
Abstract
Perovskites are mixed-metal oxides that are attracting much scientific and application interest owing to their low price, adaptability, and thermal stability, which often depend on bulk and surface characteristics. These materials have been extensively explored for their catalytic, electrical, magnetic, and optical properties. They are promising candidates for the photocatalytic splitting of water and have also been extensively studied for environmental catalysis applications. Oxygen and cation non-stoichiometry can be tailored in a large number of perovskite compositions to achieve the desired catalytic activity, including multifunctional catalytic properties. Despite the extensive uses, the commercial success for this class of perovskite-based catalytic materials has not been achieved for vehicle exhaust emission control or for many other environmental applications. With recent advances in synthesis techniques, including the preparation of supported perovskites, and increasing understanding of promoted substitute perovskite-type materials, there is a growing interest in applied studies of perovskite-type catalytic materials. We have studied a number of perovskites based on Co, Mn, Ru, and Fe and their substituted compositions for their catalytic activity in terms of diesel soot oxidation, three-way catalysis, N2O decomposition, low-temperature CO oxidation, oxidation of volatile organic compounds, etc. The enhanced catalytic activity of these materials is attributed mainly to their altered redox properties, the promotional effect of co-ions, and the increased exposure of catalytically active transition metals in certain preparations. The recent lowering of sulfur content in fuel and concerns over the cost and availability of precious metals are responsible for renewed interest in perovskite-type catalysts for environmental applications.
1. Introduction
Owing to their high thermal stability, excellent oxidation activity, and low price; perovskites have been explored for a variety of environment-related and energy-related applications, including automobile exhaust purification, fuel cells, N2O decomposition, and water–gas shift reactions [Citation1–Citation4]. In recent years, efforts have also been made to study the properties of perovskites in terms of reactions such as chemical looping combustion (CLC) and photocatalytic water splitting [Citation5, Citation6]. In this paper, perovskite-type oxides appear as a family of potential compounds for the purpose of gaining insight into the relationships between solid-state and catalytic properties. These isomorphic solids (general formula: ABO3) are highly versatile due to the flexibility in the chemical composition of perovskites with a large number of cations that can fit into both the A and B positions within the same crystalline structure (i.e., practically all the metals are stable in the perovskite lattice). Another key feature is the availability of multi-component perovskites, which can be synthesized by partial substitution of cations at either A or B sites. Perovskite oxides containing a lanthanide element at position A and a transition metal at position B are used more frequently in heterogeneous catalysis, obviously exploiting the catalytic properties of transition metals [Citation7–Citation11]. Part of the importance of perovskite-type catalysts is that a number of transition metals show excellent catalytic activity for a variety of reactions due to their electronic structure. However, most transition metals have a tendency to oxidize easily at higher temperatures in the presence of oxygen, which is why the noble metals, due to their stability under such conditions, are currently being used as catalysts. Although metal oxides are stable under oxidative conditions and are also often used as catalysts, they possess rather limited adaptability due to their simple binary composition. In contrast, perovskites, with their flexible ABO3 composition and structure, offer immense possibilities in terms of adaptation to control their properties, and this is probably one of the most important reasons for the extensive studies of perovskites.
Perovskite-type oxides ideally have a cubical crystal structure, wherein cations with a large ionic radius have 12–fold coordination with oxygen atoms, occupying A sites, and cations with a smaller ionic radius have 6–fold coordination, occupying B-sites. A and O form cubic closest packing, and B is in the octahedral voids in the packing. In an ideal structure, where the atoms are simply bonding to one another, the B–O distance is equal to a/2 (a = length of unit cell), whereas the A–O distance is equal to (a/√2) and the following relation between the ionic radii holds:
It has been found, however, that the cubic structure of perovskites can be still retained in an ABO3 structure even though this equation is not strictly obeyed. As a measure of this deviation from ideal cubical structure, Goldschmidt [Citation12] introduced a tolerance factor (t), which is used to define the range of the ionic radii of A and B defined by the following equation, which is applicable at room temperature:
For an ideal perovskite, t is unity; however, the perovskite structure is also found for lower values of t in the range 0.75 to 1. In such cases, the cubical structure can be distorted into tetragonal, rhombohedral, or other lower symmetries [Citation12]. It should be noted that perovskites with values of t greater than 1 having a hexagonal structure are also reported due to the larger ionic radii of the A ions or the smaller ionic radii of the B ions. BaNiO3 is a well-known example of a hexagonal perovskite structure where the value of t is 1.13 [Citation13, Citation14].
1.1. Synthesis of perovskites
The physical properties of perovskites are greatly dependent on synthesis methods. In general, perovskites are usually formed at elevated temperatures, and the solid-state synthesis method is quite commonly used to prepare perovskites in pure form due to the availability of impurity-free precursors. Such synthesis methods are often suitable for electronic and electrical applications, but they pose problems when perovskites are subjected to surface-related studies. Obviously, perovskites with higher surface areas are required for catalytic and adsorption-related applications. Therefore, efforts have been devoted to synthesizing perovskites at lower temperatures with improved porosity. Close contact with precursors is essential for attaining the lowest possible synthesis temperature to achieve high surface area. The concept of maintaining high homogeneity of precursors (mixed-metal form) has been exploited for the purpose of achieving close contact with a view to synthesizing perovskites in the pure phase at low temperature. Low-temperature synthesis of perovskites results in higher surface area with smaller particle size, typically between the submicron and nano levels. Co-precipitation, citrate sol–gel, and solution combustion techniques have been commonly reported as ways to synthesize perovskites for catalytic applications. Serious attempts have been made to evaluate perovskite-based catalysts on a prototype scale, and the synthesis of perovskites on commercial honeycomb substrates has been successfully demonstrated. Some of the synthesis methods commonly used are briefly discussed here.
1.1.1. Co-precipitation method
Co-precipitation is a useful technique for the synthesis of perovskites. In this technique, two or more components are precipitated by using a suitable precipitant to maintain a high degree of homogeneity of the precursors. Junwu et al synthesized nanocrystals of LaCoO3 using the co-precipitation method. In this method, La(NO3)3 and Co(NO3)2 were used as precursors and NaOH solution was used as a precipitating agent. After precipitation, the resulting product was centrifuged, washed with water and ethanol several times, dried at 100 °C, and calcined at 600 °C [Citation15]. Muneeswaran et al synthesized nano-sized BiFeO3 powder by the co-precipitation method. Bi(NO3)3 · 5H2O and Fe(NO3)3 · 9H2O were dissolved in 200 mL of distilled water and stirred for 20 min to form a clear solution. The precursor solutions were precipitated using ammonia (2.5 M) at various pH levels, and it was found that the pure phase was observed at approximately pH 10 [Citation16]. We have used the co-precipitation method extensively for the synthesis of several perovskite compositions, such as BaRuO3, La3.5Ru4.0O13, LaMnO3, La0.8Ba0.2MnO3, PrMnO3, Pr0.8Ba0.2MnO3, SrCoO3, and Sr0.8Ce0.2CoO3. It was observed that the pure crystalline phase of perovskites can be obtained by this method, but it results in a relatively moderate surface area [Citation17–Citation21]. Several precipitating agents have been reported based on the selection of A-site and B-site ions. We have observed that aqueous ammonia is one of the best precipitants, mainly due to its easy removal upon heating. It should be noted, however, that ammonia forms a complex with several metal ions and therefore it is not suitable as a precipitant in such cases. Co-precipitation is also used for preparing perovskites on ceramic structured supports, where homogeneous precipitate precursors can be easily coated onto a support for achieving a uniform perovskite on the support.
1.1.2. Citrate sol–gel method
The sol–gel method is another useful technique for preparing perovskites with a relatively high surface area. In this method, the ‘sol’ gradually leads to the formation of a gel-like diphasic system, which has both a liquid and a solid phase. Decomposition of dried gel precursor materials through calcination yields the desired perovskites. Shabbir et al [Citation22] reported a novel sol–gel process for preparing nano-sized perovskite-type LaFeO3 powder by thermal decomposition of the gel complex of LaFe–(C6H8O7 · H2O). The authors observed that optimization of the gelling conditions results in formation of the LaFeO3 perovskite phase without an explosion/combustion process and pH control. X-ray diffraction (XRD) results indicate that although the impurity phase (La2O3) appeared at ∼350 °C, the pure perovskite phase was observed above 600 °C with a particle size of approximately 25 nm. Guo et al [Citation23] synthesized a series of dually substituted perovskite catalysts, La1−xKxCo1−yPdyO3−δ (x = 0, 0.1; y = 0, 0.05) by a citrate-based sol–gel process. A 1:1 molar ratio of citric acid to total metal ions was used. In this process, the formed solution after the addition of citric acid and metal salts was transferred into a vacuum rotary evaporator to remove the excess water at 60 °C. The viscous gel thus formed was dried homogeneously in air flow at 120 °C overnight to obtain a dried precursor. After pre-calcination at 350 °C for 2 h, the precursor was further calcined at 700 °C. We have explored the synthesis of LaCoO3 and La0.9Ba0.1CoO3 perovskites using this method. It was observed that although the surface area of the substituted catalyst La0.9Ba0.1CoO3 is slightly inferior, the material shows excellent catalytic activity for CO oxidation as well for particulate matter oxidation. In a recent study, we synthesized substituted PrMnO3 with various cations (K+1, Ce+4, Ba+2) at the A site using the sol–gel method. The surface area of perovskites was observed to be strongly dependent on the nature of the substituting element [Citation24, Citation25].
1.1.3. Solution combustion synthesis (SCS)
The combustion synthesis technique takes advantage of rapid, self-sustaining exothermic chemical reactions between a metal salt and a suitable organic fuel. Since most of the heat required for the synthesis is supplied by the reaction itself, the mixture of the reactants only needs heating up to a temperature that is significantly lower than the actual phase formation temperature. In recent years, this technique has been frequently adopted to produce very fine homogeneous crystalline powder, without decomposition of the intermediates and/or calcination steps, whereas these steps are rather necessary in conventional synthesis routes. This technique is particularly suited for producing nano-size particles of catalysts. A nano-structured catalyst coating over a trap can effectively improve local catalyst–soot contact conditions [Citation26, Citation27]. The important condition that prevails in this synthesis is the generation of high localized temperatures for a very short time that nevertheless is long enough to cross the activation energy barrier for the formation of the perovskite phase. The evolution of a huge amount of gases results in the formation of a very foamy mass with smaller particles and a rough surface, which is often desired for catalytic applications. So far, little has been discussed about the gas emissions released during such synthesis methods and their suitability for the large-scale production of perovskites. This needs to be addressed before the technique can be commercially used for any number of perovskite compositions.
Russo et al synthesized a series of perovskite catalysts (LaMnO3, LaCrO3, LaFeO3, and LaNiO3) using the SCS method based on the exothermic and self-supporting reactions occurring between an aqueous solution of metal nitrates (acting as oxidizers) and urea (acting as a sacrificial fuel). The resultant perovskite crystals were between 45 and 75 nm in size with a high Brunauer–Emmett–Teller (BET) specific surface area of 15–20 m2 g−1 [Citation28]. Fino et al used combustion synthesis to prepare LaMnO3, LaFeO3, LaCrO3, LaCr0.9O3−δ, and La0.9K0.1Cr0.9O3−δ catalysts, which have a relatively high specific surface area of 18–25 m2 g−1. The authors explained that the synthesis can be split into two steps. The first is endothermic reaction, which represents the perovskite synthesis beginning with the metal nitrate precursors. The second reaction is exothermic and involves the reaction between oxygen derived from nitrate decomposition and urea [Citation29]. Nanocrystalline Th-based perovskites (BaThO3 and SrThO3) were prepared through combustion synthesis by varying the fuel-to-oxidant ratio [Citation30]. The nitrate salts of the precursors were dissolved with requisite amounts of water, and the aqueous solution of citrate with the desired stoichiometry was added. The resultant gel was calcined to get Th-based perovskites. The perovskites of iron, cobalt, and cerium (Sr0.85Ce0.15FeO3−x. La0.6Sr0.4Co0.95Fe0.05O3−x, BaCe0.9Y0.1O3−x.) were prepared through the auto-combustion method by varying the ratio of citrate and nitrate between 1 and 4. The morphology and the structural properties of these perovskites were influenced by the ratio of fuel and oxidant. No segregation of dopant was observed by maintaining a low fuel-to-oxidant ratio in this interesting study [Citation31]. A number of Ce3+-based (CeCrO3, CeScO3) [Citation32–Citation34] as well as substituted Ce3+–based perovskites (La1−xCexCrO3, 0 ≤ x ≤ 1.0) [Citation35] were prepared using the two-step synthesis method involving combustion followed by heating under a vacuum condition.
1.2. Supported perovskites
With a view to improving the surface area of perovskites, efforts have been made to synthesize perovskites directly on supports such as alumina and honeycomb supports. Among the reported methods, the supporting of perovskites as a thin layer or nanoparticles on an appropriate high-surface-area support is a potential method for practical applications [Citation36]. The main problem with this method is a solid-state reaction between the precursors of the perovskite and the support at high temperature. Such synthesis methods, however, are also inevitable if perovskites as catalytic centers need to be dispersed onto supports for practical applications.
1.2.1. Alumina-supported perovskite catalysts
We have been exploring the synthesis of various supported perovskites using both commonly available γ-alumina and ceramic cordierite honeycombs. In our previous study, we tried to synthesize LaRuO3 perovskite directly on γ-alumina through impregnation of a La3+ and Ru3+ mixed-metal solution [Citation37]. Heating of these composites resulted in the formation of different undesirable phases, including aluminates. The perovskite phase of LaRuO3 could not be detected by XRD analysis. Therefore, lanthanum pre-coated and post-heated γ-alumina was used for the synthesis of LaRuO3 on γ-alumina that was first calcined at 950 °C for 10 h. The obtained powder was subsequently impregnated with La3+ salt solution and was heated at 700 °C for 3 h. This process was repeated several times. This alumina powder pre-coated with lanthanum oxide was then impregnated repeatedly with a mixed-metal ion solution of La3+ and Ru3+, followed by oven drying at 60 °C. A total 20 wt.% loading of LaRuO3 on pre-coated γ-alumina was targeted, and the concentration of precursor solution and impregnation cycles was optimized. The powder with La3+ and Ru3+ incorporated was subsequently heated at 850 °C for 12 h in a furnace. This resulted in γ-alumina-supported LaRuO3; however, a small number of other phases were also formed along with the LaRuO3 phase, as discussed elsewhere [Citation37].
1.2.2. Ceramic honeycomb–supported perovskite catalysts
As mentioned earlier, we have systematically studied the synthesis of perovskite catalyst–coated ceramic foam filters to assess the feasibility of the developed catalysts in realistic conditions. In a recent study from our group [Citation38], we chose ceramic foam filters (CFF; 38 mm diameter, 50 mm length) made of α-Al2O3 for the preparation of perovskite-based catalyst-coated ceramic foam filters. This study was aimed at the synthesis of Pr0.8Sr0.2K0.1MnO3 on CFF. The catalyst was coated onto the CFF by the dip coating method. In this procedure, a CFF was soaked in the aqueous solution containing appropriate amounts of Pr(NO3)2 · 6H2O, Mn(CH3COO)2 · 4H2O, KNO3, and Sr(NO3)2 · 2H2O. The extra solution remaining in the pores of the ceramic foam filter was blown off by hot air, followed by drying and pre-calcination at 350 °C in air. This procedure was repeated three times, and the ceramic foam filter was calcined at 800 °C for 10 h. The perovskite catalyst coating observed in this method was approximately 3.12 wt%.
Some of the catalytic reactions of various perovskites for environment-related and energy-related applications are discussed here.
1.3. Perovskite-type catalysts for diesel soot oxidation
Diesel engines are the heart of transportation. Higher fuel economy, low maintenance requirements (especially in new-generation diesel engines), high durability, and low operating cost are the main advantages of diesel engines over gasoline engines. Diesel engines contribute, however, extensively to environmental pollution by emitting particulate matter (PM) and nitrogen-oxide (NOx) emissions. Soot has been identified as a prominent contributor to serious health and environmental issues [Citation39, Citation40]. Diesel particulate matter is composed of carbon with small amounts of organic compounds as well as inorganic materials (ash and sulfates) as a result of unburned fuel, lubricating oil, etc. Typical diesel soot is composed of approximately 41% carbon, 25% unburned oil, 27% sulfate, approximately 13% ash, and 7% unburned fuel, as shown in figure [Citation41]; however, this composition can vary significantly depending on fuel composition and engine conditions.
Figure 1. Particle matter composition of a heavy-duty diesel engine tested in a transient cycle as reported in [Citation41]. Reproduced from D B Kittelson 1998 J. Aerosol Sci. 29 575–88, with permission from Elsevier.
![Figure 1. Particle matter composition of a heavy-duty diesel engine tested in a transient cycle as reported in [Citation41]. Reproduced from D B Kittelson 1998 J. Aerosol Sci. 29 575–88, with permission from Elsevier.](/cms/asset/3f50da1c-1e0c-4770-880d-b3478741c7ac/tsta_a_11661308_f0001_oc.jpg)
PM emitted from diesel vehicles consists mainly of ultrafine particles, which can easily be inhaled inside the body and reach deep into lungs, causing serious health issues. PM is also responsible for a number of direct and indirect environmental impacts, including global warming (through the black carbon effect) and photochemical reactions leading to poor visibility [Citation42]. Control of diesel particulate matter emissions has therefore become necessary, and stringent norms for PM emissions have been stipulated for new diesel vehicles. Several techniques are promising for the reduction of emissions from diesel engines. Modified and alternative fuels have been studied in an attempt to correlate emission levels with certain fuel specifications in order to optimize fuel composition for lower PM emissions. A small but significant reduction in diesel-regulated emissions can be achieved by modifying diesel fuels but at the expense of fuel cost [Citation43]. Some engine modifications have proved very effective in reducing diesel engine emission levels and have been implemented in recent years. Diesel particulate standards were first proposed in 1981, and it was generally believed that filter-based particulate removal systems would meet future standards [Citation44]. As a consequence, the redesign of engines is currently one of the main strategies for meeting current emission standards. After-treatment techniques have been studied extensively for particulate removal from diesel exhaust gases. Options such as fuel additives, diesel oxidation catalysts, partial flow filters (PFFs), and diesel particulate filters (DPFs) have been explored as potentially successful alternatives for controlling emissions. The use of DPFs is one of the most promising approaches for reducing particulate matter (PM) from diesel vehicles; however, effective regeneration of DPFs is a major limitation to their feasibility. Catalytic regeneration has been suggested as the best approach, and intensive efforts are being made to develop suitable catalysts for this application [Citation45].
Although almost all commercial regenerative-type DPF technologies are based on noble metal catalysts, a range of other catalytic materials have been studied for their activity in terms of soot oxidation. These include noble metal–promoted transition metal oxides, mixed oxides, other metals, perovskites, bi-functional materials, eutectic mixtures, etc [Citation46–Citation49]. Perovskites have been proposed as alternative potential materials for this application, due mainly to their thermal stability, excellent oxidation activity, and low price. Studies of perovskites have confirmed their potential as catalytic materials for the regeneration of diesel particulate filters using the exhaust temperature. Their chemical stability has been a big challenge because of the significant sulfur content of diesel fuel, but perovskite catalysts can show improved stability with steep lowering of sulfur content. Russo et al have reported that Li-substituted chromite-type perovskite catalysts exhibit the highest activity for soot oxidation as a consequence of a large amount of weakly chemisorbed O- species (α–oxygen) [Citation50]. Teraoka et al have also reported that mixed-metal oxides (e.g., La–K–Mn–O system) demonstrate activity toward the simultaneous removal of NOx and soot. Activity toward soot oxidation and NOx-reduction selectivity was strongly dependent on potassium content [Citation51]. Fino and Specchia have prepared nano-structured PrCrO3 on CeO2 through combustion synthesis, which showed even better catalytic activity than Pt/Al2O3 when the catalyst was coated onto a monolith [Citation52].
Zhang et al have prepared a series of macroporous catalysts of La1−xKxCo1−yFeyO3 (x = 0, 0.1; y = 0, 0.1) by a combined method of organic ligation and solution combustion. They found that the size of the soot was smaller than the pore size of the catalyst; this allows the soot to enter into the pore of the catalyst, resulting in good contact between soot and catalyst. As a consequence, macroporous La0.9K0.1CoO3 perovskite showed better catalytic activity than nano-size catalysts. The activity of the catalysts was found to be comparable to that of supported Pt-based catalysts [Citation53].
Peng et al have reported a highly efficient catalyst (La0.8K0.2Cu0.05Mn0.95O3) for simultaneous removal of NOx and diesel soot. The catalytic properties of this catalyst were tested under simulated diesel engine exhaust using temperature programmed reduction (TPR) and it was found that this catalyst showed promise for the simultaneous removal of NOx and diesel soot [Citation54]. Ura et al have examined soot and NOx reaction on a K+-substituted SrTiO3 catalyst. The authors demonstrated that partial substitution of strontium by K+ induces oxygen vacancies that promote soot oxidation. The most efficient substitution observed by the authors was at x = 0.2. It was observed from the results of Sr0.8K0.2TiO3 that the soot ignition temperature was 380oC in a gas flow of 10% O2 in Ar and 240 °C in a gas flow containing 1500 ppm of NO2 + 10% O2 in Ar. A similar test was performed with inert material for comparison, and the soot ignition temperature was observed to be 635 °C and 570 °C in a gas flow of 10% O2 in Ar and 1500 ppm of NO2 + 10% O2 in Ar, respectively [Citation55].
Bialobok et al have studied SrTiO3 catalysts doped with alkali metals (Li, K, Cs). They prepared two types of SrTiO3-based perovskite catalysts; one was substituted in the A position of the structure (Sr1-xMexTiO3, x = 0.05–0.2), and another was impregnated with the same amount of alkali metals. Results indicate that the substituted catalysts showed improved catalytic activity over that of the impregnated catalysts [Citation56]. Cauda et al have investigated an innovative multi-functional catalyst (La0.9K0.1Cr0.9O3−δ + 1 wt.% Pt) for diesel soot combustion with combined direct and indirect (NO–NO2–NO cycle) oxidation mechanisms. This catalyst was applied over a wall-flow SiC trap and tested according to a standard protocol on an engine bench and showed excellent catalytic activity [Citation57].
Our group has also extensively examined the soot oxidation reaction using various types of perovskite materials. La-based perovskite-type catalysts (La3.5Ru4.0O13) showed very high catalytic activity for soot oxidation. We observed that this catalytic material possesses the required thermal stability in comparison with LaRuO3. This may be due to the presence of ruthenium at the stable higher oxidation state of Ru4+ in the perovskite structure. The catalyst showed soot oxidation activity through dissociative adsorption of oxygen on the surface of the catalyst. It should be noted, however, that most of the catalytic materials demonstrated their soot oxidation activity only in the presence of NOx [Citation58]. We examined the catalytic activity as a function of Ce substitution in a SrCoO3-type perovskite catalyst for diesel soot oxidation. Small amounts of substitution of cerium at the A site of SrCoO3 resulted in improved redox properties and consequently improved catalytic activity. The promotional effects of cerium substitution in SrCoO3 and the presence of a small amount of potassium ions seem to be responsible for the observed high soot oxidation activity [Citation19].
In a recent study, we reported Ba-substituted LaCoO3-type perovskite, which showed much improved catalytic activity for CO oxidation, even below 200 °C, as well as particulate matter oxidation at 370 °C. Although no clear difference in x-ray photoelectron spectroscopy studies of LaCoO3 and La0.9Ba0.1CoO3 was observed, temperature programmed desorption (TPD) experiments support the existence of small amounts of α-oxygen in La0.9Ba0.1CoO3, which may be responsible for the enhanced catalytic activity [Citation59]. Bench-scale examination of our group of Pr0.7Sr0.2K0.1MnO3 perovskite catalyst–coated ceramic foam filters indicated that Ti (the temperature at which soot combustion is initiated) decreased by 150 °C and Tf (temperature at which soot combustion completed) by 100 °C for direct soot oxidation using real diesel engine exhaust. This may be considered high catalytic activity under realistic conditions [Citation60].
1.4. N2O decomposition on perovskite catalysts
Over the last few decades, nitrous oxide has been considered one of the pollutants most responsible for the destruction of ozone in the stratosphere as well as being a relatively strong greenhouse gas [Citation61–Citation65]. It has been recognized that the migration of nitrous oxide (N2O) with chlorofluorocarbons (CFCs) into the stratosphere contributes to ozone layer depletion [Citation66]. N2O is also an absorber of infrared radiation and thus contributes to the greenhouse effect [Citation67, Citation68]. The ambient level of nitrous oxide is 324 ppb, which recently has been constantly increasing. Control of N2O emissions has attracted much interest due to the global warming potential of N2O (AR5, IPCC 2013), which is 265 times larger than that of CO2 [Citation69, Citation70]. About 18 million tons of N2O are released each year into the atmosphere from both anthropogenic and natural sources [Citation71]. The nitric and adipic acid industries are the major anthropogenic sources of N2O emissions [Citation63, Citation70–Citation73]. Nitric acid is being increasingly used for fertilizer production and is manufactured through oxidation of ammonia. During this catalytic oxidation process, nitrous oxide is produced as a byproduct. N2O equivalent to approximately 15.3 million tons of CO2 was emitted from the nitric acid industry in 2012 [Citation70]. Nylon production is also responsible for nitrous oxide emission; in nylon production, adipic acid is used as one of the major precursors. N2O is emitted as a byproduct of adipic acid production during the oxidation of a ketone–alcohol mixture with nitric acid. N2O emissions from the adipic acid industry were estimated to be the equivalent of 5.8 million tons of CO2 in 2012 [Citation70]. Hence, reduction or decomposition of N2O to control its emissions from industries is one of the important research areas in environmental catalysis.
Although a few methods for controlling N2O emissions are available, direct catalytic decomposition is considered a simple potential method for N2O removal. Different types of catalysts have been studied for the decomposition of N2O, including noble metals [Citation74–Citation77], metal oxides [Citation78–Citation82], and metal or ion exchanged zeolites [Citation83–Citation87]. At low temperatures, noble metal–based catalysts show very good activity in terms of N2O decomposition. However, these noble metal–supported systems are expensive and unstable at high temperatures as well as in the presence of oxygen [Citation75]. Most zeolite-based materials are also potential catalysts for the decomposition of N2O at low temperatures. The development of hydrothermally stable zeolite-based catalysts has significantly improved prospects relative to their natural instability in the presence of water vapor [Citation88]. Usually for industrial applications, catalysts with better chemical and thermal stability as well as having a lower cost are preferred. Perovskites are promising catalysts because of their low-cost excellent thermal stability as well as the possibility of tailoring their properties for N2O reduction and decomposition reactions. It is possible to activate N2O molecules on a transition metal associated with a perovskite structure, which can lead to decomposition of N2O into its constituents (N2 and O2), the most desirable reaction in terms of avoiding the use of reducing agents and harmful byproducts. Because perovskites offer various supra-facial and intra-facial reaction pathways, this has generated considerable interest among researchers in exploring perovskites for N2O control, and a large number of studies are therefore reported in the literature.
Russo et al have studied a LaCoO3 perovskite-type catalytic material prepared by solution combustion synthesis for N2O decomposition; they reported that 50% of N2O conversion can be achieved at 455 °C and 490 °C in the absence and presence of 5% oxygen, respectively [Citation89]. Alini et al have studied a number of alkaline metal–based perovskite materials comprising Ca and Ba for decomposition of N2O. They reported that a CaMn0.7Cu0.3O3 catalyst showed the highest activity, 95% conversion, at 550 °C as well as stable catalytic activity for a simulated industrial gas coming from an adipic acid plant for a period of 1400 h [Citation90]. This report has been considered a valuable finding, particularly regarding the stability of perovskites under an actual exhaust stream, which might be otherwise questioned due to the basic nature of many perovskite compositions, which are quite susceptible to attack by commonly present acidic gases like SO2. Dacquin et al have studied catalytic N2O decomposition on 1 wt.% Pd/LaCoO3. They reported a reaction rate of 0.0023 mmol min−1 g−1 at 460 °C [Citation91]. Ivanov et al have studied the various compositions of a La–Sr–Fe–O perovskite catalyst for high-temperature N2O decomposition, and they have also discussed the oxygen mobility of perovskites for different reactions [Citation92]. Recently our group also explored N2O decomposition using different AMnO3 (A = La and Pr) perovskite catalysts and reported the effect of partial substitution and Ag promotion on catalytic N2O decomposition using perovskites [Citation93, Citation94]. It was observed that both Ba substitution and Ag incorporation showed a promotional effect in terms of N2O decomposition activity in a LaMnO3-type perovskite catalyst. A series of Ba-substituted as well as Ag-incorporated samples were evaluated for their activity in terms of N2O decomposition and the maximum/optimized activity observed for La0.8Ba0.2MnO3 compositions promoted with La0.8Ba0.2MnO3 (20 mol %) and Ag- (1 wt.%) (figure (a)). In the case of Pr-based perovskites, the maximum catalytic activity was observed for the Pr0.8Ba0.2MnO3 composition (figure (b)) [Citation94]. O2 TPD and H2 TPR studies clearly inferred that the redox properties of Ba-substituted as well as Ag-promoted perovskites were altered, which correlates with their improved catalytic activity in terms of N2O decomposition. It is also important to study the N2O decomposition activity of supported catalysts since a number of physical properties, such as porosity and surface area, influence catalytic activity, and catalyst and support interactions can also alter catalytic activity. Hence, optimized compositions of La0.8Ba0.2MnO3, Pr0.8Ba0.2MnO3, and Ag (1 wt.%)/La0.8Ba0.2MnO3 were prepared in supported form using cordierite honeycomb support and evaluated for their catalytic activity. Interestingly, catalytic activity was observed for these supported catalyst samples that was nearly identical to that of the catalyst in powder form (figures (a) and (c)) [Citation93, Citation94]. These results convincingly inferred the possibility of coating perovskites onto commercially available ceramic honeycomb substrates for practical applications.
Figure 2. N2O decomposition as a function of temperature (0.5 vol.% N2O, 0.02 vol.% NO, and 5 vol.% O2): (a) La0.8Ba0.2MnO3, supported La0.8Ba0.2MnO3, 1 wt.% Ag/La0.8Ba0.2MnO3 and supported Ag (1 wt.%)/La0.8Ba0.2MnO3; (b) Pr0.8Ba0.2MnO3; and (c) supported Pr0.8Ba0.2MnO3 [Citation93, Citation94]. Adapted from S Kumar et al 2011 J. Mol. Catal. A: Chem. 348 42 and S Kumar et al 2012 Catal. Today 198 125, with permission from Elsevier.
![Figure 2. N2O decomposition as a function of temperature (0.5 vol.% N2O, 0.02 vol.% NO, and 5 vol.% O2): (a) La0.8Ba0.2MnO3, supported La0.8Ba0.2MnO3, 1 wt.% Ag/La0.8Ba0.2MnO3 and supported Ag (1 wt.%)/La0.8Ba0.2MnO3; (b) Pr0.8Ba0.2MnO3; and (c) supported Pr0.8Ba0.2MnO3 [Citation93, Citation94]. Adapted from S Kumar et al 2011 J. Mol. Catal. A: Chem. 348 42 and S Kumar et al 2012 Catal. Today 198 125, with permission from Elsevier.](/cms/asset/de0df4af-1aaf-407a-83bd-1f0d8e96473c/tsta_a_11661308_f0002_oc.jpg)
1.5. Perovskites for chemical looping combustion
Power generation plays a pivotal role in economic growth and is one of the most influential factors, along with globalization, in the development of any nation [Citation95]. Energy demand has increased substantially lately due to the accelerated increase in population, which is responsible for creating a gap between energy demand and supply. This demand, however, can be met by implementing high-capacity power generation plants [Citation96]. Coal is one of the world’s most abundant and most widely distributed fossil fuels, and its use for power generation is continuously increasing worldwide. Of the total global electricity production, 44% is from coal, which is expected to remain a primary fuel source for power generation in the coming years to meet electricity demand, especially in developing countries [Citation97]. Coal and methane are known as non-renewable fossil fuels and are associated with the release of an enormous amount of CO2 into the atmosphere through power generation activity [Citation98]. It has been reported that fossil fuel–based thermal power plants emit about one-third of the total CO2 emissions, which are one of the major contributors to the greenhouse gas effect [Citation98, Citation99]. It has been anticipated that installed fossil fuel-based thermal power plants around the world will most likely double by 2030 [Citation100]. Therefore, it is essential to implement new CO2 capture and storage (CCS) strategies for the economical use of fossil fuels for power generation with mitigation of CO2 emissions. CCS is one of the potential options for mitigating CO2 emissions from various fossil fuel-based sources. CCS may reduce the amount of CO2 emitted into the atmosphere while allowing the continued use of fossil fuels for power generation at thermal power plants and other large-scale industries [Citation101–Citation103]. CCS technology has been successfully implemented in small-scale industries but not in large-scale industries, owing to the technical immaturity of the components of CCS technology as well as the requirement of more energy and the high cost [Citation96]. The currently available CCS techniques capture about 85–95% of CO2; however, they require 10–40% additional energy for capturing and compressing CO2 from the large volume of flue gases [Citation104].
CLC has the inherent potential of effecting CO2 separation from flue gases after the combustion reaction of fossil fuels, as shown in figure . This makes it one of the most efficient techniques for delivering sequestration-ready CO2 at lower cost. In the CLC process, oxygen can be derived from metal oxide referred to as an oxygen carrier [Citation105–Citation108]. The virtually pure CO2 formed in the CLC process can be readily captured and stored, or it can be directed as reactants to prepare value-added products [Citation109, Citation110]. CLC possesses the potential for application in, for instance, clean energy generation or clean combustion, reforming, and hydrogen production [Citation111–Citation114]. Moreover, it has also been used for the combustion of coal with advanced techniques for coal gasification [Citation115]. It has been reported that the transportation sector alone is responsible for about one-quarter of the total global CO2 emissions, and this can be expected to increase enormously in the coming years. A promising option is the use of H2 as a fuel to reduce CO2 emissions. However, the technologies for producing pure H2 are still under development and are also expensive. Therefore, H2 production from fossil-fuel sources (e.g., steam reforming of natural gas) is currently based on existing technologies, which are expected to continue to dominate in the coming years. Currently the production of H2 comes mostly from reforming natural gas (e.g., methane), partial oxidation of heavy oils and naphtha, and gasification of coal. Natural gas is a promising source for H2 production, where the ratio of H2 to carbon is roughly twice that of oil. The chemical looping reforming (CLR) process is a promising alternative to H2 production from natural gas. In CLR, a suitable oxygen carrier is circulated between two reactors as in CLC. In a fuel reactor, the fuel (e.g., CH4) is oxidized and forms syngas from the lattice oxygen of the oxygen carrier, whereas the reduced oxygen carrier after the removal of reactive oxygen is completely re-oxidized by air in the second reactor. In this way, the products are free from flue gases (e.g., N2) [Citation116].
Figure 3. Schematic illustration of chemical looping combustion of an oxygen carrier.
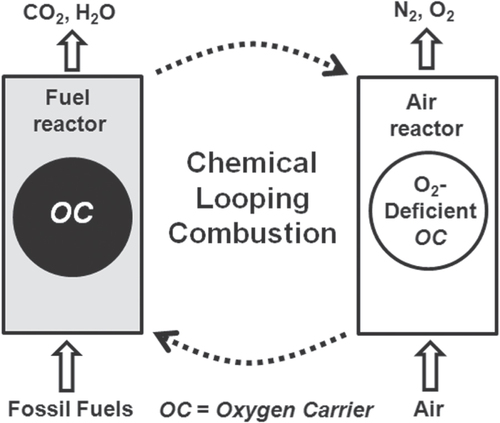
The practicability of the CLC and CLR processes strongly depends on the development of a suitable oxygen carrier, which is the key component in both chemical looping processes. Considerable efforts have been made in the last few decades regarding the development of oxygen carriers. A potential oxygen carrier should have the following characteristics: (i) abundant availability of raw materials to produce the oxygen carrier, (ii) non-toxicity, (iii) high oxygen carrying capacity, (iv) higher reactivity with fuels under harsh chemical looping conditions, (v) regeneration as well as attrition resistance properties, (vi) no carbon formation during chemical looping reactions, (vii) selectivity for product formation, (viii) low cost, and (ix) minimal environmental impacts. Individual transition metal oxides of Cu, Mn, Fe, Co, and Ni have been extensively investigated for chemical looping applications due to their favorable reductive/oxidative thermodynamics; however, single transition metal oxides pose a number of practical limitations as oxygen carriers due to their poor multi-cycle performance, carbon formation, agglomeration behavior under chemical looping conditions, etc [Citation117–Citation122]. Although individual transition oxides show inferior activity for long-term chemical looping applications, they have a number of advantages in terms of their physical and chemical properties and therefore should be exploited for their chemical looping applications, especially in mixed-oxide forms [Citation122–Citation131].
Perovskite-based mixed oxides with the general formula ABO3 have been widely exploited for catalysis applications—for instance, hydrogenation, CO oxidation, ammonia oxidation, and methane combustion. Perovskites have also been exploited recently in chemical looping applications owing to their desirable physiochemical properties—for instance, variable structural composition, excellent redox properties, high reactivity, high stability in a harsh thermal environment, high mobility of oxygen, and better mechanical properties. Perovskite-based mixed oxides have high oxygen mobility, which facilitates easy removal/insertion of oxygen from/into the structure [Citation123–Citation127].
La-based perovskite-type mixed oxides (LaBO3) have been widely exploited for chemical looping applications. LaFeO3 exhibits enhanced activity as well as selectivity for syngas formation in a fixed bed reactor, which makes it attractive as an oxygen carrier for chemical looping applications. The surface-adsorbed oxygen of LaFeO3 is highly active in completing the combustion of fuels, whereas the lattice oxygen is very selective in the formation of syngas (partial oxidation). It is well known that the substitution of cations (e.g., La3+) with a lower oxidation state (e.g., Sr) results in better chemical looping activity. LaxSr1−xFeO3−δ has shown very high selectivity for syngas formation. The substitution of Sr in La-based perovskites increases the oxygen capacity of the materials but reduces the selectivity for syngas formation [Citation116]. Partial substitution of Fe in Co sites of La1−xSrxCoyFe1−yO3 results in a slight decrease in structural stability; however, continuous oxygen supply has been observed in sequential reduction–oxidation cycles [Citation123–Citation127]. Furthermore, CO selectivity decreases dramatically with the increase in the amounts of Fe substitution and is not suitable for chemical looping reforming applications [Citation128]. Nalbandian et al have studied La1−xSrxMyFe1−yO3−δ synthesized by the citrate method as an oxygen carrier for CLR. The pure phase of La0.7Sr0.3FeO3 has shown moderate stability during multiple reduction–oxidation cycles. The addition of 5% NiO improved the yields of H2 as well as the stability of the perovskites. Among the La-based perovskites explored, La0.7Sr0.3Cr0.05Fe0.95O3 mixed with 5% NiO yielded up to 90% of H2 and also showed very good stability in multiple reduction–oxidation cycles. Thus, the addition of small quantities of NiO to La0.7Sr0.3Cr0.05Fe0.95O3 through mechanical mixing substantially increases methane decomposition as well as selectivity for the formation of syngas [Citation116]. Fang et al have also studied La1-xSrxFeO3 as oxygen carriers prepared by the combustion method for chemical looping reforming reactions of CH4. They demonstrated that Sr substitution in the range 0.3 to 0.5 resulted in better reactivity, selectivity, and oxygen-donation ability. They also concluded that substituted LaFeO3 shows promise as an oxygen carrier for CLR of methane [Citation129]. Evdou et al have studied La1−xSrxFeO3−δ as redox materials for the simultaneous production of pure hydrogen and syngas in a membrane reactor. They concluded that the calcination temperature is an important parameter that influences chemical looping performance. The addition of small quantities of NiO to La1−xSrxFeO3 substantially increases methane decomposition reactivity, selectivity for the formation of CO and H2, and water splitting activity [Citation130]. Xiao et al have studied LaFeO3/Al2O3 prepared through a wet mixing–kneading method. They examined chemical looping reforming in a fixed bed reactor and a circulating fluidized bed reactor. The addition of Al2O3 to LaFeO3 improves oxygen migration for CH4 conversion, which forms syngas selectively. The reaction temperature and bed height of the fuel reactor greatly influence CH4 conversion and CO selectivity. The higher reactor temperature and bed height are shown to be beneficial for reforming applications with bubbling fluidized fuel reactors. LaFeO3 as an oxygen carrier showed potential in terms of CLR process, but it needs further modification in the manufacturing process to improve its structural stability and chemical properties to be used as a potential oxygen carrier [Citation131].
1.6. Pervoskites in photocatalysis
In the historical background of photocatalysis [Citation132], semiconductor materials have received exceptional attention in the cleaner energy arena due to increasing energy demand and the necessity of environmental remediation. With increasing global population, the world is facing energy challenges, and there is a pressing need for desirable and sustainable growth in the energy sector. In the absence of any attractive renewable sources of energy, the conventional ways of energy production are growing several-fold, with negative environmental impact. Therefore, developments related to effective photocatalytic strategies from harvesting solar energy to producing usable chemical fuel have received their share of attention. Photovoltaic cells have been a great success in this generation and are being put to use harvesting solar energy for direct conversion to electricity. This option, however, requires the storage of electric power generated through photovoltaic reactions. Perhaps this is one of the main reasons for exploring other photocatalytic options, which involve photocatalytic and photoelectrochemical (PEC) reactions as promising approaches to harnessing solar energy. The conversion of solar to chemical energy through water splitting reactions for the generation of hydrogen has therefore been explored for some time as an ideal reaction for sustainable energy generation.
Numerous potential applications of solar photocatalysis have been reported [Citation133, Citation134]. Among these, energy and environmental issues were studied widely using a variety of photocatalysts. In general, an efficient photocatalyst, also called an ideal semiconductor, should include light harvesting and redox capabilities to facilitate the desired chemical reactions, thus achieving the targeted reaction. Inorganic semiconductor materials should have adequate capability to absorb solar energy across a broad spectrum. Because of their light harvesting property, potential photocatalysts absorb solar energy, leading to the generation of photoelectrons in the conduction band and holes in the valence band for their possible use in redox reactions. The redox nature of a photocatalyst as an intrinsic property dictates solar energy conversion efficiency. For this reason, TiO2 [Citation135], SrTiO3 [Citation136], and NaTaO3 [Citation137] are among the most explored photocatalysts for hydrogen generation because they show the desirable stability, inert nature, and thermodynamically optimal band structures to realize photocatalytic reactions. Although TiO2-based photocatalysts show light harvesting properties and stability in the UV region, they cannot absorb visible light efficiently due to their large band gap (>3.2 eV). On the other hand, gallium arsenide can absorb light more effectively but is found to be too unstable as a photocatalytic system to be of practical value [Citation138]. The continued progress driven by cleaner energy demand in this exciting research field has opened new avenues to the design and development of numerous photocatalyst compositions for a variety of applications ranging from wastewater treatment to water splitting and CO2 photoreduction. Researchers have also advanced the scientific aspects of semiconductor photocatalysis as well as those involving combinations of different mechanisms. Most semiconductors include metals/mixed metal oxides used as efficient photocatalysts (e.g., CdS [Citation139, Citation140]). Furthermore, surface tuning of oxide nanomaterials can improve photocatalytic performance, as systematically reviewed by Jing et al [Citation141]. Their efforts were mainly in the direction of the utilization of visible light in the solar spectrum. Mixed-oxide-type photocatalysts, including those of perovskite-type structures, are very prominent because of their broadly diverse properties. Recently ABO3-based photocatalysts were systematically reviewed with a view to their characteristic properties for water splitting reactions [Citation142], which further confirm their potential role in photocatalysis.
We have explored a range of ABO3-type perovskite-based materials for sacrificial donor–assisted as well as overall water splitting reactions to generate hydrogen. With the exceptional adaptability of their properties through compositional variations, perovskites show promise for solar hydrogen production among the large number of photocatalysts being explored. They are also frequently explored in combination with other oxides to carry out various steps of complex uphill reactions involved in water splitting reactions. As already discussed, the flexible composition of perovskites is based on large-size rare earth or alkaline earth metals at the A site and transition metals at the B site in an ABO3-type structure. These perovskites can be tailored through band gap engineering approaches, and photocatalysts thus designed can harvest solar light more effectively. Perovskites thus synthesized show broadband absorption over the visible to near-infrared region of the solar spectrum. Another challenge related to the charge separation between photogenerated holes and electrons has also been undertaken with perovskite compositions to achieve overall improved efficiency [Citation143].
We recently investigated metal ferrite-based photocatalysts (MFeO3, where M = La, Pr, and Ce) for water splitting reactions. LaFeO3 was synthesized in nanocrystalline form (crystallite size ∼38 nm), generating hydrogen through photocatalytic reaction [Citation144]. This photocatalyst exhibited an optical band gap of 2.07 eV with an absorption spectrum predominantly in the visible region of the spectrum with a hydrogen evolution rate of 3315 μmol g−1h−1 in the sacrificial donor–assisted water splitting reaction. Perovskites having Ce3+ ions play a key role in photocatalysis due to their allowed transitions by selection rules [Citation145]. Their photocatalytic performance was improved significantly through decreasing the optical band gap value by increasing Ce3+ in the unit cells of perovskites, thus facilitating the absorption of light in the visible region [Citation34]. Recently we also reported a PrFeO3-type perovskite composition [Citation146] with a crystallite size of 20 nm, which shows a significant hydrogen evolution rate of 2847 μmol g−1h−1. We have also synthesized CeFeO3 perovskite using different methods, showing a band gap of 2.1 eV, which suggests that the material can absorb light in the visible region. This material is being explored for its photocatalytic and PEC activity with regard to water splitting reactions. Initial PEC investigations of CeFeO3 show a significant photocurrent difference of approximately 0.6 mAcm−2 at 0.5 V between light and dark conditions. Detailed PEC evaluations of CeFeO3 and other perovskites are in progress. Most of our current efforts are being diverted toward achieving increased hydrogen yield, possibly using a visible spectrum of solar radiations.
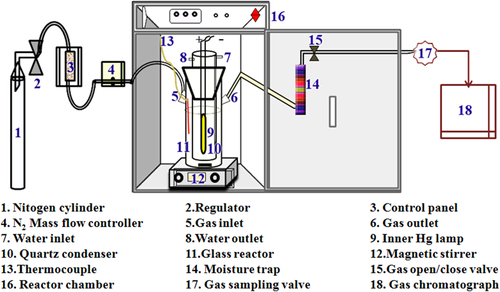
In addition to the development of improved photocatalyst compositions, other considerations need attention when it comes to photocatalytic water splitting. Undeniably a major issue is photoreactor design as well as experimental setup and conditions that can significantly affect photocatalytic activity [Citation147]. Various types of reactors are available for water splitting reactions, and a few of them are used in our studies. These include a 20 mL vial-type reactor for initial screening to optimize various parameters, including photocatalyst dose, sacrificial donors, and illumination intensity. After optimization of the parameters, photocatalytic water splitting reactions were further carried out in a tubular borosilicate/quartz photoreactor with an inlet port for inert gas purging and an outlet for automatic injection to a gas chromatograph (GC) or to collect the gas in a collector directly, as shown in figure . This setup uses typically about 200–400 mL of reaction mixture in a reactor of 750 mL volume with an efficient inner irradiation source. A high-pressure mercury lamp of 100–400 W (or any other source of illumination) is often used with a quartz inner jacket for photocatalysts with wide band gap when intensive UV light of wavelength shorter than about 300 nm is required. However, a Xe lamp with a cutoff filter is usually employed for visible light irradiation. A solar simulator that is a standard light source for evaluation of solar cells is also used in our case for simulating various wavelengths, including the so-called one-sun condition. A solar simulator with an air-mass 1.5 filter (AM-1.5) provides 100 mW cm2 of intensity, which is equivalent to normal sunlight. Prior to the exposure to the light, the reaction mixture was properly purged using nitrogen, ensuring the complete removal of oxygen/air. The gas evolved from the photocatalytic reaction was injected directly into the sample loop, which was controlled by a gas-sampling valve (GSV) equipped with a 5 Å molecular sieve column (GC Model Shimadzu-2014). The amount of H2 was analyzed by a thermal conductivity detector (TCD), and N2 was used as the carrier gas.
Figure 4. Schematic illustration of lab-scale evaluation systems for photocatalytic water splitting reactions.
Thus, perovskite-type compounds play a significant role in photocatalysis and photoelectrocatalysis, with the possibility of tailoring their semiconductor properties. Perovskite-based semiconductors have been extensively explored with respect to various photocatalytic and PEC reactions. Considering the recent progress in perovskite-based solar cell photovoltaic (PV) materials, organo-lead halide perovskite-type materials are also attracting a great deal of interest because of their consistent and high solar-to-hydrogen (STH) efficiency [Citation148]. More efforts are required to improve the kinetics of photocatalytic water splitting reactions, to achieve a hydrogen yield of practical relevance. Heterostructures (e.g., heterojunctions) with organo-halide perovskites as well as a number of other combinations of perovskite and metal oxide may show improved solar water splitting performance. The design, construction, and photocatalytic performance of semiconductor heterojunctions have been systematically reviewed by Wang et al [Citation149]. The progress reported in this field so far indicates that forming heterojunctions affords a promising option for enhancing the photocatalytic efficiencies of photocatalytic semiconductors. Because only limited heterojunction-based compositions have been explored so far, this area is likely to witness much progress, with enhanced scientific activity, in years to come [Citation150, Citation151].
1.7. Summary and future perspectives
Mixed oxides with perovskite-type structures have long been a fascinating group of materials for both academic and practical-application reasons. Because it is possible to play with ABO3-type perovskite symmetry with a very large number of cations, enormous efforts to exploit their electronic, electric, magnetic, and optical as well as catalytic properties have been reported. Even with numerous successes in applications related to electronics and oxygen permeability, however, perovskites have yet to find recognition in environmental catalysis, with few exceptions. Despite this history, the one single reason that can rejuvenate the potential of perovskite-based catalysts in environmental applications is the substantially decreasing sulfur content in fuels. Diesel sulfur content has already been lowered drastically in many parts of the world, and similar plans are under way even in countries like China and India. This may renew the possibility of using perovskite-based catalysts for vehicle exhaust applications, especially in direct diesel soot oxidation. Another interesting possibility may emerge for catalytic combustion of producer gas generated through biomass gasification. This mixture typically contains very high concentrations of carbon monoxide and hydrogen with traces of sulfur. We have already started exploring perovskites for producer gas combustion reactions, with initial results favoring for more detailed investigations, including stability studies. Photocatalytic water splitting is yet another area where perovskites, heterojunction-based, and other composite materials show good potential for application in the future. Advances in the synthesis of perovskites, particularly their successful synthesis on commercial cordierite and other supports, are some of the developments that have been successfully demonstrated and should certainly help in the discovery of more applications for this interesting group of materials in the future. Needless to say, despite hundreds of perovskite compositions explored so far, there may be even more to come. This research area is therefore poised to grow even more rapidly.
Acknowledgments
Some of the work reported was carried out under the DST-sponsored RHEES project as well as through bilateral cooperation between CSIR-NEERI and NIMS Japan. Thanks to CSIR for providing fellowship for RK.
References
- López-SuárezF EBueno-LópezAIllán-GómezM JTrawczynskiJ 2014 Appl. Catal. A 485 214 10.1016/j.apcata.2014.07.037
- WuaYCordierCBerrierENunsNDujardinCGrangerP 2013 Appl. Catal. A 151 140 10.1016/j.apcatb.2013.04.002
- GuangchuanLShuguangLChanglongLXiuqinO U 2007 J. Rare Earths 25 264 10.1016/S1002-0721(07)60485-2
- BasileFBrennaGFornasariGGallobP DGarybDVaccariaA 2010 Stud. Surf. Sci. Catal. 175 471 10.1016/S0167-2991(10)75087-4
- HeFLiXZhaoKHuangZWeiGLiH 2013 Fuel 108 465 10.1016/j.fuel.2012.11.035
- ChenWChenXYangYYuanJShangguanW 2014 Int. J. Hydrogen Energy 39 13468 10.1016/j.ijhydene.2014.03.096
- HansenK KSkouE MChristensenHJurekeJ 2001 J. Catal. 199 132 10.1006/jcat.2000.3153
- BaranE J 1990 Catal. Today 8 133 10.1016/0920-5861(90)87015-U
- ViswanathanB 1992 Catal. Rev. Sci. Engg. 34 337 10.1080/01614949208016316
- KeshavarajaARamaswamyA V 1994 Indian J. Eng. Mater. 1 229
- TejucaL GFierroJ LTasconJ M DEleyD DPinesHWeiszP B 1989 Adv. Catal. 36 237 10.1016/S0360-0564(08)60019-X
- GoldschmidtV M 1926 Strifter Norske Videnskaps Akad. (Oslo), Mat-Natur. 14 477
- JohnssonMLemmensP 2006 Novel Materials vol 4 Chichester Wiley pp 2098 2106 2098–106
- NagaiTItoWSakonT 2007 Solid State Ion. 177 3433 10.1016/j.ssi.2006.10.022
- JunwuZXiaojieSYanpingWXujieY 2007 J. Rare Earths 25 601 10.1016/S1002-0721(07)60570-5
- MuneeswaranMJegatheesanPGiridharanN V 2013 J. Exp. Nanosci. 8 341 10.1080/17458080.2012.685954
- DoggaliPGrassetFCadorORayaluSTeraokaYLabhsetwarN 2014 J. Environ. Chem. Eng. 2 340 10.1016/j.jece.2014.01.002
- LabhsetwarNDoggaliPChankapurePValechhaDLokhandeSWatanabeARayaluSHanedaHMitsuhashiT 2009 Top. Catal. 52 1909 10.1007/s11244-009-9362-6
- DhakadMRayaluSKumarRDoggaliPBakardjievaSSubrtJDevottaSLabhsetwarN 2008 Catal. Letters 121 137 10.1007/s10562-007-9310-7
- KumarSVinuASubrtJSnejanaSRayaluSTeraokaYLabhsetwarN 2012 Catal. Today 198 125 10.1016/j.cattod.2012.06.015
- KumarSTeraokaYJoshiA GRayaluSLabhsetwarN 2011 J. Mol. Catal. A: Chem. 348 42 10.1016/j.molcata.2011.07.017
- ShabbirGQureshiA HSaeedK 2006 Mater. Lett. 60 3706 10.1016/j.matlet.2006.03.093
- GuoXMengaMDaiFLiQZhangZJiangZZhangSHuangcaTianjinY 2013 Appl. Catal. B 142 278 10.1016/j.apcatb.2013.05.036
- DoggaliPKusabaHTeraokaYChankapurePRayaluSLabhsetwarN 2010 Catal. Comm. 11 665 10.1016/j.catcom.2010.01.019
- DoggaliPRayaluSTeraokaYLabhsetwarN 2015 J. Environ. Chem. Eng. 3 420 10.1016/j.jece.2014.11.019
- FinoDSpecchiaV 2004 Chem. Eng. Sci. 59 4825 10.1016/j.ces.2004.07.012
- TyagiA K 2007 J. Mater. Res. 22 587 10.1557/jmr.2007.0101
- RussoNFinoDSaraccoGSpecchiaV 2008 Catal. Today 137 306 10.1016/j.cattod.2007.09.010
- FinoDRussoNSaraccoGSpecchiaV 2003 J. Catal. 217 367 10.1016/S0021-9517(03)00143-X
- PurohitR DTyagiA KMathewsM DSahaS 2000 J. Nucl. Mater. 280 51 10.1016/S0022-3115(00)00026-X
- DeganelloFMarcìGDeganelloG 2009 J. Eur. Ceram. Soc. 29 439 10.1016/j.jeurceramsoc.2008.06.012
- BurangeA SJayaramR VShuklaRTyagiA K 2013 Catal. Commun. 40 27 10.1016/j.catcom.2013.05.019
- ShuklaRBeraA KYusufS MDeshpandeS KTyagiA KHermesW 2009 J. Phys. Chem. C 113 12663 10.1021/jp903013u
- ShuklaRManjannaJBeraA KYusufS MTyagiA K 2009 Inorg. Chem. 48 11691 10.1021/ic901735d
- ShuklaRAryaATyagiA K 2010 Inorg. Chem. 49 1152 10.1021/ic9020096
- ShaoZXiongGShengSChenHLiL 1998 Stud. Surf. Sci. Catal. 118 431 10.1016/S0167-2991(98)80209-7
- LabhsetwarN KWatanabeAMitsuhashiT 2003 Appl. Catal. B 40 21 10.1016/S0926-3373(02)00123-6
- DoggaliPKusabaHTeraokaYLabhsetwarN 2013 Top. Catal. 56 457 10.1007/s11244-013-9996-2
- HeckRFarrautoR 2001 Appl. Catal. A 221 443 10.1016/S0926-860X(01)00818-3
- LuPLiCZengGXieXCaiZZhouYZhaoYZhanQZmZ 2012 J. Hazard. Mater. 272 199 10.1016/j.jhazmat.2012.02.052
- KittelsonD B 1998 J. Aero. Sci. 29 575 10.1016/S0021-8502(97)10037-4
- WichmannH-ESpixCTuchTWölkeGPetersAHeinrichJKreylingW GHeyderJ 2000 Daily mortality and fine and ultrafine particles in Erfut, Germany. Part I: role of particle number and particle mass Research Report 98 Cambridge, MA Health Effects Institute
- ZelenkaPKrieglerWHetzogP LCartellieriW P 1990 Ways Toward the Clean Heavy-Duty Diesel Warrendale, PA SAE International
- NeeftJ P AMakkeeMMoulijnJ A 1996 Fuel Process. Technol. 47 115 10.1016/0378-3820(96)01002-8
- LinFWuXLiuSWengDHuangY 2013 Chem. Eng. J. 226 105 10.1016/j.cej.2013.04.006
- Van SettenB A A LSchoutenJ MMakkeeMMoulijnJ A 2000 Appl. Catal. B 28 253 10.1016/S0926-3373(00)00182-X
- LoxE SVan den TillaartJLeyrerLEckhoffS 1996 Appl. Catal. B 10 53 10.1016/0926-3373(96)00023-9
- FinoDRussoNSaraccoGSpecchiaV 2006 J. Catal. 242 38 10.1016/j.jcat.2006.05.023
- ShangguanW FTeraokaYKagawaS 1996 Appl. Catal. B 8 21 10.1016/0926-3373(95)00070-4
- RussoNFinoDSaraccoGSpecchiaV 2005 J. Catal. 229 459 10.1016/j.jcat.2004.11.025
- TeraokaYKanadaKKagawaS 2001 Appl. Catal. B 34 73 10.1016/S0926-3373(01)00202-8
- FinoDSpecchiaV 2004 Chem. Eng. Sci. 59 4825 10.1016/j.ces.2004.07.012
- ZhangGZhaoZLiuJXuJJingYDuanAJiangG 2009 J. Rare Earths 27 955 10.1016/S1002-0721(08)60369-5
- PengXLinHShangguanWHuangZ 2007 Catal. Commun. 8 157 10.1016/j.catcom.2006.04.015
- UraBNskiJ TZawadzkiMGomezM JLopezA BLopez SuarezF E 2011 Catal. Today 176 169 10.1016/j.cattod.2010.11.097
- BialobokBTrawczynskiJRzadkiTMistaWZawadzkiM 2007 Catal. Today 119 278 10.1016/j.cattod.2006.08.024
- CaudaEFinoDSaraccoGSpecchiaV 2004 Top. Catal. 30 31 10.1023/b:toca.0000029766.91816.d2
- LabhsetwarN KDhakadMRayaluaSKumarRSubrtJHanedaHDevottaSMitsuhashiT 2007 Top. Catal. 299 42 10.1007/s11244-007-0195-x
- DoggaliPKusabHTeraokYChankapurePRayaluSLabhsetwarN 2010 Catal. Comm. 11 665 10.1016/j.catcom.2010.01.019
- DoggaliPKusabHRayaluSTeraokaYLabhsetwarN 2013 Top. Catal. 56 457 10.1007/s11244-013-9996-2
- CrutzenP L 1971 J. Geophys. Res. 76 7311 10.1029/JC076i030p07311
- KramlichJ CLinakW P 1994 Progr. Energy Combust. Sci. 29 149 10.1016/0360-1285(94)90009-4
- WojtowiczM APelsJ RMoulijnJ A 1993 Fuel Process. Technol. 34 1 10.1016/0378-3820(93)90061-8
- RodheH 1990 Science 248 1217 10.1126/science.248.4960.1217
- vanA A RStewardR J 1994 Fertilizer Res. 37 213 10.1007/bf00748940
- JacobD J 1999 Introduction to Atmosphere Chemistry Princeton, NJ Princeton University Press
- SeinfeldJ HPandisS N 1998 Atmospheric Chemistry and Physics 2nd edn New York Wiley Interscience
- IPCC 2013 Observations: atmosphere and surface forcing Climate Change 2013: The Physical Science Basis Cambridge Cambridge University Press
- US EPA 2014 Inventory of US Greenhouse Gas Emissions and Sinks Washington, DC US Environmental Protection Agency
- http://www.ghgonline.org/
- ReimerR ASlatenC SSeapanMLowerM WTomlinsonP E 1994 Environ. Progr. 13 134 10.1002/ep.670130217
- SoeteG G D 1993 Revue de l’Institute Français du Pétrole 48 413 10.2516/ogst:1993026
- Perez-RamirezJKapteijnFSchoffelKMoulijnJ A 2003 Appl. Catal. B: Environ. 44 117 10.1016/S0926-3373(03)00026-2
- KapteijnFMirasolJ RMoulijnJ A 1996 Appl. Catal. B: Environ. 9 25 10.1016/0926-3373(96)90072-7
- ReddyP S SPashaNRaoM G V CLingaiahNSuryanarayana I PrasadP S S 2007 Catal. Commun. 8 1406 10.1016/j.catcom.2006.11.039
- TzitziosV KGeorgakilasV 2005 Chemosphere 59 887 10.1016/j.chemosphere.2004.11.021
- BurchRDaniellsS TBreenJ PHuP 2004 J. Catal. 224 252 10.1016/j.jcat.2004.03.027
- SatsumaAMaeshimaHWatanabeKSuzukiKHattoriT 2000 Catal. Today 63 347 10.1016/S0920-5861(00)00478-8
- AlonsoF J PCabreraI MGranadosM LF KapteijnFFierroJ L G 2006 J. Catal. 239 340 10.1016/j.jcat.2006.02.008
- GrangerPP EstevesPS KiegerSL NavascuesLG LeclercqG 2006 Appl. Catal. B: Environ. 62 236 10.1016/j.apcatb.2005.07.015
- CentiGPerathonerU SVazzanaFMarellaMTomaselliMMantegazzaM 2000 Adv. Environ. Res. 4 325 10.1016/S1093-0191(00)00032-0
- ChellamUXuZ PZengH C 2000 Chem. Mater. 12 650 10.1021/cm990355l
- PieterseJBooneveldSBinkR 2004 Appl. Catal. B: Environ. 51 215 10.1016/j.apcatb.2004.02.013
- RamirezJ PKapteijnFMulGMoulijnJ A 2001 Chem. Comm. 8 693 10.1039/b100953m
- KondratenkoE VKondratenkoV ASantiagoMRamírezJ P 2008 J. Catal. 256 248 10.1016/j.jcat.2008.03.016
- KumarSRayaluSRussoNKanadeG SKusabaHTeraokaYLabhsetwarN 2009 Catal. Lett. 132 248 10.1007/s10562-009-0092-y
- LabhsetwarN 2009 Catal. Today 141 205 10.1016/j.cattod.2008.03.032
- vandenB R WBooneveldSPelsJ RBakkerD FVerhaakM J F M 2001 Appl. Catal. B: Environ. 32 73 10.1016/S0926-3373(00)00294-0
- RussoNMesciaDFinoDSaraccoGSpecchiaV 2007 Ind. Eng. Chem. Res. 46 4226 10.1021/ie0612008
- AliniSBasileFBlasioliSRinaldiCVaccariA 2007 Appl. Catal. B: Environ. 70 323 10.1016/j.apcatb.2005.12.031
- DacquinJ PDujardinCGrangerP 2008 Catal. Today 137 390 10.1016/j.cattod.2008.01.016
- IvanovD VPinaevaL GIsupovaL ASadovskayaE MProsvirinI PGerasimovE YYakovlevaI S 2013 Appl. Catal. A: Gen. 457 42 10.1016/j.apcata.2013.03.007
- KumarSTeraokaYJoshiA GRayaluSLabhsetwarN 2011 J. Mol. Catal. A: Chem. 348 42 10.1016/j.molcata.2011.07.017
- KumarSVinuASubrtJSnejanaSRayaluSTeraokaYLabhsetwarN 2012 Catal. Today 198 125 10.1016/j.cattod.2012.06.015
- RaghuvanshiS PChandraARaghavA K 2006 Energy Conversation Manag. 47 427 10.1016/j.enconman.2005.05.007
- Chandra1HPaliwalSTripathiA 2013 Int. J. Eng. Sci. 2 1
- IEA 2010 Power Generation From Coal. Measuring and Reporting Efficiency Performance of and CO2 Emissions Paris International Energy Agency
- OlivierJ G JJanssens-MaenhoutGMunteanMPetersJ A H W 2013 Trends in Global CO2 Emissions PBL Netherlands Environmental Assessment Agency
- HoughtonH 2004 Global Warming. The Complete Briefing 3rd Edn New York Cambridge University Press
- ItoOChinoKMarushimaE S SBerginsCWuS 2008 Hitachi Rev. 571 66
- RichardV N 2010 Nature 463 871 10.1038/463871a
- FolgerP 2014 Carbon capture and sequestration: research, development, and demonstration at the US Department of Energy September 30 CRS Report R42496
- IEA 2010 Carbon capture and storage: progress and next steps 2010 Report to the Muskoka 2010 G8 Summit Paris International Energy Agency
- IPCC 2005 IPCC Special Report on Carbon Dioxide Capture and Storage Cambridge Cambridge University Press
- IshidaMZhengDAkehataT 1987 Energy 12 147 10.1016/0360-5442(87)90119-8
- HossainM MDe LasaH I 2008 Chem. Eng. Sci. 63 4433 10.1016/j.ces.2008.05.028
- FangHHaibinLZengliZ 2009 Int. J. Chem. Eng. 2009 710515 10.1155/2009/710515
- MoghtaderiB 2012 Energy Fuel 26 15 10.1021/ef201303d
- OlajireA A 2013 J. CO2 Utilization 4 74 10.1016/j.jcou.2013.10.004
- SaeidiSSaidinaA N A RezaR M 2014 J. CO2 Utilization 5 66 10.1016/j.jcou.2013.12.005
- TianLZhaoX HLiuB SZhangW D 2009 Energy Fuel 23 607 10.1021/ef800647n
- FooS YChengC KNguyenT HAdesinaA A 2010 Ind. Eng. Chem. Res. 49 10450 10.1021/ie100460g
- NajeraMSolunkeRGardnerTVeserG 2011 Chem. Eng. Res. Des. 89 1533 10.1016/j.cherd.2010.12.017
- OrtizMGayánPDe DiegoL FGarcía-LabianoFAbadAPansM AAdánezA 2011 J. Power Sources 196 4370 10.1016/j.jpowsour.2010.09.101
- MarkstromPLinderholmCLyngfeltA 2013 Int. J. Greenhouse Gas Con. 15 150 10.1016/j.ijggc.2013.01.048
- NalbandianLEvdouAZaspalisV 2011 Int. J. Hydrogen Energy 36 6657 10.1016/j.ijhydene.2011.02.146
- MattissonTJohanssonMLyngfeltA 2003 Energ. Fuel 17 643 10.1021/ef020151i
- De DiegoL FGarcia-LabianoFAdanezJGayanPAbadACorbellaB MPalaciosJ M 2004 Fuel 83 1749 10.1016/j.fuel.2004.03.003
- RouxSBensakhriaAAntoniniG 2009 Int. J. Chem. React. Eng. 4 38A
- SiriwardaneRTianHRichardsGSimonyiTPostonJ 2009 Energ. Fuel 23 3885 10.1021/ef9001605
- AbadAAdanezJGarcia-LabianoFDe DiegoL FGayanPCelayaJ 2007 J. Chem. Eng. Sci. 62 533 10.1016/j.ces.2006.09.019
- ChoPMattissonTLyngfeltA 2005 Ind. Eng. Chem. Res. 44 668 10.1021/ie049420d
- DaiX PLiR JYuC CHaoZ P 2006 J. Phys. Chem. B 110 22525 10.1021/jp063490b
- DaiX PYuC CWuQ 2008 J. Nat. Gas Chem. 17 415 10.1016/S1003-9953(09)60019-0
- DaiX PYuC CLiR JWuQHaoZ P 2008 J. Rare Earth 26 76 10.1016/S1002-0721(08)60041-1
- DaiX PYuC CLiR JWuQShiK JHaoZ P 2008 J. Rare Earth 26 341 10.1016/S1002-0721(08)60041-1
- DaiX PYuC CWuQ 2008 Chin. J. Catal. 29 954 10.1016/s1872-2067(08)60077-7
- RydenMLyngfeltAMattissonTChenDHolmenABjorgumE 2008 Int. J. Greenhouse Gas Con. 2 21 10.1016/S1750-5836(07)00107-7
- HeFLiXZhaoKHuangZWeiGLiH 2013 Fuel 108 465 10.1016/j.fuel.2012.11.035
- EvdouAZaspalisVNalbandianL 2010 Fuel 89 1265 10.1016/j.fuel.2009.09.028
- Ping DaiXLiJTao FanJSheng WeiWXuJ 2012 Ind. Eng. Chem. Res. 51 11072 10.1021/ie300033e
- CoronadoJ MFresnoFHernández-AlonsoM DPortelaR 2013 Design of Advanced Photocatalytic Materials for Energy and Environmental Applications London Springer
- ChenXShenSGuoLMaoS S 2010 Chem. Rev. 110 6503 10.1021/cr1001645
- IbhadonA OFitzpatrickP 2013 Catalysts 3 189 10.3390/catal3010189
- MaedaK 2013 Chem. Commun. 49 8404 10.1039/c3cc44151b
- TownsendT KBrowningN DOsterlohF E 2013 Energy Environ. Sci. 5 9543 10.1039/c2ee22665k
- KudoAKatoH 2000 Chem. Phys. Lett. 331 373 10.1016/S0009-2614(00)01220-3
- QuYDuanX 2013 Chem. Soc. Rev. 42 2568 10.1039/C2CS35355E
- ChenXShangguanW 2013 Front. Energy 7 111 10.1007/s11708-012-0228-4
- XuYZhaoWXuRShiYZhangB 2013 Chem. Commun. 49 9803 10.1039/c3cc46342g
- JingLZhouWTianGFuH 2013 Chem. Soc. Rev. 42 9509 10.1039/c3cs60176e
- ShiJGuoL 2013 Prog. Nat. Sci. Mater. Int. 22 592 10.1016/j.pnsc.2012.12.002
- CastelliI EThygesenK SJacobsenK W 2013 MRS Proc. 1523 mrsf12-1523-qq07-06 10.1557/opl.2013.450
- TijareS NJoshiM VPadoleP SMangrulkarP ARayaluS SLabhsetwarN K 2012 Int. J. Hydrogen Energy 37 10451 10.1016/j.ijhydene.2012.01.120
- WatrasAPązikRDereńP J 2013 J. Lumin. 133 35 10.1016/j.jlumin.2011.12.031
- TijareS NBakardjievaSSubrtJJoshiM VRayaluS SHishitaSLabhsetwarN K 2014 J. Chem. Sci. 126 517 10.1007/s12039-014-0596-x
- MangrulkarP AKotkondawarA VMukherjeeSJoshiM VLabhsetwarN KSarmaD DRayaluS S 2014 Energy Environ. Sci. 7 4087 10.1039/c4ee02444c
- HamannT 2014 Science 345 1566 10.1126/science.1260051
- WangHZhangLChenZHuJLiSWangZLiuJWangX 2014 Chem. Soc. Rev. 43 5234 10.1039/C4CS00126E
- LiuDGangishettyM KKellyT L 2014 J. Mater. Chem. A 2 19873 10.1039/C4TA02637C
- KamatP V 2014 J. Am. Chem. Soc. 136 3713 10.1021/ja501108n