Abstract
Members of the Sphingomonas-related genera (Sphingomonas, Sphingobium, Novosphingobium and Sphingopyxis) are dominant in bacterial isolates from the floodwater of Japanese paddy fields. Fifty-eight Sphingomonas/Novosphingobium bacteriophages (phages) were isolated to elucidate their morphology, host range and phylogenetic diversity based on the capsid gene (g23) sequence. All of the phages were siphoviruses with isometric or elongated, icosahedral capsids and a long, non-contractile tail. The genomes were double-stranded DNA measuring either 40, 60, 100 or 160 kb. The host range of the phages was examined by infecting 16 bacterial isolates from the floodwater, belonging to Sphingomonas, Novosphingobium, Sphingopyxis and Porphyrobacter. The host range was widely different and varied between infection of only the host used for isolation and infection of hosts belonging to the three genera of Sphingomonas, Novosphingobium and Porphyrobacter. All phages had g23, indicating the ubiquity of the g23 gene among Myoviridae and Siphoviridae members. Every g23 sequence of the phages belonged to one of the six uncharacterized Paddy Groups proposed by CitationFujii et al. (2008). The g23 sequences were identical at the nucleotide level for several phages with isometric and elongated capsids with 60 and 160 kb genomes, and between some phages and the clones that were retrieved from distant paddy fields. This indicates the common occurrence of horizontal transfer of g23 in the paddy fields. The g23 sequence does not correlate with the host range of those phages. In addition, a larger degree of divergence of g23 from coliphage T4 in paddy fields compared to marine environments was estimated from the present study.
INTRODUCTION
Viruses are the most abundant biological entities in marine & freshwater environments, and they are generally threefold to 25-fold more numerous than bacteria in those environments (CitationWeinbauer 2004; CitationWommack & Colwell 2000). Viruses are also abundant in the floodwaters of paddy fields, a unique man-made aquatic environment that appears during the rice cultivation period in summer. In the floodwater of a Japanese paddy field, viral abundance ranged from 5.6 × 106 to 1.2 × 109 virus-like particles (VLP) mL−1 with a mean abundance of 1.5 × 108 VLP mL−1. The virus : bacterium ratio (VBR) fluctuated from 0.11 to 72, with a mean value of 8.1 (CitationNakayama et al. 2007a).
Notwithstanding the huge populations of viruses in aquatic environments, the abundance of bacteriophages (phages) infectious to the respective bacterial strains is generally so low (commonly less than 1 plaque forming unit (PFU) mL−1) that pre-concentration of water samples is necessary prior to inoculation and enumeration by culture methods (CitationHavelaar et al. 1993; CitationPaul et al. 1997). This is because phages possess extremely strict host specificities (CitationMoebus 1992), and the narrow host range of the respective phages might ensure the co-existence of diverse bacterial and viral communities in aquatic environments (CitationKimura et al. 2008). Although many studies have elucidated the host range of the isolated phages (e.g. CitationMoebus 1992), the susceptibility and immunity of bacteria to those phages have not been examined in terms of the phylogenetic relatedness of the bacteria.
Floodwater is also unique in terms of the abundance of phages of heterotrophic bacteria. In our previous study, 41 out of 44 floodwater samples, which were collected throughout the flooding period of rice cultivation, harbored more than 100 PFU mL-1 that were infectious to at least one of 18 heterotrophic bacterial strains (CitationNakayama et al. 2007b). In addition, among four Sphingomonas strains, phages of the Sphingomonas strain 070409 often formed more than 100 PFU mL−1 of floodwater (16 of 44 water samples). In contrast, the abundances of phages of the other three Sphingomonas strains were considerably lower (< 100 PFU mL−1), indicating that phages of Sphingomonas spp. were strain specific in the floodwater (CitationNakayama et al. 2007b).
The diversity of biological entities is evaluated from their phylogenetic diversity, but until recently it was common knowledge among virus taxonomists that there is no single gene that is commonly present in all virus genomes. Therefore, the total diversity of uncultured virus communities cannot be evaluated by approaches analogous to ribosomal DNA for microbial communities (CitationRohwer & Edwards 2002). However, recent developments in viral genomics have shown that comparable genomic information is preserved among viral subsets and this information can be used as a tool for the phylogenetic classification of viruses and the evaluation of viral diversity in the environment (CitationWeinbauer & Rassoulzadegan 2004). The most popular candidate genes for the evaluation of viral diversity are the capsid gene g23 for the T4-type phage subset in Myoviridae (CitationFilée et al. 2005), the DNA polymerase gene pol for the T7-type phage subset in Podoviridae (CitationBreitbart et al. 2004) and the photosynthesis gene psbA for cyanophages (CitationMillard et al. 2004; CitationZeidner et al. 2003). Thus, the recent development of viral genomics makes the evaluation of viral diversity in the environment possible for viral subsets for which the appropriate primers are available.
Recently, CitationJia et al. (2007) and CitationFujii et al. (2008) extracted soil DNA from Japanese paddy fields and obtained g23 clones by applying g23-specific primers to the DNA extracts. The clones had quite distinct deduced amino acid sequences from those obtained from marine environments and most of them formed six phylogenetically novel groups in the T4-type phage family (Paddy Groups I to VI; CitationFujii et al. 2008). This finding indicates that T4-type phage communities in paddy soil consist of previously uncharacterized members that are phylogenetically distant from those in marine environments.
The majority of viruses in the floodwater are not derived from irrigation water. These viruses are specific to the floodwater and exist by adsorbing on suspended soil particles, indicating their close relationship to soil viruses (CitationNakayama et al. 2007a). Based on the above overview, the present study aimed to isolate Sphingomonas phages from floodwater, examine their host ranges in relation to the phylogenetic positions of the bacteria and compare the phylogenetic position and diversity of the g23 sequences with those of soil and marine viral clones. We isolated Sphingomonas phages from the plaques that formed in a viral enumeration experiment using the most probable number (MPN) method from the floodwater by infecting the three Sphingomonas strains described above (CitationNakayama et al. 2007b). The morphology and host range of the phages for the Sphingomonas-related strains were examined. Members of the Sphingomonas-related genera (Sphingomonas, Sphingobium, Novosphingobium and Sphingopyxis) were chosen to be hosts in the present study because they dominated the bacterial communities isolated from the floodwater of a Japanese paddy field (CitationNakayama et al. 2006).
MATERIALS AND METHODS
Sphingomonas strains
Sixteen Sphingomonas strains were selected from 44 Sphingomonas isolates that were obtained previously from a paddy field (E2 field) located at Aichi-ken Anjo Research and Extension Center, Central Japan (latitude 34.8°N, longitude 137.5°E), considering the phylogenetic variety of the isolates based on the information of partial 16S rDNA sequences (CitationNakayama et al. 2006). As the genus Sphingomonas has now been reclassified into four genera, Sphingomonas, Sphingobium, Novosphingobium and Sphingopyxis (CitationTakeuchi et al. 2001), we sequenced almost the entire 16S rDNA sequence of the selected 16 Sphingomonas strains to understand the phylogenetic relationships among them in more detail.
Preparation of the DNA template and polymerase chain reaction (PCR) amplification were as described by CitationNakayama et al. (2006). The primer pairs were 27f (5′-AGA GTT TGA TCC TGG CTC AG-3′) and 1492r (5′-GGC TAC CTT GTT ACG ACT T-3′). All sequences were checked for their closest relatives by BLAST search within the National Center for Biotechnology Information and neighbor-joining trees were constructed using ClustalX 1.81 software (CitationThompson et al. 1997). The sequences were affiliated to the four genera: Sphingmonas (eight strains), Novosphingobium (six strains), Sphingopyxis (one strain) and Porphyrobacter (one strain). Sequences of 16S rDNA were deposited in the DNA Data Bank of Japan (DDBJ) under the accession numbers AB202144, AB202173, AB202176, AB202180, AB202184, AB202189, AB202193, AB202196, AB202200, AB202203, AB202204, AB202209, AB202211, AB202217, AB202227 and AB202236.
Isolation of virulent phages of the Sphingomonas strains
We had enumerated phages infectious to Sphingomonas (072822) and Novosphingobium (070409, 081631) strains in the floodwater of a Japanese paddy field during a long-term fertilizer trial over the 2005 rice cultivation period using the MPN method (CitationNakayama et al. 2007b). The long-term fertilizer trial had been conducted since 1925 and the field consisted of four plots: one without fertilizer (NF plot), one with the chemical fertilizers N, P and K applied (CF plot), one with the chemical fertilizers N, P, K and Ca applied (Ca plot) and one plot with both chemical fertilizers of N, P, K and Ca and compost applied (CM plot). The floodwater was collected 11 times over the rice cultivation period. The field management and the methods of floodwater sampling and MPN enumeration of virulent phages have been described previously (CitationNakayama et al. 2007a,Citationb). Virulent phages were isolated from the wells forming plaques in MPN multiple-well plates that had been used for the enumeration of phages (CitationNakayama et al. 2007b) and purified by plaque isolation on double-agar layer plates (CitationAdams 1959) at least three times. Only one phage was isolated from each floodwater sample as a general rule to avoid isolation of the same phages.
Determination of host range/infectivity
Host range infection experiments were carried out using the disk method. Sterile disks (Advantec filter paper 8 mm; Advantec Toyo, Tokyo, Japan) were placed on agar plates (0.9% nutrient agar; Eiken Chemical Company, Tokyo, Japan) that had been inoculated with bacterial strains before the agar medium had become solidified. Phage lysates (30 µL) were inoculated on the disk and incubated at 30°C for 7 days. The infection potential was determined from the halo development around the disk.
Induction assay of prophages
An aliquot of every precultured bacterial strain was inoculated into 10 mL of 9 g L−1 nutrient broth in a test tube and 0.1 mL of 0.1 g L−1 mitomycin C solution was added when the bacterial strains reached logarithmic growth at 30°C. After 1 week of further incubation, the cultures were passed through a 0.22-µm Nuclepore filter (Whatman Japan KK, Tokyo, Japan) and the filtrates were examined for prophage with a transmission electron microscope ([TEM] H-7500AMT Advantage HR; Hitachi High-Technologies, Tokyo, Japan). For the bacterial strains containing prophages, the absence of phages in the filtrates was checked with cultures without mitomycin C.
Electron microscopy
The size and morphology of the phages were examined with TEM as explained in CitationNakayama et al. (2007c). Phage lysates in 9 gL−1 nutrient broth were sonicated for 3 min (38 kHz, 80 W), vortexed for 30 s and passed through a 0.22-µm Nuclepore filter. After the filtrates were appropriately diluted, phage particles were collected on formvar-coated 400-mesh Cu grids by ultracentrifugation at 100,000 g for 60 min (Beckman TL-100 tabletop ultracentrifuge; Beckman Coulter, Tokyo, Japan), according to the method of CitationBørsheim et al. (1990). The phage samples were observed with a TEM and photographed after negative staining with 20 g L−1 uranyl acetate. As variation in the size of the capsid of the phages was less than 5 nm, we measured the size of the capsid of one phage. However, for phage tail length, we measured several individuals. We also measured the width of the tails of the phages to check whether it was less than 10 nm (a criterion for separating myoviruses from siphoviruses).
DNA extraction from phages
Extraction of phage DNA was carried out using the method of CitationWilson et al. (1993) with little modification. In brief, a phage suspension was inoculated into a 10-mL culture of exponentially growing host cells (9 g L−1 nutrient broth) and incubated at 30°C until the culture became clear (approximately 1 week). Host cell debris was precipitated by centrifugation at 5,400 g for 10 min. DNase and RNase were added to the supernatant to a final concentration of 10 µg mL−1 and incubated at 37°C for 60 min to degrade any bacterial DNA and RNA released during lysis. After the lysate was kept on ice with 1 mol L−1 NaCl for 1.5 h, the cell debris was removed by centrifugation at 5,400 g for 10 min followed by filtration of the supernatant with a 0.22-µm Nuclepore filter. Polyethylene glycol (grade 8000) was added to the supernatant (final concentration 10%[w/v]) and left at 4°C overnight. The phage was precipitated by centrifugation at 5400 g for 20 min and the pellet was suspended again in 500 µL of TE buffer (10 mM Tris-HCl, PH7.5 and 1 mM EDTA). The lysate was incubated with proteinase K (20 µg mL−1) in lysis buffer (10 mmol L−1 Tris–HCl, pH 7.6, 10 mmol L−1 ethylenediaminetetraacetic acid [EDTA] and 0.5% sodium dodecylsulfate) at 50°C overnight. After the addition of 10 µL of 5 mol L−1 NaCl, the DNA was extracted two or three times by phenol–chloroform–isoamyl alcohol (25:24:1, v/v/v) and once by chloroform–isoamyl alcohol (24:1, v/v), followed by precipitation with 10% (v/v) 3 mol L−1 NaOAc at pH 5.2 and isopropyl alcohol. The pellet was washed with 70% ethanol, dried and then resuspended in TE buffer.
Pulsed-field gel electrophoresis
The genome size of the phages was estimated by pulsed-field gel electrophoresis (PFGE). The extracted DNA was loaded onto a 1.0% agarose gel (Agarose NA; Amersham Biosciences, NJ, USA) in 0.5× TBE gel buffer (45 mmol L−1 Tris-borate and 1 mmol L−1 EDTA, pH 8.0) and CHEF DNA Size Standard (Bio-Rad, Tokyo, Japan) and Loading Quick λ/Hind III (Toyobo, Osaka, Japan) were used as size markers. The PFGE was carried out with a CHEF-DRII gel (Bio-Rad) under the following conditions: 0.5× TBE tank buffer, 0–40-s pulse rump, 200 V, 14°C and 16.5 h. Visualization of the bands was achieved by staining with SYBR Green I (Molecular Probes, Eugene, OR, USA) for 45 min and photographing under ultraviolet illumination.
Polymerase chain reaction amplification of g23 in the phage genomes
The primers used for the g23 amplification were MZIA1bis (5′-GAT ATT TGI GGI GTT CAG CCI ATG A-3′) and MZIA6 (5′-CGC GGT TGA TTT CCA GCA TGA TTT C-3′) (CitationFilée et al. 2005). The 50-µL PCR reaction mixture contained target DNA, 5 µL of 10× Ex Taq Buffer (20 mmol L−1 Mg2+; TaKaRa Bio, Otsu, Japan), 5 µL of 2.5 mmol L−1 deoxynucleoside triphosphates (dNTP), 0.5 µL of each 50 µmol L−1 primer, 0.3 µL of ExTaq DNA polymerase (TaKaRa Bio) and milli-Q water. Cycle conditions for the amplification were as follows: initial denaturation at 94°C for 1.5 min, 30 cycles of denaturation at 94°C for 1 min, annealing at 50°C for 1 min and extension at 72°C for 1 min and a final extension at 72°C for 5 min. The PCR product was electrophoresed on a 2% agarose gel and bands of the expected length for g23 fragments were cut out from the agarose gel. The DNA fragments were purified from the gel pieces using the QIAEX Gel Extraction Kit (Qiagen, Tokyo, Japan), suspended in 35 µL of TE buffer and then used as templates for cloning.
Sequencing and phylogenetic analysis of g23 fragments based on g23 amino acid sequences
Cloning was carried out for the purified PCR products before sequencing. The PCR products were ligated into the pT7 Blue T-vector (Novagen, Darmstadt, Germany) and the plasmids were transformed into Escherichia coli XLI blue competent cells (TaKaRa Bio). Extracted plasmid DNA was sequenced bi-directionally with an ABI PRISM 310 Genetic Analyzer or a 373S DNA Automated Sequencer (Applied Biosystems) using the DYEnamic ET Terminator Cycle Sequencing Kit (GE Healthcare Bio-Sciences, Piscataway, NJ, USA). The obtained nucleotide sequences were translated to deduced amino acid sequences using the SeqConv (for Windows) v0.81 software (European Bioinformatics Institute, Cambridge, UK). The identities of the deduced amino acid sequences were analyzed using the ClustalW program available from the DDBJ. The deduced amino acid residues of the partial g23 fragments were aligned with the representative sequences of T-evens, PseudoT-evens, SchizoT-evens and ExoT-evens, and with the marine g23 clones obtained by CitationFilée et al. (2005) and the soil g23 clones of CitationJia et al. (2007) and CitationFujii et al. (2008), using the ClustalX 1.81 software (CitationThompson et al. 1997). A neighbor-joining tree was constructed using the Molecular Evolutionary Genetics Analysis software (MEGA3.0) (CitationKumar et al. 2004). Partial g23 DNA sequences were deposited in the DDBJ under accession numbers AB331966–AB332023.
RESULTS
Phage morphology
In total, 58 virulent phages were isolated from the floodwater; of these, 45, 5 and 8 phages were obtained from strains 070409, 072822 and 081631, respectively (). All of the phages had an isometric or elongated icosahedral capsid with a long and thin non-contractile tail (diameter 8–9 nm; ). As Myoviridae and Siphoviridae have thick (16–20 nm) and thin (7–10 nm) tails, respectively, the morphology of the tails indicated that all of the phages belonged to the family Siphoviridae in the order Caulovirales (CitationFauquet et al. 2005).
Genome size of the phages
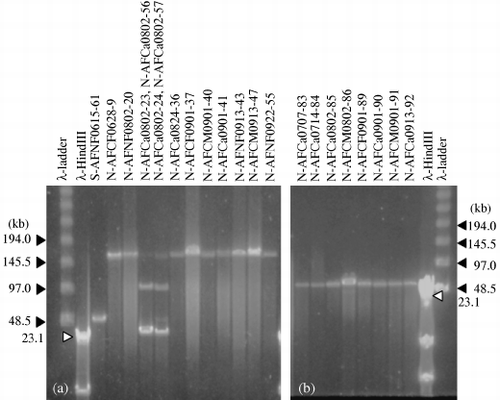
All of the phages had double-stranded DNA genomes and the genome sizes were 40, 60, 100 or 160 kb (). Of the 45 phages isolated with strain 070409 as the host, 37 phages had 160 kb genomes and four phages from the floodwater collected on 15 June 2005 had 60 kb. On 2 August 2005, two phages had 40 kb and the other two phages had 100 kb. Phages of strain 072822 had 60 or 160 kb genomes, and all of the phages of strain 081631 had 60 kb genomes ().
Host range of the phages
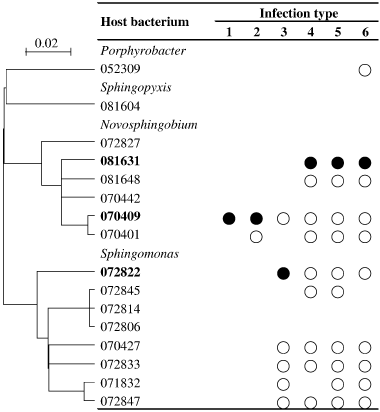
The host ranges of the phages were grouped into six infection types (), and the types of infection of the phages are summarized in . Phages N-AFCa0615-6 and N-AFNF0913-43 lysed only strain 070409, the original host strain used for isolation of those phages. In contrast, seven phages induced lysis in 10 of 16 bacterial strains (), and the host range covered three genera, including the genus Porphyrobacter for four phages (N-AFCa0707-83, N-AFCa0714-84, N-AFCa0802-85 and N-AFCM0802-86).
Table 1 Morphology genome size, host range and phylogenetic affiliation of the g23 genes of respective isolated phages
Figure 1 Pulsed-field gel electrophoresis images of the genomes of 21 phages. (a) Phages with genomes of 40, 60, 100 and 160 kb. Phages N-AFCa0802-23, N-AFCa0802-24, N-AFCa0802-56 and N-AFCa0802-57 were purified individually and subjected to sequence analyses. (b) Phages with genomes of 60 kb.
The host ranges of the phages isolated with strain 070409 were narrow, infecting one or two bacterial strains. Host ranges of the phages of strain 072822 were intermediate, infecting bacteria belonging to two genera (six bacteria). The host ranges of the phages of strain 081631 were broad, infecting bacteria belonging to two or three genera (9–10 bacteria), although strains 070409 and 081631 showed close phylogenetic relationships with each other. Phages infecting bacterial strains 081604, 07827, 070442, 072806 and 072814 were not isolated in the present study ().
Induction assay of the prophages
Prophage induction with mitomycin C was observed for three bacterial strains; strain 072833 was polylysogenic and had two prophages with an isometrically icosahedral, no-tail capsid (diameter 90–100 nm) and a filamentous form (380–3,500 × 10–20 nm) (CitationNakayama et al. 2007b). Prophages of the other two strains were also polylysogenic and had two prophages with an isometrically icosahedral, no-tail capsid (diameter 50 nm) and a filamentous form (12,00–4,050 × 15 nm) for strain 072806 and three prophages with an isometrically icosahedral, no-tail capsid (diameter 50 nm) and two filamentous forms (160–210 × 20 nm and 1,940–4,200 × 15 nm) for strain 072814. These filamentous phages were microscopically similar to those observed by CitationBradley (1967) and CitationYu et al. (2006).
Figure 2 Host range patterns of the phages isolated in the present study. Bacterium from which the phage was isolated (•) and bacterium that the phage can infect (○). The phylogenetic relationships among the hosts are shown on the left-hand side. The scale bar represents the number of nucleotide substitutions per residue.
Phylogenetic positions of the phages estimated from the g23 sequence
The PCR amplification of the g23 gene was successful for all of the isolated phages, indicating that it is useful for phylogenetic studies. The DNA sequences of the PCR fragments revealed that g23 had two highly conserved segments of amino acid residues (111–127 and 261–304 of the sequence of phage T4 [CitationFilée et al. 2005]). The sequence of g23 was completely the same among 13 phages (VI*) and between two phages (N-AFCa0615-6 and N-AFCM0913-47 in the III*** group) at the nucleotide level, and another two sequences were also identical among three and two phages (III*** and VI**, respectively) at the amino acid level ().
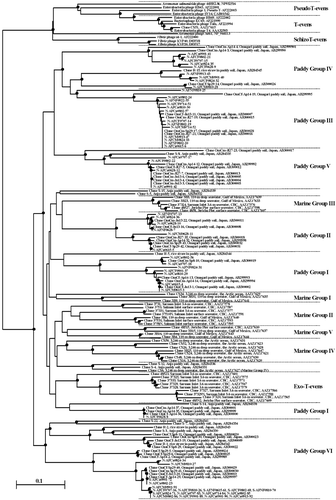
A phylogenetic tree was constructed () using g23 sequences of the isolated phages and those of T-evens, PseudoT-evens, SchizoT-evens and ExoT-evens from the culture collection, clones of marine origins (CitationFilée et al. 2005; CitationHambly et al. 2001; CitationTétart et al. 2001) and soil clones (CitationFujii et al. 2008; CitationJia et al. 2007). All of the g23 sequences from the phages were grouped into the six Paddy Groups proposed by CitationFujii et al. (2008). Phages of strain 070409 were distributed into all six Paddy Groups, whereas all of the phages of strains 072822 and 081631 belonged only to Paddy Group VI.
The g23 sequences of 10 phages were completely identical to clones found in another Japanese paddy soil at the nucleotide and amino acid levels (CitationFujii et al. 2008). In addition, g23 sequences of N-AFCa0615-5, AFCa0615-6 and N-AFCM0913-47, and S-AFCa0615-72 were identical to clones OmCf-Sp29-42 and OmCM-Sp29-43, clones OmCm-0727-21 and OmCm-Sp29-17, and clones OmCf-Ap14-29 and OmCm-Sp29-41, respectively, at the nucleotide level. In addition, N-AFNF0802-20, N-AFCMAg24-33, N-AFNF0628-7, N-AFCF0628-8, N-AFCa0802-23 and N-AFCa0901-42 showed sequences identical to several clones of CitationFujii et al. (2008) at the amino acid level ().
DISCUSSION
Morphology and genome size of the phages isolated from the floodwater
Although the isolated phages had isometric or elongated icosahedral capsids with long and non-contractile tails, indicating their affiliation with Siphoviridae (), their genome sizes were relatively large, ranging from 40 to 160 kb. CitationWichels et al. (1998) studied the genome sizes of 22 marine phages and found that siphoviruses had genomes ranging from 25 to 35 kb, podoviruses from 38 to 48 kb and myoviruses from 32 to 100 kb. Therefore, all of the phages obtained in the present study appear to be siphoviruses with unusually large genomes. All of the phages with genome sizes of 40 and 60 kb were isometrically icosahedral and smaller in capsid size (45 × 45 to 70 × 70 nm) than those with genome sizes of 100 and 160 kb (either isometric of 80 × 80 nm or elongated of 100 × 50 to 160 × 75 nm) (). These results agreed with the findings of CitationAuguet et al. (2006) that small viral-like genome sizes (< 100 kb) were correlated with small viral-like particle sizes (< 65 nm) in the Charente estuary (France).
The observation of only four genome sizes (40, 60, 100 and 160 kb) in the isolated phages in the present study does not imply that the phage communities in the floodwater are simple and is likely to be a result of the method of phage isolation. In the present study, only one phage was isolated from each floodwater sample, except for two phages from the 2 August 2005 sample (). Therefore, the dominant phages in the floodwater at the respective sampling time might have been isolated preferentially.
Figure 3 Neighbor-joining tree showing the relationships of the g23-segment amino acid sequences of the isolated phages with those from GenBank. Internal nodes with at least 50% bootstrap support (•). CBC, Coast of British Columbia. The scale bar represents the number of amino acid substitutions per residue.
Host range of the phages and the effect of prophage presence on subsequent phage infection
The host range was primarily determined by the host strain used for the isolation of the phages (; ). The host range was very narrow for phages from strain 070409 and broad for phages from strain 081631, covering three genera. However, because all of the phages had the potential to infect strain 070409 (), phages with a broad host range (Types 3–6 in ) could be isolated from strain 070409. A similar unexplainable result was that the phages isolated from strain 081631 as the host (Types 4–6) could infect strain 072822 (). Some physiological traits of the phages related to infection might be different in the floodwater from those after culture incubation with strain 081631. It is important to note that the five phages that were isolated from strain 072822 had 60-kb and 160-kb genomes and belonged to the Type 3 group (), indicating that host range is determined by the host strain used for isolation. In addition, the phages belonging to infection types 3–6 () infected two or three bacterial genera, but they could not infect some bacterial strains of the same genera with which they were isolated. This indicates that phylogenetic relatedness to the host bacterium was not a decisive factor in determining the infection of the phage.
Prophage induction was observed for strains 072806, 072814 and 072833. Many phages infected strain 072833, whereas no phage could infect strains 072814 and 072806 (). Thus, the effect of prophage(s) in phage infection was not recognized for strain 072833. A similar finding of no prophage effect on virulent phage infection was reported for bacterial strains in broad phylogenetic positions in our previous study (CitationNakayama et al. 2007b). It is generally believed that lysogenic bacteria are immune to superinfection by the same or related phage types (CitationDuckworth 1987; CitationMann 2003; CitationMarsh & Wellington 1994). All of the prophages detected by mitomycin C induction had tail-less capsids or filamentous forms and were different from the phages that were subjected to host range analysis at the family level (CitationFauquet et al. 2005). Therefore, the difference in sensitivity to phage lysis among those three bacterial strains appeared to be unrelated to superinfection immunity by prophages.
Diversity of the phages estimated from the g23 sequence
We believe that the isolated phages belong to the family Siphoviridae according to their morphology. CitationJenkins and Hayes (2006) recently compared the amino acid sequences of g23 fragments among 17 cyanophage isolates of Nodularia spumigena belonging to Myoviridae and Siphoviridae using CAP primers and found that the encoded proteins were 99% identical to T4 g23 homologues across all cyanophages. This finding indicates that g23 might be shared among Myoviridae and Siphoviridae members. Therefore, we amplified the capsid genes of isolated phages with g23-specific primers. Although all of the phages were siphoviruses in morphology, g23 was successfully amplified. Therefore, the present finding reconfirmed that g23 is shared among Myoviridae and Siphoviridae members.
Despite the indication from the host range and genome size that the isolated phages were not diverse, the diversity of phage communities estimated from g23 sequences was comparably higher than that of the clones obtained from paddy soils (). The phages of strain 070409 distributed into the six Paddy Groups of CitationFujii et al. (2008), which were proposed from unique sequences of g23 clones of paddy soil origins.
Identical g23 sequences at the nucleotide and amino acid levels were observed in three cases among the phages (shown by III***, VI* and VI** in ). The same g23 sequences were shared among several phages with genome sizes of 60 kb and 160 kb, as well as among some phages with isometric and elongated capsids in III*** and VI*, which is strongly indicative of a horizontal transfer of g23 among these phages in the paddy field. Furthermore, several identical g23 sequences at the nucleotide and amino acid levels were also observed between this study field (34.8°N, 137.5°E) and CitationFujii et al.'s (2008) paddy field (39°49′N, 140°50′E) (). CitationBreitbart et al. (2004) found that some DNA pol genes of T7-like phages were > 99% identical at the nucleotide level in multiple different environments (marine, freshwater, sediment, terrestrial, extreme and metazoan-associated environments). CitationShort and Suttle (2005) also reported that nearly identical (> 99%) g20 sequences (another capsid protein gene of phages) were recovered from marine and freshwater environments. These results indicate that closely related hosts and the phages infecting them are distributed widely across environments or that the horizontal transfer of genes occurs among phage communities in very different/distant environments. The horizontal transfer of g23 indicated for Paddy Groups III and VI () might suggest that it is not extraordinary among phage groups in paddy fields, although the remaining sequences in g23 need to be compared in the future to obtain definitive evidence of horizontal transfer of g23 among these sequences.
CitationShort and Suttle (2002) also demonstrated that closely related algal viruses occur in distant geographical locations. They found similar pol gene fragments of virus origin (> 98% sequence identity) in algal–virus communities from coastal sites in the Pacific Ocean in British Columbia, Canada and the Southern Ocean near the Antarctic Peninsula. Thus, sequence analyses of structural genes and the pol gene in various viral communities in aquatic environments suggest that viral diversity could be high on a local scale, but relatively low on a global scale (CitationBreitbart & Rohwer 2005). The present finding that g23 of Sphingomonas/Novosphingobium phages in the floodwater consisted of the six Paddy Groups of CitationFujii et al. (2008) and that there was no difference in phage members between distant paddy fields, supports the view of CitationBreitbart & Rohwer (2005).
Phylogenetic grouping of g23 into the Paddy Groups did not correlate to the infection type grouping in because the phages belonging to Paddy Group VI showed five infection types (2–6), whereas those of Paddy Group III showed two infection types (1 and 2). The nine phages whose g23 genes were indicative of horizontal gene transfer over long distances belonged to infection types 1 and 2 (the narrow type of infection range), except for S-AFCa0615-72 (type VI). Thus, a wide host range of phages might not be favorable for carriers of genes in horizontal transfer.
Every Paddy Group except for Paddy Group III consisted of two or more clades with large bootstrap values in , indicating further division of the present Paddy Groups into subgroups and/or new Paddy Group(s).
Phage communities in floodwater during the rice cultivation period
The results of the genome size, host range and g23 distribution in the phylogenetic tree indicate the stability of phage communities in floodwater during the rice cultivation period (; ), although the method of phage isolation used in the present study might have biased the evaluation of phage communities in the floodwater. Nevertheless, virulent phages of strain 070409 consistently appeared with a genome size of 160 kb and this might be the first clue of the phylogenetic stability of phage communities in floodwater. The phages of strain 070409 belonging to infection type 2 in were also isolated throughout the stages of rice growth. In addition, all Paddy Groups in generally consisted of phages isolated in the early, middle and late stages of rice growth.
Divergence of g23 from the prototype, coliphage T4, between paddy fields and marine environments
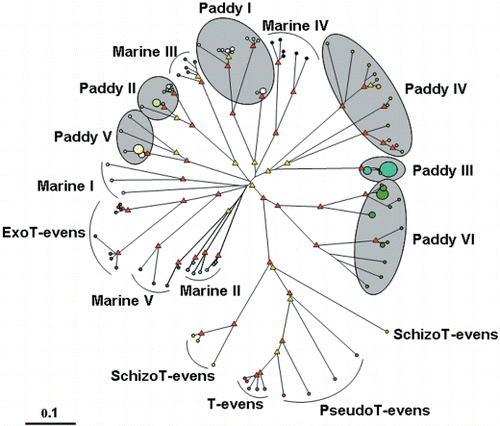
As the majority of g23 sequences in paddy field phages were clearly phylogenetically different from those in marine environments (), g23 divergence from coliphage T4 was compared between paddy fields and marine environments using an unrooted diagram (). To make the comparison clear, clones that did not belong to any groups and those that grouped marginally in respective groups (e.g. OmCf-Ap14-19, OmCm-Jl27-23, R-5 and CS50) were excluded (). As a result, some of the relatedness between the groups differed between and . For example, separated clusters of Paddy Group I in formed a cluster in . ExoT-evens, five Marine Groups and six Paddy Groups diverged differently from T-evens, PseudoT-evens and SchizoT-evens. ExoT-evens and Marine Groups I, II and V appeared to have diverged from the same stem, probably because they were all clones/isolates of marine origin (CitationFilée et al. 2005; CitationHambly et al. 2001). In addition, Marine Group III and Paddy Group II, Marine Group IV and Paddy Group I, and Paddy Group III and IV were estimated to have diverged from the same stems, respectively. In contrast, Paddy Groups V and VI diverged from their own stems. Thus, the characteristic divergence of marine g23 genes was from three stems in comparison to five stems for paddy field g23 genes (). Paddy Group VI, Paddy Groups III and IV and Paddy Group V also formed independent clusters in . And Marine Groups I and III and Paddy Groups I and II appeared to form a cluster consisting of three remote subclusters of Marine Group III with Paddy Group II, paddy Group I and Marine Group I. In addition, Marine Groups II, IV and V formed another cluster, and ExoT-evens and Paddy Group I formed the other cluster in . Thus, the stem for divergence of marine g23 genes was estimated to be from two or three in comparison to five or six for paddy field g23 genes in . Therefore, the g23 divergence in paddy fields was evaluated to be at least comparable to or greater than that in marine environments.
Figure 4 Unrooted diagram showing the relationships between the g23-segment amino acid sequences among phages and clones of T-evens, PseudoT-evens, SchizoT-evens, ExoT-evens and five Marine Groups and six Paddy Groups. The red and yellow triangles indicate internal nodes with at least 90% and 50% bootstrap support, respectively. The sizes of the circles at the ends of branches are proportional to the number of clones/phages and the smallest and largest circles represent, respectively, one and nine clones/phages. The scale bar represents the number of amino acid substitutions per residue.
Conclusions
All of the Sphingomonas phages isolated from the floodwater were estimated to be affiliated with Siphoviridae according to their morphology. However, their genome sizes were unusually large (40–160 kb) in comparison with those of common siphoviruses (25–35 kb). Several phages showed wide host ranges that cut across three genera, and the host range appeared to be determined by the host strain used for the phage isolation. All of the phages had g23, which indicates that g23 is commonly shared among myoviruses and siphoviruses in the floodwater. The g23 sequences of the phages in the floodwater were completely different from those in other aquatic environments, such as marine environments, and all of them belonged to one of the six Paddy Groups of CitationFujii et al. (2008), which strongly indicates that phages in the floodwater are of soil origin. In addition, the horizontal transfer of g23 appears to occur commonly among phages in the floodwater and between paddy fields over long distances.
ACKNOWLEDGMENTS
We thank Dr Hiroshi Miyake, Professor of the Graduate School of Bioagricultural Sciences, Nagoya University, for TEM technical assistance and staff members of the Aichi-ken Anjo Research and Extension Center for allowing us to sample the floodwater. We sincerely thank Dr Saman Bowatte, AgResearch-Grasslands, New Zealand, for proofreading and providing valuable comments on this manuscript. This work was funded by a Grant-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science (No. 18380051).
REFERENCES
- Adams , MH . 1959 . Bacteriophages , 450 – 451 . New York : Interscience Publishers .
- Auguet , JC , Montanié , H and Lebaron , P . 2006 . Structure of virioplankton in the Charente estuary (France): transmission electron microscopy versus pulsed field gel electrophoresis . Microb. Ecol , 51 : 197 – 208 .
- Børsheim , KY , Bratbak , G and Heldal , M . 1990 . Enumeration and biomass estimation of planktonic bacteria and viruses by transmission electron microscopy . Appl. Environ. Microbiol , 56 : 352 – 356 .
- Bradley , DE . 1967 . Ultrastructure of bacteriophages and bacteriocins . Bacteriol. Rev , 31 : 230 – 314 .
- Breitbart , M , Miyake , JH and Rohwer , F . 2004 . Global distribution of nearly identical phage-encoded DNA sequences . FEMS Microbiol. Lett , 236 : 249 – 256 .
- Breitbart , M and Rohwer , F . 2005 . Here a virus, there a virus, everywhere the same virus? . Trends Microbiol , 13 : 278 – 284 .
- Duckworth , DH . 1987 . “ History and basic properties of bacterial viruses ” . In Phage Ecology , Edited by: Goyal , SM , Gerba , CP and Bitton , G . 1 – 43 . New York : John Wiley and Sons .
- Fauquet , CM , Mayo , MA , Maniloff , J , Desselberger , U and Ball , KA , eds. 2005 . Virus Taxonomy: Eighth Report of the International Committee on Taxonomy of Viruses , London : Elsevier Academic Press .
- Filée , J , Tétart , F , Suttle , CA and Krisch , HM . 2005 . Marine T4-type bacteriophages, a ubiquitous component of the dark matter of the biosphere . Proc. Natl Acad. Sci. USA , 102 : 12471 – 12476 .
- Fujii , T , Nakayama , N Nishida , M . 2008 . Novel capsid genes (g23) of T4-type bacteriophages in a Japanese paddy field . Soil Biol. Biochem , 40 : 1049 – 1059 .
- Hambly , E , Tétart , F , Desplats , C , Wilson , WH , Krisch , HM and Mann , NH . 2001 . A conserved genetic module that encodes the major virion components in both the coliphage T4 and the marine cyanophage S-PM2 . Proc. Natl Acad. Sci. USA , 98 : 11411 – 11416 .
- Havelaar , AH , van Olphen , M and Drost , YC . 1993 . F-specific RNA bacteriophages are adequate model organisms for enteric viruses in freshwater . Appl. Environ. Microbiol , 59 : 2956 – 2962 .
- Jenkins , CA and Hayes , PK . 2006 . Diversity of cyanophages infecting the heterocystous filamentous cyanobacterium Nodularia isolated from the brackish Baltic Sea . J. Mar. Biol. Assoc. UK , 86 : 529 – 536 .
- Jia , Z , Ishihara , R , Nakajima , Y , Asakawa , S and Kimura , M . 2007 . Molecular characterization of T4-type bacteriophages in a rice field . Environ. Microbiol , 9 : 1091 – 1096 .
- Kimura , M , Jia , Z , Nakayama , N and Asakawa , S . 2008 . Viral ecology in soil: past, present and future perspectives . Soil Sci. Plant Nutr , 53 : 1 – 32 .
- Kumar , S , Tamura , K and Nei , M . 2004 . MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment . Brief. Bioinform , 5 : 150 – 163 .
- Mann , NH . 2003 . Phages of the marine cyanobacterial picophytoplankton . FEMS Microbiol. Rev , 27 : 17 – 34 .
- Marsh , P and Wellington , EMH . 1994 . Phage–host interactions in soil . FEMS Microbiol. Ecol , 15 : 99 – 108 .
- Millard , A , Clokie , RJ , Shub , DA and Mann , NH . 2004 . Genetic organization of the psbAD region in phages infecting marine Synechococcusstrains . Proc. Natl Acad. Sci. USA , 101 : 11007 – 11012 .
- Moebus , K . 1992 . Further investigations on the concentration of marine bacteriophages in the water around Helgoland, with reference to the phage–host systems encountered . Helgol. Meeresunters , 46 : 275 – 292 .
- Nakayama , N , Okabe , A , Toyota , K , Kimura , M and Asakawa , S . 2006 . Phylogenetic distribution of bacteria isolated from the floodwater of a Japanese paddy field . Soil Sci. Plant Nutr , 52 : 305 – 312 .
- Nakayama , N , Okumura , M , Inoue , K , Asakawa , S and Kimura , M . 2007a . Seasonal variations in abundances of virus-like particles and bacteria in the floodwater of a Japanese paddy field . Soil Sci. Plant Nutr , 53 : 420 – 429 .
- Nakayama , N , Okumura , M , Inoue , K , Asakawa , S and Kimura , M . 2007b . Abundance of bacteriophages of common heterotrophic bacteria in the floodwater of a Japanese paddy field . Soil Sci. Plant Nutr , 53 : 595 – 605 .
- Nakayama , N , Okumura , M , Inoue , K , Asakawa , S and Kimura , M . 2007c . Morphological analysis of viral communities in the floodwater of a Japanese paddy field . Soil Biol. Biochem , 39 : 3187 – 3190 .
- Paul , JH , Rose , JB , Jiang , SC , London , P , Xhou , X and Kellogg , C . 1997 . Coliphage and indigenous phage in Mamala Bay, Oahu, Hawaii . Appl. Environ. Microbiol , 63 : 133 – 138 .
- Rohwer , F and Edwards , R . 2002 . The phage proteomic tree: a genome based taxonomy for phage . J. Bacteriol , 184 : 4529 – 4535 .
- Short , CM and Suttle , CA . 2005 . Nearly identical bacteriophage structural gene sequences are widely distributed in both marine and freshwater environments . Appl. Environ. Microbiol , 71 : 480 – 486 .
- Short , SM and Suttle , CA . 2002 . Sequence analysis of marine virus communities reveals that groups of related algal viruses are widely distributed in nature . Appl. Environ. Microbiol , 68 : 1290 – 1296 .
- Takeuchi , M , Hamana , K and Hiraishi , A . 2001 . Proporsal of the genus Sphingomonas sensu stricto and three new genera, Sphingobium, Novosphingobium and Sphingopyxis, on the basis of phylogenetic and chemotaxonomic analysis . Int. J. Syst. Evol. Microbiol , 51 : 1405 – 1417 .
- Tétart , F , Desplats , C , Kutateladze , M , Monod , C , Ackermann , HW and Krisch , HM . 2001 . Phylogeny of the major head and tail genes of the wide-ranging T4-type bacteriophages . J. Bacteriol , 183 : 358 – 366 .
- Thompson , JD , Gibson , TJ , Plewniak , F , Jeanmougin , F and Higgins , DG . 1997 . The Clustal_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools . Nucleic Acids Res , 25 : 4876 – 4882 .
- Weinbauer , MG . 2004 . Ecology of prokaryotic viruses . FEMS Microbiol. Rev , 28 : 127 – 181 .
- Weinbauer , MG and Rassoulzadegan , F . 2004 . Are viruses driving microbial diversification and diversity? . Environ. Microbiol , 6 : 1 – 11 .
- Wichels , A , Biel , SS , Gelderblom , HR , Brinkhoff , T , Muyzer , G and Schütt , C . 1998 . Bacteriophage diversity in the North Sea . Appl. Environ. Microbiol , 64 : 4128 – 4133 .
- Wilson , WH , Joint , IR , Carr , NG and Mann , NH . 1993 . Isolation and molecular characterization of five marine cyanophages propagated on Synechococcus sp. strain WH7803 . Appl. Environ. Microbiol , 59 : 3736 – 3743 .
- Wommack , KE and Colwell , RR . 2000 . Virioplankton: viruses in aquatic ecosystems . Microbiol. Mol. Biol. Rev , 64 : 69 – 114 .
- Yu , MX , Slater , MR and Ackermann , HW . 2006 . Isolation and characterization of Thermus bacteriophages . Arch. Virol , 151 : 663 – 679 .
- Zeidner , G. , Preston , CM Delong , EF . 2003 . Molecular diversity among marine picophytoploankton as revealed by psbA analyses . Environ. Microbiol , 5 : 212 – 216 .