
Abstract
Different structural chemistries resulting from the Pb2+ lone-pair electrons in the PbMO3 perovskites are reviewed. The Pb2+ lone-pair electrons enhance the ferroelectric transition temperature in PbTiO3, stabilize vanadyl formation in PbVO3, and induce a disproportionation reaction of CrIV in PbCrO3. A Pb2+ + NiIV = Pb4+ + NiII reaction in PbNiO3 stabilizes the LiNbO3 structure at ambient pressure, but an A-site Pb4+ in an orthorhombic perovskite PbNiO3 is stabilized at modest pressures at room temperature. In PbMnO3, a ferroelectric displacement due to the lone pair electron effect is minimized by the spin–spin exchange interaction and the strong octahedral site preference of the MnIV/III cation. PbRuO3 is converted under pressure from the defective pyrochlore to the orthorhombic (Pbnm) perovskite structure where Pb–Ru interactions via a common O −2p orbital stabilize at low temperature a metallic Imma phase at ambient pressure. Above Pc a covalent Pb–Ru bond is formed by Pb2+ + RuIV = Pb4+ + RuII electron sharing.
1. Introduction
The PbMO3 perovskites manifest an interplay between the Pb–6s,6p lone-pair hybrids and the transition-metal MO3 perovskite framework. The classic case of ferroelectricity in PbTiO3 has been well-discussed and understood in the literature. The application of high-pressure synthesis in recent years has made available PbMO3 with M covering almost all 3d transition metals and revived interest in the study of PbRuO3. The relevant features of this MO3 framework with which the Pb2+ lone pairs interact are reviewed in the discussion of the properties of each PbMO3 compound.
The geometric perovskite tolerance factor1 where (A–O) and (M–O) are the equilibrium bond lengths, is a useful parameter. Compounds AMO3 with a t < 1 can be stabilized in the perovskite structure by a cooperative rotation of the MO6/2 octahedra. The versatility of the MO3 framework has provided a wealth of information on the properties of the d electrons in the transition-metal AMO3 perovskites [Citation1]. However, with a t > 1, the (M–O) bonds are either stretched beyond their equilibrium length or the structure transforms to a hexagonal polytype [Citation2]. Where the (A–O) bond is more compressible than the (M–O) bond, high pressure lowers the effective tolerance factor to stabilize the cubic perovskite structure, and the cubic perovskite is commonly retained at ambient pressure and temperature. Most of the PbMO3 perovskites are synthesized under high pressure at high temperature.
In a cubic AMO3 perovskite, the oxide-ion orbitals that π-bond with the M-cation d orbitals also σ-bond with the larger A cation in the body center of the MO3 framework. Competition for covalent bonding with those O–2p electrons means that the stronger is the covalent component of the A–O bond, the weaker is the covalent component of the M–O pπ bonding and therefore the weaker the interatomic M–O–M interactions via the O–2pπ bonding of the MO3 framework. This competition is particularly significant for PbMO3 perovskites where the d electrons of the MO3 framework are at the crossover from localized to itinerant behavior as a result of the M–O–M interactions; the equilibrium (M–O) bond length is larger for localized than for itinerant d electrons.
The large mass of the Pb atom separates the energies of the 6s and 6p electrons; the 6s electrons are more stable than the 6p electrons because the 6s wave functions have a finite density at the nucleus whereas the 6p wave functions have a node at the nucleus. The larger the mass of the atom, the greater the difference in the screening of the outer s and p electrons from the nuclear charge by the core electrons. As a result, two cation valences are stable: Pb2+: 6s26p0 and Pb4+:6s06p0. Hybridization of the 6s and 6p orbitals creates an electron density with its center displaced from the nucleus; and in an oxide, the energy cost to form a 6s,6p hybrid can be more than compensated by the gain in bond energy by the covalent component of the Pb–O bond with the empty hybrid orbital on the opposite side of the nucleus. This hybridization is the origin of the lone-pair phenomenon of a Pb2+ ion in an oxide. However, in a PbMO3 perovskite, the increased Pb–O covalent bonding competes with π-bonding d-orbital covalent bonding, which introduces an interplay between the lone pair on a Pb2+ ion and orbital ordering or the strength of the M–O–M interactions in the MO3 framework. All data of crystal structure and physical properties of PbMO3 (M = Ti, V, Cr, Mn, Fe, Ni, Ru) reviewed in this article are listed in table
Table 1. Crystal structure and lattice parameters at room temperature and physical properties of PbMO3.
2. Data and discussion
2.1. PbTiO3
A t > 1 in BaTiO3 stretches the Ti–O bond lengths to make the symmetric O–Ti–O bonds along the 〈100〉 axes unstable relative to a cooperative, ferroelectric displacement of the TiIV towards one of its two neighbors: first along one principal axis, then along two, and finally along all three axes as the temperature is lowered [Citation4]. With a room-temperature t 1, SrTiO3 remains cubic to below room temperature, undergoing a cooperative rotation of the TiO6/2 octahedra about a principal axis below 110 K to increase the Sr–O bond energy [Citation15]. Since the ground state is nearly degenerate with a ferroelectric phase, an 18O isotope substitution induces a soft-mode phase transition [Citation16]. Although the size of the Pb2+ ion (1.63 Å) is comparable to that of the Sr2+ ion (1.58 Å) (tabulated value for XII coordination), nevertheless the difference is large enough to create a t > 1 that induces a ferroelectric displacement of the Pb2+ by a hybridization of the Pb–6s,6p orbitals. As a results, PbTiO3 undergoes a cooperative ferroelectric displacement of the TiIV along a single principal axis at a Curie temperature Tc = 764 K [Citation17, Citation18], significantly higher than Tc = 393 K of BaTiO3 [Citation19]. PbTiO3 has a room-temperature axial ratio c/a = 1.064 and exhibits a negative thermal expansion to Tc because the cation displacements that enhance the bonding also increase the volume [Citation17, Citation18]. In PbTiO3, ordering of the lone pair on the Pb2+ ions along a principal axis stretches the O–Ti–O bond lengths in this direction to induce the ferroelectric TiIV displacement. The c-axis O–2pπ orbitals are not impacted by the high-temperature transition of the Pb2+ lone-pair hybrid orbitals; only those in the basal plane. The interplay between the lone-pair ordering on the Pb2+ and the ferroelectric TiIV displacements of the TiO3 framework yields a high ordering temperature and restricts the TiIV displacement to a single axis. The ferroelectric displacement in PbTiO3 has been fully justified by the density functional theory (DFT); readers are referred to [Citation20] for more detailed information.
2.2. PbVO3
An octahedral-site VIV has a single d electron in a threefold-degenerate manifold of π-bonding orbitals. In the perovskites CaVO3 and SrVO3, the V–O–V interactions are strong enough to create an itinerant-electron band 1/6-filled; these perovskites are metallic [Citation21]. On the other hand, the single d electron per TiIII in the RTiO3 perovskites (R = Y or a rare earth) are localized [Citation22]. The smaller ΔE to transfer an O–2p electron to an empty π-bonding t2 orbital of VIV enhances the covalent contribution to the V–O bond, thereby increasing the interatomic V–O–V interactions.
Whereas the orbital degeneracy of a localized-electron manifold is removed by a cooperative Jahn–Teller orbital ordering, as is illustrated in the RTiO3 perovskites, the itinerant-electron orbital degeneracy of a VIV may be removed by formation of the vanadyl ion (V = O)2+ in which the d electron is ordered into a plane perpendicular to the V = O bond axis. The vanadyl formation places the VIV in a square-pyramidal site with five rather than six near-neighbor oxide ions.
This situation is well-illustrated by VO2, which crystallizes in the rutile structure, but undergoes a first-order metal-insulator transition below 340 K. The low-temperature monoclinic phase is characterized by a cooperative shift of the VIV ions perpendicular to the c-axis to form short V = O bonds with an ordering of the single d electron per VIV into a narrow c-axis band that is split in two by a charge-density wave of V–V dimers containing V–V bonding across shared site edges [Citation1].
With this background, we now look at the structure of PbVO3 prepared under high pressure at high temperature [Citation5]. The lattice instabilities associated with the lone-pair ordering on the Pb2+ together with lattice instabilities associated with orbital degeneracies stabilize competitive phases in attempts at ambient-pressure synthesis. Nevertheless, under high pressure at high temperature, the PbVO3 phase is preferred and is retained on removal of the pressure at room temperature. Like PbTiO3, the high-pressure PbVO3 phase is tetragonal under ambient conditions with the V atoms displaced from symmetric O–V–O bonding along the c-axis; but where PbTiO3 has a room-temperature axial ratio c/a = 1.062, PbVO3 has a much larger c/a = 1.229 as a result of formation of the vanadyl (V = O)2+ ion [Citation5]. The tetragonal PbVO3 structure is shown in figure . The single d electron per VIV is localized and ordered into the xy orbital in the basal plane; the in-plane interatomic V–O–V interactions between the localized-electron spins give a broad maximum in the paramagnetic susceptibility near 200 K [Citation6] typical of 2D antiferromagnetic interactions [Citation24].
Figure 1. Crystal structure of perovskite PbVO3 (based on the CIF data from [Citation5]).
![Figure 1. Crystal structure of perovskite PbVO3 (based on the CIF data from [Citation5]).](/cms/asset/08e3ebb3-7a4d-43eb-ac96-31da6521bdfa/tsta_a_11661309_f0001_oc.jpg)
Since the high-pressure synthesis at high temperature favors the PbVO3 phase, it provides a means of obtaining a nearly pure PbVO3 phase. Once this phase is quenched to ambient pressure and room-temperature, a natural next step is to investigate what happens to this tetragonal phase when it is subjected to high pressure at room temperature. High pressure can be expected to change the VIV coordination from square-pyramidal to octahedral by a reduction of both the c-axis V displacements from an asymmetric to a symmetric bond and by suppression of lone-pair 6s, 6p hybridization until a cubic PbVO3 perovskite structure is attained. It took 10 years for this investigation to be performed, and then the motivation was a search for a new high-temperature superconductor. However, this investigation proves to be of fundamental importance for our understanding of the transition in a single-valent oxide from localized to itinerant d-electron behavior. In the Mott–Hubbard scenario, the dynamics of electron lattice interactions is neglected; orbital degeneracies are removed by the itinerant-electron dispersion curve of width W, and the electron–electron correlation energy U associated with excitations of electrons to atomic states of lower positive valence is able to open a gap in the density of one-electron states where a U > W is realized with a reduced bandwidth W. This scenario is the basis of the DFT + U calculations in which U appears as an adjustable parameter. The alternative view for a mixed-valent system is to invoke strong electron–lattice interactions that trap electrons in M-cation sites with (M–O) bond lengths appropriate to the M-cation valence. Alternatively, it has been argued from the Virial Theorem for central-force fields [Citation25] that the equilibrium (M–O) bond length for itinerant electrons (Mott–Hubbard scenario) should be smaller than that for localized electrons and, therefore, that there can be a first-order bond contraction at a transition from localized to itinerant electrons in a single-valent system like PbVO3. The DFT + U calculations work well for a single-valent MO3 configuration with itinerant or localized d electrons because a periodic potential is preserved in a single-valent compound. Figure (a) shows the anticipated reduction in the axial ratio c/a of the tetragonal PbVO3 phase under pressure at room temperature over the pressure range 0 < P ≤ 2.5 GPa and figure (b) shows a first-order phase change between the tetragonal and cubic phase in the interval 1.16 < P < 3.82 GPa. However, despite a drop in resistivity by 5 orders of magnitude across the transition, this itinerant-electron cubic phase is still semiconductive between 2 and 300 K up to 11.3 GPa, indicating a U > W. Since cubic SrVO3 is metallic, the data indicate that the Pb2+ ion is narrowing the π∗ bandwidth compared to its width in SrVO3. The availability of the Pb–6p0 orbitals for covalent bonding with the O–2p orbitals in competition with the V–O2pπ bond covalence reduces the interatomic V–O–V interactions in cubic PbVO3 relative to its value in cubic SrVO3; this reduction appears to render a W < U in PbVO3, but there is no evidence for either polaronic conduction in cubic PbVO3 or localized-electron orbital order. On removal of the high pressure, the cubic perovskite PbVO3 reverts back to the tetragonal phase in the interval 1.11 < P < 2.02 GPa, which has prevented the measurement of the magnetic properties of the strongly correlated itinerant-electron phase with U > W. Of particular interest would be the extent to which the itinerant-electron dispersion width W suppresses the intraatomic orbital angular momentum and spin relative to that for a localized-electron phase. The RTiO3 system contains a single localized t2 electron per TiIII ion for comparison [Citation22].
Figure 2. Lattice parameters as a function of pressure for perovskite PbVO3, data are after [Citation26].
![Figure 2. Lattice parameters as a function of pressure for perovskite PbVO3, data are after [Citation26].](/cms/asset/ccb83600-effa-432d-9249-5efd78af90ec/tsta_a_11661309_f0002_ob.jpg)
2.3. PbCrO3
Octahedral-site CrIV has two d electrons per CrIV, which prevents removal of the d-state degeneracy by the formation of asymmetric O–Cr–O bonds; therefore, a c-axis ordering of the Pb2+ lone pairs can only be stabilized by a single-atom removal of a localized-electron orbital degeneracy on the (CrO3)2− framework. The intra-atomic spin–spin interactions responsible for Hund’s highest multiplicity rule on a free CrIV ion can be expected to narrow the bandwidth W of a cubic (CrO3)2− framework and to enlarge the correlation energy U to create a U > W; but a π∗ band 1/3-filled would be at the crossover from antiferromagnetic to ferromagnetic spin ordering, which can be unstable relative to a charge-disproportionation as is found in metallic Mn [Citation27]. Nature resolves this problem in ferromagnetic CrO2 by providing a c-axis band as well as a π∗ band; the π∗ band is ¼-filled, and the remaining electrons are ordered into the c-axis band as localized electrons. The localized-electron spins are aligned parallel to the spins of a ferromagnetic π∗ band ¼-filled [Citation27]. In a cubic- perovskite (CrO3)2− framework, this option is not available. A remaining option for removal of the orbital degeneracy at an octahedral-site CrIV is a charge disproportionation (CD).
A CD of octahedral-site CrIV is illustrated by the spinel compound Li[MnCr]O4 [Citation28]. As Li is removed, the CrIII are progressively transformed to CrIV and the Li vacancies create empty tetrahedral sites in the interstitial space that do not share a face with a neighboring Li+ ion. These tetrahedral sites can accept a CrVI cation, which triggers the CD reaction 3CrIV = CrVI + 2CrIII. In a PbCrO3 perovskite, lone-pair hybridizations on the Pb2+ can perturb the (CrO3)2− framework so as to allow a CD reaction within the framework. The t3 configurations on the CrIII have a localized spin S = 3/2.
A cubic PbCrO3 perovskite phase was first stabilized under high pressure at high temperature by Roth and DeVries in 1967 [Citation29, Citation30], but the structure exhibits several unusual properties. First, its lattice constant a = 4.00 Å is significantly larger than that expected based on the equilibrium (Pb–O) bond length for a Pb2+ ion. Second, it exhibits a Type-G antiferromagnetic order (all nearest-neighbor spins antiparallel) with a TN = 240 K [Citation29], nearly three times higher than the TN = 90 K of CaCrO3; CaCrO3 has localized CrIV d electrons (its tolerance factor is t < 1) that are ordered to give Type-C antiferromagnetic order (ferromagnetic c-axis interactions) [Citation31]. PbCrO3 also has a much higher resistivity and energy gap than are found in CaCrO3 and SrCrO3 [Citation31]. In addition, PbCrO3 has a highly irregular peak profile in the powder x-ray diffraction pattern [Citation32] and an unusually high thermal factor at the Cr site characteristic of a static disorder of the Cr atoms. By allowing the Cr atoms to occupy the general Wickoff position 8g (x,x,x) with x = 0.035, the overall intensity and peak profile matched well with the structural refinements. Although neutron diffraction does not show evidence of long-range order of the Cr displacements, the refinement of the oxygen positions shows a thermal parameter 10 times larger than is found in other CrIV-containing perovskites: [Citation33] viz. 2 Å3 versus 0.2 and 0.44 Å3. These observations are important experimental clues in support of a CD model, and a DFT + U calculation gave an independent prediction of the CD model [Citation7].
By allowing the cubic phase of PbCrO3 to expand its volume at room temperature by 10% on removing the high pressure, the calculation showed the volume expansion leads to significant atomic displacements from the ideal cubic positions. Two CD reactions were shown to be close in energy: 3+ + 3+ + 6+ and 3+ + 5+. The optimized model structure with the 3+ + 3+ + 6+ configuration is shown in figure . Pb atoms are displaced substantially towards CrIII, which are displaced slightly from the ideal cubic positions. The CrIII and CrVI are coordinated, respectively, by six and by four oxygen nearest neighbors. In the distorted tetrahedron, there are two short Cr=O bonds and two relatively long Cr–O bonds. The two bond lengths, 1.65 and 1.78 Å are comparable to the 1.59 and 1.78 Å for the calculated Cr=O and Cr–O bonds in a tetrahedral CrO2(OH)2 cluster in the gas phase. The oxygen bridging to a neighboring CrIII moves towards the CrVI to give a long 2.40 Å CrIII–O bond length while the other five Cr–O bonds of CrIII are 2.00 ± 0.05 Å, close to a typical CrIII–O bond length of 2.0 Å in the RCrO3 perovskites [Citation34]. The oxygen bridging from the CrIII to the Pb2+ form Pb–O bond lengths of 2.43–2.56 Å, substantially shorter than the 2.85 Å estimated for the ideal cubic phase. These shorter Pb–O bonds indicate the Pb–6s,6p hybridization plays an important role in stabilizing the CD of the (CrO3)2− framework.
Figure 3. The optimized CD model structure with bond length in Å together with the ideal cubic structure shown as faded structure at the background; the data are from [Citation7].
![Figure 3. The optimized CD model structure with bond length in Å together with the ideal cubic structure shown as faded structure at the background; the data are from [Citation7].](/cms/asset/1a3b8dce-bdff-4a2b-b8dd-d0796a02525c/tsta_a_11661309_f0003_oc.jpg)
Figure shows the calculated relative energies E per formula unit for the ideal cubic perovskite (blue) and the CD (red) phases of PbCrO3 as a function of the cell volume V. The application of hydrostatic pressure at room temperature should suppress the CD model in favor of the ideal cubic model. Indeed, with a sample synthesized with laser heating under the pressure of a diamond anvil cell, an earlier report [Citation35] signaled a large (9.8%) volume and resistivity collapse in a reversible cubic-cubic first-order transition at a Pc = 1.4 ± 0.2 GPa at room temperature. The abrupt transition was found at a Pc near 3 GPa in a sample synthesized with a large-volume press [Citation7]. In this latter experiment, the large thermal parameters of the diffraction profiles were also found to drop precipitously in the high-pressure phase. The resistivity drops by over two orders of magnitude at Pc; the conductivity of the high-pressure polycrystalline phase was characteristic of a narrow-band conductor with grain-boundary scattering. The magnetic properties of the high-pressure phase have not been determined because this phase is not retained on removal of the pressure.
Figure 4. The relative energies (E) per formula unit (f.u.) for the ideal cubic perovskite (blue) and the CD (red) phases of PbCrO3 as a function of cell volume (V); the data are from [Citation7].
![Figure 4. The relative energies (E) per formula unit (f.u.) for the ideal cubic perovskite (blue) and the CD (red) phases of PbCrO3 as a function of cell volume (V); the data are from [Citation7].](/cms/asset/90b8ac78-654f-4565-a621-713c697d9a46/tsta_a_11661309_f0004_oc.jpg)
2.4. Solid solutions
Arevalo-Lopez and Alario-Franco [Citation36] have investigated the three solid-solution systems PbTi1-xVxO3, PbTi1-xCrxO3, PbV1−xCrxO3. The system PbTi1−xVxO3 remains tetragonal for all x with a continuous increase with x of the c axis and volume. The tetragonal to cubic transition decreases from Tc = 763 K for x = 0–730 K for x = 0.2, but for x ≥ 0.3, the samples decompose on heating at ambient pressure. The authors suggest there is a percolation threshold for the PbVO3 phase near x = 0.3. Similarly, a percolation threshold for the PbCrO3 appears near x = 0.3 in PbTi1−xCrxO3 and PbV1−xCrxO3, a tetragonal/cubic two-phase region appearing at x = 0.3 in the former and an abrupt tetragonal to cubic transition occurring in the interval 0.3 < x < 0.4 in PbV1−xCrxO3. The Pb2+ displacements become disordered at the percolation threshold in the two Cr substituted systems.
2.5. PbMnO3
The octahedral-site MnIV ions of stoichiometric PbMnO3 should have a non-degenerate t3 manifold with a localized-electron spin S = 3/2 if there is no overlap of the MnIV/MnIII redox energy by the Pb–6s states. With no orbital degeneracy to be removed, the (MnO3)2− framework of a cubic PbMnO3 can be expected to resist deformation of the cubic framework by ordering of a lone-pair on the Pb2+ ions. Except for a reduction of the Mn–O–Mn antiferromagnetic interactions compared to SrMnO3 by a larger competition of the O–2p orbitals for covalent bonding to the Pb2+–6p orbitals, the behavior of PbMnO3 might be similar to that of SrMnO3if the t > 1 does not stabilize a hybridization of the Pb2+−6s,6p electrons.
Both SrMnO3 and PbMnO3 have a tolerance factor t > 1, and both form a perovskite polytype. Under pressure at high temperature, both compounds crystallize in the 6H polytype in which the AO3 (111) planes of a cubic phase are stacked cubic, cubic, hexagonal along the c-axis. Application of higher pressure at high temperature normally converts 6H polytypes to the cubic 3C perovskite structure [Citation2]. Oka et al [Citation9] converted the 6H PbMnO3 polytype to the cubic perovskite structure by treating oxygen-stoichiometric 6H PbMnO3 under 15 GPa at 1000 °C. Unfortunately the cubic phase obtained was not oxygen-stoichiometric; iodometric titration showed a composition PbMnO2.94(1). SrMnO3 is notoriously sensitive to loss of oxygen [Citation37], which stabilizes a mixed-valent phase by the introduction of Mn3+; the Mn3+ introduces a ferromagnetic component into the MnO3 framework. The small tetragonal (c/a = 1.017) distortion of their 3C PbMnO3−δ and the reduction of the TN to 20 K compared to 233 K for oxygen-stoichiometric 3C SrMnO3 [Citation37] cannot be properly interpreted in terms of the relative properties of the Pb2+ and Sr2+ ions. As a matter of fact, refinement of the synchrotron x-ray diffraction pattern cannot distinguish the polar structure P4mm versus the non-polar structure P4/mmm of the 3C phase. A DFT calculation [Citation38] indicates that the P4mm phase has a slightly lower energy of the ground state than that in the P4/mmm phase. However, the calculation also predicts the P4mm phase is metallic. It remains to be clarified whether the lone-pair electrons would induce a ferroelectric displacement in oxygen stoichiometric PbMnO3.
2.6. PbFeO3
Tsuchiya et al [Citation10] have reported the synthesis of PbFeO3 under high pressure. In comparison with other PbMO3 perovskites, the study of PbFeO3 is incomplete. The report just shows that the x-ray diffraction pattern of the high-pressure product can be indexed by an orthorhombic perovskite cell, indicating a reduction of the tolerance factor to a t < 1 and therefore a transfer of electrons from Pb2+ to FeIV. The x-ray photoemission spectra indicate that the valance of iron in PbFeO3 is close to that of the FeIII in LaFeO3. The FeIV/FeIII couple has an energy that favors the reaction Pb2+ + 2FeIV = Pb4+ +2FeIII, but FeIII is stable in the presence of Pb2+. This situation is consistent with the observation of FeIII, but it leaves the valence state of Pb ambiguous: Pb2+ + Pb4+ versus Pb3+−6s1.
2.7. PbNiO3
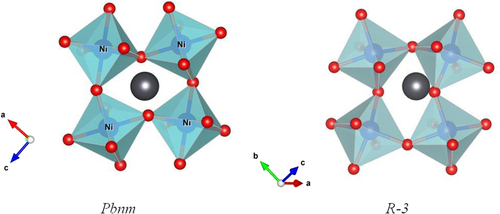
The NiIV/NiIII and NiIII/NiII redox energies are at the top of the O–2p bands [Citation39], and removal of Li from LiNiO2 results in loss of O2 as nearly all the Li is removed [Citation40]. It is easier to synthesize PbO2 than NiO2, which favors the reaction Pb2+ + NiIV → Pb4+ + NiII in PbNiO3. Although Pb4+ is stable in octahedral coordination, Inagauma et al [Citation11] have synthesized an orthorhombic (tolerance factor t < 1) perovskite PbNiO3 at 3 GPa and 800°C; the Pb4+ is displaced from the center of a (NiO3)4− framework to lower the number of nearest-neighbor oxygen atoms from 12. However, on removal of the pressure at room temperature and heating to only 400 K, the perovskite PbNiO3 transforms irreversibly to the LiNbO3 structure, which also consists of a framework of corner-shared octahedra. In the LiNbO3structure, the framework is rhombohedral (R-3); the Pb4+ are displaced along a [111] axis into an octahedral site. As pointed out [Citation11] and illustrated in figure , this transformation only requires a rearrangement of the cooperative rotations of the corner-shared octahedra of the framework that is triggered by a different displacement of the Pb4+ ions. The LiNbO3 structure of PbNiO3 is more stable at ambient pressure, which is why the transformation is irreversible on thermal cycling. However, under pressure the ambient-pressure PbNiO3 can be converted back to the perovskite structure in which the Pb4+ ion has a larger oxygen coordination. The geological literature has recognized that the LiNbO3 structure commonly transforms to perovskite under high pressure.
Figure 5. Two structural models of PbNiO3.
2.8. PbRuO3
A large cubic-field splitting of the 4d states stabilizes the low-spin configuration t4e0 at an octahedral-site RuIV. The Sr1−xCaxRuO3 system can be prepared as perovskites under ambient pressure; the Sr1-xBaxRuO3 perovskites with x > 0 have a tolerance factor t > 1 and require a high-pressure synthesis to form as cubic perovskites [Citation41–Citation43]. In ferromagnetic SrRuO3, the 4d electrons of the low-spin t4e0 manifold are at the crossover between localized and itinerant behavior; [Citation42] the RuO3 framework of the system Sr1−xCaxRuO3 evidences a Griffiths-type ferromagnetism with an eventual vanishing of long-range magnetic order in CaRuO3 [Citation41]. The high-pressure Sr1−xBaxRuO3 cubic phase exhibits strong itinerant-electron ferromagnetism; in BaRuO3, there is an abrupt loss of long-range magnetic order at a critical pressure Pc 8.6 GPa [Citation44] indicative of a first-order transition of the majority-spin electrons to itinerant behavior like the minority-spin electrons.
Although PbRuO3 has a perovskite tolerance factor t ≤ 1, PbRuO3 crystallizes at ambient pressure in the oxygen-deficient pyrochlore structure Pb2Ru2O7-x with in which Pb–6s, 6p hybrids project electron density into the oxygen vacancies. However, PbRuO3 can be stabilized as an orthorhombic (Pbnm) perovskite at 9 GPa and 1400 °C;[Citation45] but this phase transforms at ambient pressure to another orthorhombic (Imma) perovskite phase below 90 K [Citation12]. Since cracks are created in the sample during the first-order transition from Pnma to the Imma phase, the Imma phase was initially reported to be an insulator [Citation12]. By synthesizing the PbRuO3 perovskite phase in a Walker-type multianvil module, Cheng et al [Citation13, Citation14] were able to obtain a sample with large enough grains to measure the transport properties on a single grain; they showed that the perovskite PbRuO3 becomes a metal in the Imma phase as a result of broadening of the π∗ band and that the phase change below 90 K under ambient pressure is not caused by changes in the perovskite tolerance factor, but by an interaction between the Pb–6s, 6p hybrids and the Ru–4d electrons that broadens the π∗ band. In the perovskite system Sr1−xPbxRuO3, this interaction broadens the Ru–4d band to suppress systematically with increasing x the ferromagnetism of SrRuO3, figure [Citation14]. Plots of the Weiss constant θCW and the μeff obtained from the paramagnetic susceptibility, figures (a) and (b), show that suppression of the long-range magnetic order occurs where θCW passes through zero. A power-law analysis of the low-temperature resistivity ρ (T) = ρo + aTn to obtain the x dependence of n is shown in figure (c); the minimum n at x = 0.6 indicates that suppression of the magnetism is associated with the existence of a quantum critical point (QCP); the second minimum of n(x) at x
0.9 is a QCP associated with the appearance of the low-temperature Imma phase of perovskite PbRuO3. The low-temperature Imma phase can be suppressed by applying hydrostatic pressure [Citation13, Citation46]. Kusmartseva et al [Citation46] have monitored the transition by measuring resistivity ρ(T) under different pressures. They have found that the phase transition temperature Tt lowers gradually as pressure increases; it vanishes at about 5 GPa. The power-law analysis of ρ(T) under different pressures also confirms a QCP at around 5 GPa.
Figure 6. Phase diagram of Sr1−xPbxRuO3; the data are from [Citation14].
![Figure 6. Phase diagram of Sr1−xPbxRuO3; the data are from [Citation14].](/cms/asset/306472ad-058e-428b-81ae-a935a9d4bd34/tsta_a_11661309_f0006_oc.jpg)
Figure 7. (a), (b) Parameters from fitting the paramagnetic susceptibility to a Curie–Weiss law and (c) temperature dependence of resistivity ρ of Sr1−xPbxRuO3; the inset: n versus x obtained from the power-law analysis of ρ(T); the data are from [Citation14].
![Figure 7. (a), (b) Parameters from fitting the paramagnetic susceptibility to a Curie–Weiss law and (c) temperature dependence of resistivity ρ of Sr1−xPbxRuO3; the inset: n versus x obtained from the power-law analysis of ρ(T); the data are from [Citation14].](/cms/asset/64294b99-a14e-4851-aa2d-ba13658f88b0/tsta_a_11661309_f0007_oc.jpg)
Application of hydrostatic pressure P at room temperature to the PbRuO3 orthorhombic (Pbnm) perovskite phase suppresses the transition to the metallic Imma phase at low temperature. More interesting, the room-temperature lattice parameters and volume V of the Pbnm phase show a systematic contraction with P (figure ) until, at a Pc ≈ 32 GPa,the lattice parameter a jumps and the lattice parameters b, c and volume V drop abruptly at a first-order transition to a lower-symmetry Pbn21 phase. The volume decrease is about 6%. This behavior contrasts with that of a normal Pbnm perovskite, which transforms under pressure to a higher symmetry R-3c phase [Citation47].
Figure 8. Lattice parameters as a function of pressure for PbRuO3; the data are from [Citation48].
![Figure 8. Lattice parameters as a function of pressure for PbRuO3; the data are from [Citation48].](/cms/asset/b3e8d75e-3479-4dce-bd50-54b2222b32e5/tsta_a_11661309_f0008_ob.jpg)
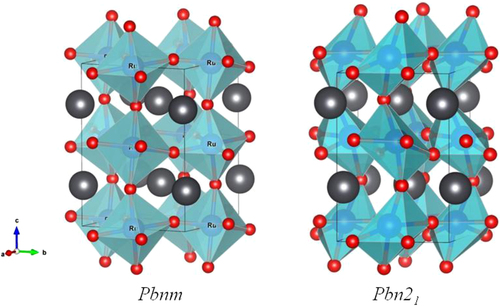
The PbRuO3 perovskite has an ambient-pressure tolerance factor t < 1, and a cooperative rotation of the RuO6/2 octahedra about a cubic [1-1 0] axis (b-axis of orthorhombic Pbnm) shifts the Pb atoms from the A-site center to coordinate it with fewer, but closer oxygen-atom nearest-neighbors. The small shift of the Pb atoms results in four Pb–Ru distances across a shared site face about 0.05 Å shorter than the average value. A distortion of the octahedral sites accompanies the cooperative rotation of the octahedra. In the Pbnm structure with a tolerance factor t near unity, the principal site distortion is a reduction from 90° of the O2–Ru–O2 bond angle that subtends the rotation axis [Citation14]. In contrast, the cooperative rotation of highly distorted RuO6/2 octahedra in the Pbn21 phase of figure creates eight different Pb–Ru distances [Citation48]. Most striking is a single Pb–Ru distance as short as 2.6 Å, about 0.6 Å shorter than the average value, which is reduced only slightly on traversing Pc. Kusmartseva et al [Citation46] have also found independently the pressure-induced phase transition at P ≈ 30 GPa by their structural study under high pressure. They have given only the change of lattice parameters on crossing the phase boundary.
Figure 9. Two structural models of PbRuO3.
A DFT calculation has shown [Citation48] that the short Pb–Ru bond is the result of an interaction of the Pb2+ lone-pair electrons with the two holes per Ru in the 4d-electron π∗ band in a shared-electron reaction Pb2+ + RuIV = Pb4+ + RuII shifted to the left. Figure shows the variations with volume in the relative internal energies per formula unit as obtained by the DFT calculation. By fitting the E–V data to the third-order Birch–Murnaghan equation, the enthalpy difference ΔH between the two phases (figure inset) clearly demonstrates the phase crossover. A density of states plot of the Pbnm phase at ambient pressure shows an overlap of the Pb–6s, Ru–4d, and O–2p states in the energy range EF−3 eV < E < EF + 1 eV. The covalent-like Pb–Ru bond formation in the Pbn21 phase leaves the corresponding antibonding orbitals completely empty; EF is located at the leading edge of the bonding states, which accounts for the observation that the Pbn21 phase remains a bad metal.
Figure 10. Variations in the relative internal energies (E) per formula unit (f.u.). for the Pbnm and Pbn21 phases of PbRuO3 as a function of volume (V) from the LDA calculation. Inset shows the enthalpy difference between the Pbnm and Pbn21 phases (ΔH = HPbn21 − HPbnm) as a function of pressure (P). The data are from [Citation48].
![Figure 10. Variations in the relative internal energies (E) per formula unit (f.u.). for the Pbnm and Pbn21 phases of PbRuO3 as a function of volume (V) from the LDA calculation. Inset shows the enthalpy difference between the Pbnm and Pbn21 phases (ΔH = HPbn21 − HPbnm) as a function of pressure (P). The data are from [Citation48].](/cms/asset/1803050d-11fa-4a8e-812f-842a200b3774/tsta_a_11661309_f0010_oc.jpg)
3. Conclusions
This review of the observed properties of the PbMO3 perovskites with M = Ti, Cr, Mn, Fe, Ni, and Ru leads to the following conclusions.
| (1) | A perovskite tolerance factor t > 1 for PbMO3 with M = Ti, Cr, Mn, Fe, Ni makes it necessary to use high-pressure synthesis to obtain a cubic 3C perovskite phase. Although PbRuO3 has a t < 1, the Pb2+ lone pair stabilizes an oxygen-deficient pyrochlore, so here also high-pressure synthesis is needed to obtain a perovskite phase. | ||||
| (2) | Where the cubic phase is stable at ambient temperature and pressure, a t > 1 stretches the Pb–O bonds from their equilibrium length to make the phase unstable with respect to a Pb2+ displacement from the center of symmetry of its site with an associated hybridization of the Pb–6s,6p orbitals, but creation of a polar state is confined by symmetry to a transition from a cubic to a tetragonal phase. In PbTiO3 and PbVO3 where the MIV cation has a | ||||
| (3) | As the atomic number of the M atoms increases, the energies of the MIV/MIII and MIII/MII redox energies decrease; where they cross the top of the Pb−6s band, electron transfer from the Pb2+−6s to the MIV/MIII or even to the MIII/MII redox couple occurs. This electron transfer reduces the perovskite tolerance factor to a t < 1 and an orthorhombic structure in which the Pb-atom displacements are non-ferroic. | ||||
| (4) | With three half-filled π-bonding d orbitals on MIV:d3 ions in octahedral sites, ferroic MIV displacements, charge disproportionation, and orbital ordering cannot occur. Moreover, Pb2+ displacements along a tetragonal axis are opposed by M–O–M interactions, which limits a displacement of the Pb2+ ion as has been argued by Hill [Citation49]. | ||||
| (5) | Competition between the Pb2+−6p and the empty MIV−3d orbitals for covalent bonding with the O−2pπ orbitals of the MO3 sublattice narrows any π∗ band in a single-valent MO3 perovskite sublattice; this competition may induce a transition from itinerant to localized d-electron behavior. Where the MIV:d2 electrons are localized by the band narrowing, applying hydrostatic pressure can induce a first-order transition to itinerant d electrons with a Mott–Hubbard U > W; and applying additional pressure may give a smooth transition to metallic conduction with a W > U. | ||||
Acknowledgments
The work was supported by NSF (DMR 1122603) and the Robert A Welch Foundation (F-1106).
References
- GoodenoughJ B 1971 Prog. Solid State Chem. 5 145 10.1016/0079-6786(71)90018-5
- GoodenoughJ BKafalasJ ALongoJ M 1972 Preparative Methods in Solid State Chemistry HagenmullerP New York Academic chapter 1
- ShahzadKNasir KhanMShabbirGBashirJ 2011 Ferroelectrics 414 155 161 155–61 10.1080/00150193.2011.577332
- KayH FVousdenP 1949 Phil. Mag. 40 1019 10.1080/14786444908561371
- ShpanchenkoR VChernayaV VTsirlinA AChizhovP SSklovskyD EAntipovE V 2004 Chem. Mater. 16 3267 10.1021/cm049310x
- OkaKYamadaIAzumaMTakeshitaSSatohK HKodaAKadonoRShimakawaY 2008 Inor. Chem. 47 7355 7359 7355–9 10.1021/ic800649a
- ChengJ-G 2015 Proc. Natl Acad. Sci. 112 1670 4 1670–4 10.1073/pnas.1424431112
- Arevalo-LopezA MDos Santos-GarciaA JAlario-FrancoM A Inorg. Chem. 48 92009
- OkaKAzumaMHiraiSBelikA AKojitaniHAkaogiMTakanoMShimakawaY 2009 Inorg. Chem. 48 2285 2288 2285–8 10.1021/ic802081f
- TsuchiyaTSaitoHYoshidaMKatsumataTOhbaTInagumaYTsuruiTShikanoM 2007 Mater. Res. Soc. Symp. Proc. 988 QQ09 QQ16 QQ09–Q16
- InagumaYTanakaKTsuchiyaTMoriDKatsumataTOhbaTHirakiKTakahashiTSaitohH 2011 J. Am. Chem. Soc. 133 16920 10.1021/ja206247j
- KimberS A JRodgersJ AWuHMurrayC AArgyriouD NFitchA NKhomskiiD IAttfieldJ P 2009 Phys. Rev. Lett. 102 046409 10.1103/PhysRevLett.102.046409
- ChengJ-GZhouJ-SGoodenoughJ B 2009 Phys. Rev. B 80 174426 10.1103/PhysRevB.80.174426
- ChengJ-GZhouJ-SGoodenoughJ B 2010 Phys. Rev. B 81 134412 10.1103/PhysRevB.81.134412
- CowleyR A 1964 Phys. Rev. 134 A981 10.1103/PhysRev.134.A981
- ItohMWangRInagumaYYamaguchiTShanY-JNakamuraT 1999 Phys. Rev. Lett. 82 3540 10.1103/PhysRevLett.82.3540
- ShiraneGHoshinoS 1951 J. Phys. Soc. Japan 6 265 10.1143/JPSJ.6.265
- ShiraneGSuzukiKTakedaA 1952 J. Phys. Soc. Japan 7 12 10.1143/JPSJ.7.12
- KayH FVousdenP 1949 Phil.Mag. 40 1019 10.1080/14786444908561371
- RabeK MAhnC HTrisconeJ-M 2007 Physics of Ferroelectrics, A Modern Perspective Berlin Springer
- FujimoriAHaseINamatameHFujishimaYTokuraYEisakiHUchidaSTakegaharaKde GrootF M F 1992 Phys. Rev. Lett. 69 1796 10.1103/PhysRevLett.69.1796
- InoueI HHaseIAiuraYFujimoriAHaruyamaYMaruyamaTNishiharaY 1995 Phys. Rev. Lett. 74 2539 10.1103/PhysRevLett.74.2539
- OkimotoYKatsufujiTOkadaYArimaTTokuraY 1995 Phys. Rev. B 51 9581 10.1103/PhysRevB.51.9581
- BonnerJ CFisherM E 1964 Phys. Rev. 135 A640 10.1103/PhysRev.135.A640
- GoodenoughJ B 2001 Structure & Bonding vol 98 ed J B Goodenough Berlin Springer
- BelikA AYamauchiTUedaHUedaYYusaHHiraoNAzumaM 2014 J. Phys. Soc. Japan 83 074711 10.7566/JPSJ.83.074711
- GoodenoughJ B 1963 Magnetism and the Chemical Bond New York Wiley-Interscience
- GoodenoughJ BManivannanV 1998 Denki Kagaku oyobi Kogyo Butsuri Kagaku 66 1173
- RothW LDeVriesR C 1967 J. Appl. Phys. 38 951 10.1063/1.1709698
- DeVriesR CRothW L 1968 J. Am. Ceram. Soc. 51 72 10.1111/j.1151-2916.1968.tb11839.x
- Komarekac 2011 Phys. Rev. B 84 125114 10.1103/PhysRevB.84.125114
- Arevalo-LopezA MAlario-FrancoM A 2007 J. Solid State Chem. 180 3271 10.1016/j.jssc.2007.09.017
- Castillo-MartínezEDuranAAlario-FrancoM A 2008 J. Solid State Chem. 181 895 904 895–904 10.1016/j.jssc.2008.01.022
- ZhouJ-SAlonsoJ APomjakushinVGoodenoughJ BRenYYanJ-QChengJ-G 2010 Phys. Rev. B 81 214115 10.1103/PhysRevB.81.214115
- XiaoWTanDXiongXLiuJXuJ 2010 Proc. Natl Acad. Sci. 107 14026 10.1073/pnas.1005307107
- Arevalo-LopezA MAlario-FrancoM A 2011 Inorg. Chem. 50 7136 7141 7136–41 10.1021/ic200680u
- ChmaissemODabrowskiBKolesnikSMaisJBrownD EKrukRPriorPPylesBJorgensenJ D 2001 Phys. Rev. B 64 134412 10.1103/PhysRevB.64.134412
- SubramanianS SNatesanB 2014 Adv. Mater. Res. 895 420 423 420–3 10.4028/www.scientific.net/AMR.895.420
- GoodenoughJ BKimY 2010 Chem. Mater. 22 587 10.1021/cm901452z
- ChebiamR VPradoFManthiramA 2001 Chem. Mater. 13 2951 7 2951–7 10.1021/cm0102537
- JinC-Q 2008 Proc. Natl Acad. Sci. 105 7115 10.1073/pnas.0710928105
- ChengJ-GZhouJ-SGoodenoughJ BJinC-Q 2012 Phys. Rev. B 85 184430 10.1103/PhysRevB.85.184430
- ChengJ-GZhouJ-SGoodenoughJ B 2013 Proc. Natl Acad. Sci. 110 13312 13315 13312–5 10.1073/pnas.1311871110
- ZhouJ-SMatsubayashiKUwatokoYJinC-QChengJ-GGoodenoughJ BLiuQ QKatsuraTShatskiyAItoE 2008 Phys. Rev. Lett. 101 077206 10.1103/PhysRevLett.101.077206
- KafalasJ ALongoJ M 1970 Mater. Res. Bull. 5 193 10.1016/0025-5408(70)90005-X
- KusmartsevaA FSinclairARodgersJ AKimberS A JAttfieldJ P 2013 Phys. Rev. B 87 165130 10.1103/PhysRevB.87.165130
- ZhouJ-SAlonsoJ AMuonzAFernandez-DiazM TGoodenoughJ B 2011 Phys. Rev. Lett. 106 057201 10.1103/PhysRevLett.106.057201
- ChengJ-G 2013 Proc. Natl Acad. Sci. 110 20003 10.1073/pnas.1318494110
- HillN A 2000 J. Phys. Chem. B 104 6694 6709 6694–709 10.1021/jp000114x