 40 791 10.1016/S0008-6223(02)00039-8
- LiLXingY 2008 J. Power Sources 178 75 10.1016/j.jpowsour.2007.12.002
- KongkanandAKuwabataSGirishkumarGKamatP 2006 Langmuir 22 2392 10.1021/la052753a
- ShaoYYinGGaoYShiP 2006 J. Electrochem. Soc. 153 A1093 10.1149/1.2191147
- WangXLiWChenZWajeMYanY 2006 J. Power Sources 158 154 10.1016/j.jpowsour.2005.09.039
- LiLXingY 2006 J. Electrochem. Soc. 153 A1823 10.1149/1.2234659
- EbbesenT W 1996 Adv. Mater. 8 155 10.1002/adma.19960080212
- XuHZengLXingSShiGXianYJinL 2008 Electrochem. Commun. 10 1839 10.1016/j.elecom.2008.09.030
- de PaulaC CGarcia RamosAda SilvaA CCocchieri BotelhoERezendeM C 2002 Carbon 40 787 10.1016/S0008-6223(01)00136-1
- GuoD-JLiH-L 2005 J. Colloid Interface Sci. 286 274 10.1016/j.jcis.2004.12.042
- ChaoGWenwenLYi ZhengJHaoK 2006 Nanotechnology 17 2882 10.1088/0957-4484/17/15/053
- WuGChenY SXuB Q 2005 Electrochem. Commun. 7 1237 10.1016/j.elecom.2005.07.015
- CheGLakshmiB BMartinC RFisherE R 1999 Langmuir 15 750 10.1021/la980663i
- ZhangSShaoYYinGLinY 2010 J. Mater. Chem. 20 2826 10.1039/b919494k
- GrzelczakMCorrea-DuarteM ASalgueiriño-MaceiraVRodríguez-GonzálezBRivasJLiz-MarzánL M 2007 Angew. Chem. Int. Edn 46 7026 10.1002/anie.200701671
- WuBZhangYKuangYYuYZhangXChenJ 2012 Chem. Asian. J. 7 190 10.1002/asia.201100564
- HsuC-HLiaoH-YKuoP-L 2010 J. Phys. Chem. C 114 7933 10.1021/jp100328f
- HeDZengCXuCChengNLiHMuSPanM 2011 Langmuir 27 5582 10.1021/la2003589
- ZhaoYYangXTianJWangFZhanL 2010 J. Power Sources 195 4634 10.1016/j.jpowsour.2010.02.023
- WangSJiangS PWhiteT JGuoJWangX 2009 J. Phys. Chem. C 113 18935 10.1021/jp906923z
- ZhangSShaoYYinGLinY 2011 Appl. Catal. B 102 372 10.1016/j.apcatb.2010.11.029
- SelvarajVAlagarMKumarK S 2007 Appl. Catal. B 75 129 10.1016/j.apcatb.2007.03.012
- OhH-SKimKKimH 2011 Int. J. Hydrogen Energy 36 11564 10.1016/j.ijhydene.2011.06.079
- AsensioJ ASanchezE MGomez-RomeroP 2010 Chem. Soc. Rev. 39 3210 10.1039/b922650h
- OzakiJ-ITanifujiS-IKimuraNFuruichiAOyaA 2006 Carbon 44 1324 10.1016/j.carbon.2005.12.026
- XiongWDuFLiuYPerezASuppMRamakrishnanT SDaiLJiangL 2010 J. Am. Chem. Soc. 132 15839 10.1021/ja104425h
- SidikR AAndersonA BSubramanianN PKumaraguruS PPopovB N 2006 J. Phys. Chem. B 110 1787 10.1021/jp055150g
- ZhangYFuganeKMoriTNiuLYeJ 2012 J. Mater. Chem. 22 6575 10.1039/c2jm00044j
- QuLLiuYBaekJ-BDaiL 2010 ACS Nano 4 1321 10.1021/nn901850u
- PengCZhangSJewellDChenG Z 2008 Prog. Nat. Sci. 18 777 10.1016/j.pnsc.2008.03.002
- SunYShiG 2013 J. Polym. Sci., B. Polym. Phys. 51 231 10.1002/polb.23226
- AntiohosDFolkesGSherrellPAshrafSWallaceG GAitchisonPHarrisA TChenJMinettA I 2011 J. Mater. Chem. 21 15987 10.1039/c1jm12986d
- LiQLiuJZouJChunderAChenYZhaiL 2011 J. Power Sources 196 565 10.1016/j.jpowsour.2010.06.073
- LiP 2014 ACS Appl. Mater. Interfaces 6 5228 10.1021/am500579c
- SharmaR KKarakotiASealSZhaiL 2010 J. Power Sources 195 1256 10.1016/j.jpowsour.2009.08.093