Abstract
Up to 70% of human genes are associated with regions of nonmethylated DNA called CpG islands (S. Saxonov, P. Berg, and D. L. Brutlag, Proc. Natl. Acad. Sci. U. S. A. 103:1412–1417, 2006). Usually associated with the 5′ end of genes, CpG islands are thought to impact gene expression. We previously demonstrated that the histone demethylase KDM2A is specifically recruited to CpG islands to define a unique chromatin architecture and highlight gene regulatory regions in large and complex mammalian genomes. This targeting relies on a zinc finger CXXC DNA binding domain (ZF-CXXC), but how this demethylase interfaces with CpG island chromatin in vivo remains unknown. Here we demonstrate, using defined chromatin templates in vitro and chromatin profiling in vivo, that nucleosomes are a major barrier to KDM2A binding and that CpG islands are directly interpreted by the ZF-CXXC domain through specific interaction with linker DNA. Furthermore, KDM2A appears to be constrained to CpG islands not only by their nonmethylated state but also by a combination of methylated DNA and nucleosome occlusion elsewhere in the genome. Our observations suggest that both DNA sequence and chromatin structure are defining factors in interpreting CpG island chromatin and translation of the CpG signal. More generally, these features of CpG island recognition suggest that chromatin structure and accessibility play a major role in defining how transcription factors recognize DNA and regulatory elements genome-wide.
INTRODUCTION
Genomic DNA information in eukaryotes is organized within the nucleus by packaging into repeated units called nucleosomes that consist of roughly 147 bp of DNA wrapped around an octamer of histone proteins. Packaging of DNA into nucleosomes permits compaction of the genome into the relatively small confines of the nucleus and can also significantly impact the function of DNA sequences with which it associates. For example, specific positioning of nucleosomes over gene regulatory elements can inhibit initiation of transcription (Citation25Citation–Citation27) and nucleosomes in transcribed regions of genes can act as a barrier to transcriptional elongation (Citation14, Citation15, Citation31, Citation36). More recently it has become clear that modification of both DNA and histones can provide an additional layer of information that is specifically interpreted and alters the function of surrounding regions of the genome (Citation21, Citation43). In the case of histone proteins, marking of N-terminal tails by a diverse complement of posttranslational modifications can lead to direct effects on chromatin packing (Citation38) and specific nucleation of effector proteins that translate these modification signals into a functional outcome (Citation1, Citation8, Citation22, Citation43). The DNA component of the nucleosome can also be modified on the five position of the cytosine base by enzymatic addition of a methyl group which occurs mostly within the context of CpG dinucleotides (Citation20). This methylated dinucleotide signal acts as a binding site for proteins containing a methyl-CpG binding domain (MBD) to create transcriptionally repressive chromatin states. Together these chromatin modification systems appear to have important roles in regulating the function of transcription and other nuclear processes.
Since the majority of CpG dinucleotides in the mammalian genome are methylated, a significant effort has been placed on understanding how the DNA methylation signal impacts genome function. Interestingly, there are short contiguous regions of the genome that remain free of DNA methylation and are characterized by increased CpG content and GC percentage (Citation2). These elements are called CpG islands and are found associated with up to 70% of genes (Citation37), but how they remain free of DNA methylation and function remain poorly understood (Citation3). Based on their association with gene promoter elements, it has been hypothesized that CpG islands contribute to gene regulation, but how this is achieved mechanistically remains enigmatic. Recently we and others have shown that the zinc Finger CXXC DNA binding domain (ZF-CXXC)-containing proteins can specifically recognize nonmethylated CpG islands, where they recruit chromatin-modifying enzymes to create a CpG island-specific chromatin modification profile (Citation4, Citation45). This targeting mechanism appears to rely on both specific recognition of nonmethylated CpG dinucleotides by the ZF-CXXC domain and inhibition of binding to other CpG dinucleotides outside CpG islands by DNA methylation. This simple yet elegant system relies on hardwired DNA sequence and epigenetic DNA methylation profiles to create unique chromatin architecture at CpG islands in contributing to gene regulatory potential.
The majority of our understanding of CpG island recognition by ZF-CXXC proteins comes from in vitro studies on naked DNA and descriptive work from genome wide chromatin immunoprecipitation sequencing (ChIP-seq) analysis (Citation4, Citation45). Given the intimate relationship between CpG island function and chromatin, we sought to investigate how ZF-CXXC proteins interpret CpG islands in a chromatin context. To this end we have taken advantage of a completely recombinant chromatin reconstitution system to define how the ZF-CXXC protein KDM2A interacts with CpG island chromatin. Surprisingly, KDM2A has a very specific requirement for recognition of nucleosome-free linker DNA in vitro, and this binding mode is required for specific nucleation of KDM2A at CpG islands in vivo. Furthermore, we demonstrate that KDM2A binding is seemingly restricted to CpG islands even in the absence of genome methylation, suggesting that both DNA methylation and nucleosome occupancy function to restrict the promoter CpG island recognition system to these specific regions of the genome.
MATERIALS AND METHODS
DNA constructs.
The vector used for prokaryotic expression of His-KDM2A-StrepII (AA 1-747) and mutant derivatives were generated as previously described (Citation4). A mutation in the plant homeodomain (PHD domain) was introduced using the QuikChange mutagenesis XL kit (Stratagene).The pBlu2SKP 2×601 +48 plasmid (a gift from Tom Owen-Hughes) was amplified by mismatch PCR to mutate all CpG dinucleotides in the 48-bp linker region (pBlu2SKP 2×601 +48 0CG L).
Protein expression and purification.
Expression and purification of KDM2A was performed as previously described (Citation4). In brief, cells were lysed by sonication and the lysate cleared by centrifugation. The resulting lysate was loaded onto a prewashed Ni-NTA (nitrilotriacetic acid) column (GE Healthcare) and the eluate was further purified using a prewashed StrepTactin Superflow column (IBA). Purified protein was dialyzed overnight in BC100 (50 mM HEPES, pH 7.9, 100 mM KCl, 10% glycerol, 0.5 mM dithiothreitol [DTT]) and stored at −80°C until use.
Reconstitution and purification of nucleosomes.
Reconstitution of nucleosomes was performed following a previously described protocol (Citation9). In brief, recombinant Xenopus histones were expressed in Escherichia coli and purified via Sephacryl S-200 gel filtration (GE Healthcare). Stoichiometric amounts of each core histone were incubated together under high-salt conditions, and the resulting histone octamer was purified using a Superdex 200 gel filtration column (GE Healthcare). DNAs (147 bp and 216 bp) carrying high-affinity nucleosome positioning sequences (601 sequence) were PCR amplified from the pGEM-3Z 601 plasmid (a gift from Tom Owen-Hughes and Jonathan Widom). The 378-bp DNA carrying two 601 sequences and a 48-bp linker region was PCR amplified from the pBlu2SKP 2×601 +48 plasmid. All PCR products were then purified using a Resource Q anion-exchange column (GE Healthcare). Purified PCR DNA was then end labeled with [γ-32P]ATP (Perkin Elmer) using T4 polynucleotide kinase (Fermentas). Unincorporated label was removed using a nucleotide removal kit (Qiagen). Equimolar ratios of purified labeled DNA and octamers were mixed together in 2 M NaCl and diluted stepwise with 10 mM Tris-HCl, pH 7.6, to reach a final concentration of 100 mM NaCl. The reconstituted nucleosome was then purified on a 5 to 20% sucrose gradient, and the resulting fractions were analyzed on a 0.8% agarose gel. Fractions containing only reconstituted mono- or dinucleosomes were then pooled together and used for the electrophoretic mobility shift assay (EMSA).
EMSA.
EMSA reactions were assembled as previously described (Citation4). Briefly, purified, labeled nucleosome substrates were incubated with the KDM2A protein in the presence of poly(dA-dT) competitor DNA for 20 min at room temperature and then loaded onto a 0.8% agarose gel in 0.2× Tris-borate (TB). The gel was run at 4°C, dried onto a DE81 anion exchanger paper (Whatman), and exposed to a phosphorimager screen.
MNase chromatin immunoprecipitation.
For chromatin preparation, V6.5 mouse embryonic stem (ES) cells were fixed for 10 min in 0.37% formaldehyde and then quenched by the addition of glycine to a final concentration of 125 mM. Cells were then resuspended in 1 ml RSB buffer (10 mM Tris-HCl, pH 8, 10 mM NaCl, 3 mM MgCl2) before being lysed by the addition of RSB buffer supplemented with 0.1% NP-40. Nuclei were then collected by centrifugation at 1,500 × g for 5 min, resuspended in RSB supplemented with 10 mM CaCl2 and complete protease inhibitors (Roche) at a density of approximately 5 × 107 cells per ml, and divided into 1-ml aliquots. To each 1-ml aliquot, 200 U of micrococcalnuclease (MNase; Worthington) was added, and the reaction mixtures were then incubated at 37°C for 2.5 min (polynucleosome chromatin preparations) or 1 h (mononucleosome chromatin preparations). The reactions were stopped by adding 20 mM EDTA, and 0.5% SDS was added to ensure complete lysis of nuclei. Chromatin immunoprecipitations were performed overnight at 4°C with approximately 3 μg of antibody and 100 μl of chromatin (corresponding to 5 × 106 cells) diluted in 900 μl of ChIP dilution buffer (1% Triton X-100, 1 mM EDTA, 20 mM Tris-HCl, pH 8, and 150 mM NaCl). Antibodies used for ChIP were anti-KDM2A (Citation4) and anti-histone H3K4me3 (histone trimethylated at lysine 4; Abcam ab8580). Antibody-bound proteins were isolated on protein A agarose beads (Repligen), washed extensively, and eluted, and cross-links were reversed according to the Upstate protocol. Samples were then sequentially treated with RNase and proteinase K before being purified on a PureLink PCR microcolumn (Invitrogen). Real-time quantitative PCR (qPCR) was performed using Sybr green (Quantace) on a Rotor-Gene 6000 (Corbett). Primer sets used for qPCR are available upon request.
Sucrose gradient separation of native nucleosome preparations.
Native nucleosome preparations and sucrose gradients were performed as previously described (Citation16), with minor modifications. Briefly, approximately 1 × 108 purified nuclei from V6.5 cells were resuspended in 1 ml RSB buffer supplemented with 0.25 M sucrose, 3 mM CaCl2, and complete protease inhibitors. Digestions were performed with 10 units (polynucleosome preparation) or 500 units (mononucleosome preparation) of MNase for 15 min at 37°C, and reactions were then stopped by adding EDTA and EGTA, each to a final concentration of 3.5 mM. After centrifugation at 5,000 rpm for 5 min, the supernatant (S1) was collected. The remaining nuclear pellet was resuspended in 300 μl of nucleosome release buffer (10 mM Tris-HCl, pH 7.5, 10 mM NaCl, 0.5 mM EDTA, and protease inhibitor cocktail) and gently mixed at 4°C for 1 h. Following centrifugation at 5,000 rpm for 5 min, the second supernatant (S2) was collected. Nucleosome preparations comprising 700 μl S1 and 300 μl S2 were then fractionated through an 11-ml sucrose density gradient (5%-to-25% sucrose, 10 mM Tris-HCl, pH 7.4, 0.25 mM EDTA, 300 mM NaCl) at 30,000 rpm for 16 h at 4°C. Each 500-μl fraction had 50 μl removed for DNA purification and agarose gel analysis, and the remainder of each fraction was concentrated by trichloroacetic acid precipitation and used for protein analysis by Western blotting. Antibodies used for Western blotting were anti-KDM2A (Citation4), anti-histone H3 (Ab1791), anti-histone H1 (Millipore AE-4), and anti-DNMT3B (Enzo Life Sciences 52A1018).
RESULTS
KDM2A binds extranucleosomal DNA.
We recently demonstrated that recombinant KDM2A binds to nonmethylated CpG dinucleotides via its ZF-CXXC DNA binding domain (Citation4). The capacity of KDM2A to recognize the nonmethylated fraction of the genome results in nucleation of KDM2A at CpG island elements in vivo, where it demethylates histone H3 lysine 36 (H3K36). These experiments demonstrated that KDM2A could bind nonmethylated CpG on naked DNA templates, but given that KDM2A is a chromatin-modifying enzyme, we were interested to investigate how KDM2A interrogates more physiological nucleosome substrates and understand if DNA alone is sufficient for KDM2A nucleation on chromatin.
To address these questions, we first used a fully recombinant nucleosome reconstitution system based on the 601 nucleosome positioning sequence (Citation28). We chose to use the 601 nucleosome positioning sequence, as it will house a histone octamer at a well-defined position on the DNA template and recapitulates both the CpG content and GC nucleotide composition characteristics of mammalian CpG islands. Histones were assembled on a 147-bp 601 sequence by salt dilution, and the reconstituted nucleosomes were purified by density gradient centrifugation, yielding a highly pure mononucleosome fraction (data not shown). KDM2A was then used in an electromobility shift assay (EMSA) comparing binding to the naked 147-bp 601 sequence or the 147-bp 601 mononucleosome. As observed previously, KDM2A bound robustly to the naked CpG-containing DNA sequence (A, left panel). However, addition of KDM2A did not lead to a mobility shift of the mononucleosome (A, right panel). This interesting observation suggests that deposition of histones on DNA may occlude KDM2A binding to the CpG dinucleotides. The lack of binding to the 147-bp nucleosome suggested that perhaps KDM2A may require extranucleosomal DNA for efficient binding to chromatin. To test this possibility, we generated a second 216-bp 601 mononucleosome that has an additional 60 bp of DNA that extend out from the nucleosome positioning sequence (B and C). KDM2A was incubated with the 216-bp nucleosome and analyzed by EMSA. The addition of extranucleosomal DNA to the mononucleosome resulted in a dose-dependent shift of the nucleosome substrate by KDM2A. Importantly, this interaction was dependent on DNA binding, as a point mutation in the ZF-CXXC domain abrogates interaction with the 216-bp nucleosome.
Fig 1 KDM2A interacts specifically with linker DNA in vitro. (A) KDM2A efficiently binds to the naked 147-bp 601 nucleosome positioning DNA (left-hand panel) but binding is completely inhibited when a nucleosome is positioned on this same DNA sequence (right-hand panel), as analyzed by EMSA. The K601A mutant version of KDM2A lacks DNA binding activity. The positions of free DNA and nucleosomal DNA are indicated to the left of the panel with arrows. (B) Nucleosomes were reconstituted using a DNA substrate with 216 bp of DNA. The cartoon indicates nucleosomes in which the extranucleosomal DNA has CpG dinucleotides (top, open lollypop), methylated CpG dinucleotides (middle, closed lollypop), or mutated CpG dinucleotides (bottom, with x). (C) KDM2A interacts specifically with nucleosomes that have CpG dinucleotides in the extranucleosomal DNA but not in the same fragments when methylated or mutated. (D) Dinucleosome substrates that have 48 bp of linker DNA were reconstituted with the CpG dinucleotides in the linker DNA either intact (left panels) or mutated (right panels). KDM2A bound specifically to the dinucleosome with intact linker CpG dinucleotides, as indicated by the protein concentration-dependent nucleosome mobility shift and capacity to supershift this species with a KDM2A-specific antibody (left-hand panels). Mutation of the CpG dinucleotide in the linker DNA resulted in a loss of binding (right-hand panels).
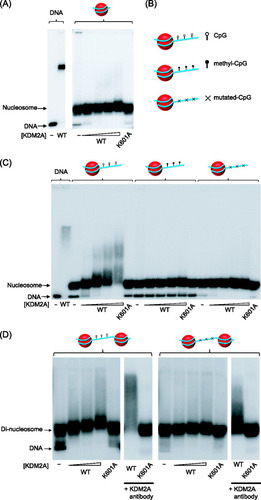
We have previously demonstrated that methylation of CpG dinucleotides abrogates KDM2A binding. To determine whether KDM2A binding to the 216-bp mononucleosome was dependent on nonmethylated CpG DNA, we reconstituted nucleosomes where all CpG dinucleotides were in vitro methylated prior to reconstitution (B and C). KDM2A binding to the methylated substrate was completely abrogated, as analyzed by EMSA, indicating that KDM2A binding to the 216-bp nucleosome is dependent on nonmethylated CpG. The main difference between the 147- and 216-bp nucleosomes is the presence of extranucleosomal DNA. This suggests that KDM2A likely recognizes nucleosome-free DNA in the context of a mononucleosome. To investigate this possibility, a 216-bp nucleosome was reconstituted in which only the CpG dinucleotides in the extranucleosomal DNA were mutated (B and C). Despite the fact that the nucleosome-occupied portion of the 216-bp nucleosome has an abundance of nonmethylated CpG dinucleotides, specific mutation of CpGs in the extranucleosomal DNA was sufficient to abrogate binding. Together, these observations demonstrate that KDM2A specifically interrogates extranucleosomal DNA in the 216-bp nucleosome.
KDM2A binding is restricted to linker DNA.
KDM2A will never encounter an isolated mononucleosome in vivo, but instead extranucleosomal DNA will largely occur between nucleosomes on the chromatin polymer. Therefore, to examine whether KDM2A can bind to linker DNA between nucleosomes, we reconstituted dinucleosomes which consist of two 601 nucleosome positioning sequences separated by 48 bp of linker DNA (Citation10). The only extranucleosomal DNA in this substrate exists between the positioned nucleosomes. KDM2A was then presented with this substrate in an EMSA (D). Much like the 216-bp nucleosome, KDM2A bound efficiently to the dinucleosome substrate. The retardation of the KDM2A/dinucleosome complex (D) compared to the KDM2A/mononucleosome complex (C) is lesser in magnitude, as the relative increase in mass due to KDM2A binding to the KDM2A/dinucleosome complex is smaller. To ensure that this modest mobility shift was dependent on KDM2A, we used a DNA binding mutant that abrogated the mobility shift and antibodies against KDM2A to supershift the KDM2A/nucleosome complex. To verify that KDM2A interacts with the linker region, a dinucleosome was reconstituted in which the CpG dinucleotides in the linker region were mutated. Importantly, this results in a loss of nucleosome binding (D, right-hand panel). The very small amount of residual binding in the dinucleosome lacking linker CpGs (D right hand panels) appears to result from a fraction of the reconstituted dinucleosome being imperfectly phased over 601 nucleosome positioning sequences (Citation9), resulting in accessible CpG dinucleotides. This mode of interaction is supported by the fact that the ZF-CXXC mutant (K601A) does not cause a mobility shift of either dinucleosome substrate (D). Together, these observations demonstrate that KDM2A interacts in the linker region between nucleosomes.
Our in vitro binding studies with reconstituted nucleosomes indicate that KDM2A interacts with linker DNA. To try and understand why nucleosomes might be a barrier to interaction of the ZF-CXXC domain with DNA, the structures of two recently solved ZF-CXXC/CpG DNA structures (MLL1 and DNMT1) were analyzed () (Citation7, Citation41). In both cases the ZF-CXXC domain forms a crescent-shaped fold that coordinates two zinc ions. Side chain residues in a single loop at the tip of the zinc finger interrogate the major groove of DNA where they recognize CpG bases and are sterically occluded by the presence of methyl CpG. Interestingly, in both of these structures a distinct portion of the ZF-CXXC domain also wraps around the DNA and interrogates the minor groove on the opposite side of the double helix. Mutations that disrupt this interaction also affect DNA binding, suggesting that simultaneous interaction with major- and minor-groove DNA is required for efficient DNA binding (Citation7). In the context of a nucleosome, when the major groove is facing outwards, the minor groove points inwards and is often engaged by arginine residues from the histone octamer (Citation29). This observation likely explains the lack of KDM2A binding to nucleosomal DNA, as access to information in both the major and minor groove would be sterically constrained in nucleosomal DNA compared to naked DNA. Therefore, it seems likely that ZF-CXXC domain proteins are occluded from binding nucleosomal DNA by the inability to easily interface with both major- and minor-groove DNA.
Fig 2 ZF-CXXC domain proteins bound to nonmethylated CpG DNA. (A) A cartoon depicting the structure of the DNMT1 ZF-CXXC domain bound to nonmethylated CpG DNA (Protein Data Bank [PDB] number, 3PT6). The green ribbon indicates the protein backbone and the residues that specifically interact with CpG dinucleotides in the major groove. The DNA is colored light brown and is viewed looking down the DNA double helix. The gray shading indicates the protein surface, and the red spheres correspond to the two zinc ions coordinated by the ZF-CXXC domain. (B) The same representation as in panel A of the MLL1 ZF-CXXC domain bound to nonmethylated CpG DNA (PDB number, 2KKF). From both panels it is apparent that the ZF-CXXC domain interacts specifically with CpG DNA in the major groove and interacts with minor-groove DNA on the opposite side of the DNA double helix.
![Fig 2 ZF-CXXC domain proteins bound to nonmethylated CpG DNA. (A) A cartoon depicting the structure of the DNMT1 ZF-CXXC domain bound to nonmethylated CpG DNA (Protein Data Bank [PDB] number, 3PT6). The green ribbon indicates the protein backbone and the residues that specifically interact with CpG dinucleotides in the major groove. The DNA is colored light brown and is viewed looking down the DNA double helix. The gray shading indicates the protein surface, and the red spheres correspond to the two zinc ions coordinated by the ZF-CXXC domain. (B) The same representation as in panel A of the MLL1 ZF-CXXC domain bound to nonmethylated CpG DNA (PDB number, 2KKF). From both panels it is apparent that the ZF-CXXC domain interacts specifically with CpG DNA in the major groove and interacts with minor-groove DNA on the opposite side of the DNA double helix.](/cms/asset/1bb924bb-1768-429b-8790-df7225347a01/tmcb_a_12273712_f0002_oc.jpg)
Neither H3K4me3 nor the KDM2A PHD domain is required for 216-bp nucleosome binding.
It has recently been demonstrated that the plant homeodomains (PHD domains) in many proteins can function as lysine or methylated lysine binding modules (Citation43, Citation50). KDM2A has a highly conserved PHD domain immediately adjacent to the ZF-CXXC DNA binding domain. The close proximity of the PHD and DNA binding domains suggested that perhaps together these domains may form a chromatin binding interface that interrogates both DNA and histone. To test this possibility, we generated a mutation in a conserved cysteine within the KDM2A PHD domain (C645A) that has been shown in other PHD domains to disrupt the structure and function (A) (Citation5, Citation19). Although this mutation disrupts the PHD domain, the DNA binding capacity of the mutant protein was unaffected (B). The C645A mutant protein was then used in an EMSA to examine the effect of this mutation on substrate binding (B). As with binding to naked DNA, the KDM2A PHD mutant did not affect interaction with the 216-bp nucleosome, suggesting that any potential interaction with histone by the PHD domain does not significantly contribute to binding. Our reconstitution system uses unmodified recombinant histones. Although some PHD proteins interact with unmodified lysines, others interact specifically with methylated lysine residues. We have recently shown that KDM2A binds specifically to CpG islands in vivo. Interestingly, the majority of CpG islands are modified on the tail of histone H3 at position 4 (H3K4me3) (Citation45), suggesting that perhaps the KDM2A PHD domain could function in concert with ZF-CXXC-mediated recognition of CpG DNA to create a high-affinity CpG island binding module. To specifically test this possibility, we used a recently developed semisynthetic technique to install H3K4me3 into our recombinant histone H3 (C) (Citation39). This histone was then incorporated into a 216-bp nucleosome to create a homogenously H3K4me3 modified nucleosome. This substrate was used in an EMSA with either the KDM2A wild-type (WT) or PHD mutant protein (D). In both instances KDM2A bound to the modified nucleosome with the same efficiency as the unmodified substrate and mutation of the PHD domain did not impact this binding. Therefore, nucleosome binding by KDM2A does not appear to rely on the function of the PHD domain nor on H3K4me3.
Fig 3 The KDM2A PHD domain does not contribute significantly to nucleosome binding. (A) KDM2A proteins encoding a wild-type (WT) or mutant PHD domain (C645A) were expressed and purified. (B) Both the WT and C645A KDM2A proteins bind to naked 216-bp nucleosome positioning DNA with the same level of efficiency, as determined by EMSA (left-hand lanes). Mutating the PHD domain did not inhibit binding to the 216-bp mononucleosome, whereas mutation of the ZF-CXXC DNA biding domain (K601A) completely abrogated binding. (C) Histone H3K4me3 was installed specifically into histone H3 and the incorporation verified by Western blotting of the recombinant histone with antibodies against H3K4me3 or histone H3. (D) Mononucleosomes (216 bp) were reconstituted with histones containing H3K4me3 as indicated by the green dots on the nucleosome cartoon above the EMSA panels. The addition of H3K4me3 to the mononucleosome did not increase binding of KDM2A, nor did mutation of the PHD domain inhibit binding to the nucleosome, indicating that H3K4me3 does not contribute significantly to nucleosome recognition by KDM2A. (E) Multiple sequence alignment of PHD domains that bind either to H3K4me3 (BPTF, ING2, and PHF8) or H3K4me0 (BHC80 and DNMT3L) and KDM2A. Gray boxes indicate conserved zinc-coordinating cysteines/histidines, and orange boxes indicate methyl-lysine- or lysine-interacting residues. Although KDM2A has conserved zinc-coordinating residues, lysine interaction residues are absent.
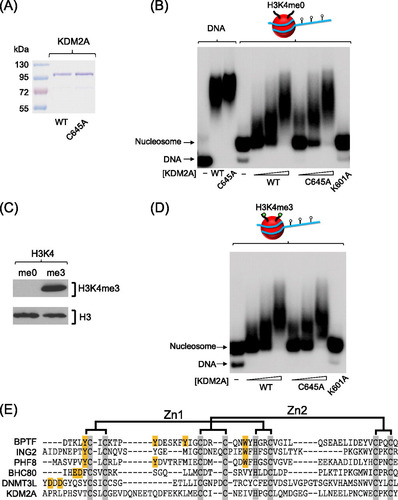
As the KDM2A PHD domain did not appear to contribute to nucleosome binding using the substrates tested, we hypothesized that the KDM2A PHD domain may interact with histone modifications on other residues of H3 or other core histones. To test this possibility, we applied the KMD2A PHD domain to a peptide array containing 600 different variations of modified and unmodified peptides corresponding to all four core histones (Citation30). Perhaps somewhat surprisingly, in this screen we observed no specific binding of the KDM2A PHD domain to any peptide on the array, suggesting that the function of this domain is not to recognize the histone component of the nucleosome (unpublished observations). Given that the structure of several PHD domains bound to their cognate substrates has been solved, we carried out a multiple sequence alignment of the KDM2A PHD domain, comparing it to PHD domains that bind either nonmethylated or methylated lysine residues (E). Importantly, the structural cysteines required to form the PHD fold are conserved among all PHD domains in the alignment, but the specific aromatic cage that is utilized for methyl-lysine binding and the conserved residues used for lysine binding are completely absent from KDM2A. Together these data suggest that although the PHD domain is conserved across KDM2A orthologues, the function of the PHD domain in KDM2A is not to recognize histone.
KDM2A binds linker DNA in vivo.
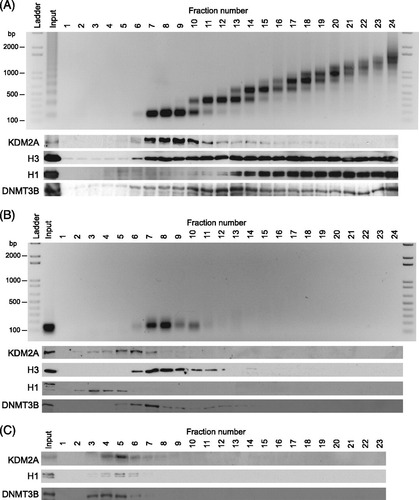
Our in vitro nucleosome binding assays indicate that KDM2A interacts with chromatin through recognition of linker DNA via the ZF-CXXC DNA binding domain. To understand if this binding mechanism is utilized in vivo, nuclei were isolated from mouse embryonic stem cells and digested with micrococcal nuclease. Digestion was done either under limiting reaction conditions that produced a polynucleosomal ladder of approximately 1 to 10 nucleosomes or under complete conditions that resulted in exclusively mononucleosome fragments (A and B) (Citation16). If endogenous KDM2A associates with linker DNA in vivo it would remain associated with polynucleosomal chromatin that has intact linker regions but not with more completely digested mononucleosomal chromatin that lacks linker DNA. To examine whether this was the case, chromatin was isolated using limited digestion and fractionated by density sucrose gradient centrifugation. A nucleosome repeat length DNA ladder was observed in gradient fractions that have histone H3, indicating intact chromatin (A). When the same fractions were analyzed by Western blotting, KDM2A was found exclusively in the chromatin-containing fractions. This suggests KDM2A is intimately associated with chromatin that contains linker DNA. Interestingly, KDM2A was enriched in the mono-trinucleosome fractions, suggesting that it may predominantly associate with nuclease-sensitive chromatin, a feature that is characteristic of CpG island elements (Citation35). In agreement with previous observations, histone H1 is preferentially enriched in the oligonucleosomal fractions, suggesting that these regions of the gradient contain the rapidly sedimenting, compact and more nuclease-resistant chromatin (Citation23). In contrast, when chromatin was digested to a homogenous mononucleosome population that lacks linker DNA and resolved on the same density gradient, a single mononucleosome DNA band was observed that cofractionated with histone H3, again indicating intact chromatin (B). Importantly, at this point, when KDM2A was analyzed by Western blotting, it was no longer associated with the chromatin-containing fraction but instead migrated in the same position on the gradient as is observed when chromatin-free nuclear extracts are fractionated in a similar manner (C). This indicates that KDM2A chromatin binding is abrogated when linker DNA is removed. In support of the contention that this binding is dependent on linker DNA, these observations mirrored the behavior of histone H1, which also interacts with linker DNA (A to C). The loss of KDM2A from the mononucleosomal fraction appears to be due to the loss of linker DNA as DNMT3B, a protein previously shown to interact with nucleosomes (Citation16), remains associated with the mononucleosomal fraction (C). Together these observations demonstrate that KDM2A in bulk chromatin relies on linker DNA to bind chromatin.
Fig 4 KDM2A interacts with linker DNA in vivo. (A) Nuclei were partially digested with micrococcal nuclease to produce polynucleosomes that contain linker DNA. Chromatin was separated by sucrose density gradient centrifugation. The gradient was fractionated from the top into 24 fractions. DNA was purified from each fraction and resolved by agarose gel electrophoresis (top panel), and proteins from each fraction were visualized by Western blotting (bottom panels). DNA and protein analyses include an unfractionated input sample representing 10% of material loaded onto the sucrose gradient. (B) As above, except that native chromatin was digested to completion with micrococcal nuclease to produce mononucleosomes lacking linker DNA. (C) Fractionation of chromatin-free nuclear extract.
KDM2A binds linker DNA in CpG islands.
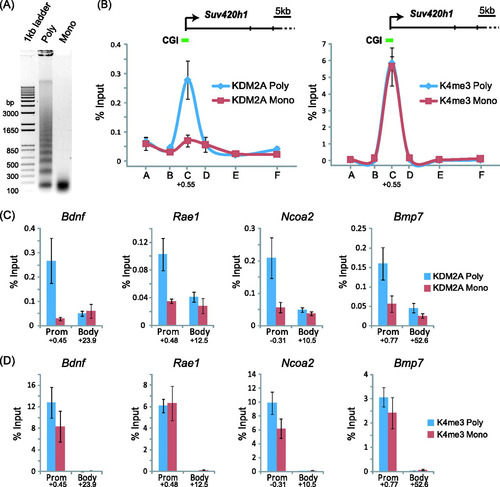
KDM2A binding to bulk chromatin relies on intact linker DNA. Our recent work demonstrated that KDM2A binds to CpG islands in vivo (Citation4). Therefore, we wanted to know whether targeted interaction of KDM2A with promoter-associated CpG islands relies on linker DNA. To address this possibility, a modified chromatin immunoprecipitation (ChIP) approach was designed whereby nonhistone factors were mildly fixed to chromatin using formaldehyde and then nuclei were isolated and digested under limiting or complete conditions using micrococcal nuclease (A). This resulted in chromatin that either contains linker DNA or lacks linker DNA. KDM2A was then immunoprecipitated, and the isolated DNA was analyzed by quantitative real-time PCR. Initially amplicons spanning the CpG island containing the Suv420H1 gene were analyzed in either the limited or complete digestions (B). The KDM2A signal in the limited digestion revealed a normal KDM2A ChIP signal that peaked over the CpG island and was absent from the rest of the gene. However, when the same regions were analyzed in the complete digestions, the KDM2A ChIP signal was completely lost (B, left panel). This loss of signal is not due to preferential overdigestion of CpG island chromatin in these complete digestions, as the amount of H3K4me3 ChIP signal at the CpG island regions was the same in both chromatin preparations (B, right panel). A further four CpG island genes were analyzed at both the CpG island and body for both KDM2A and H3K4me3 (C and D). In all instances, removal of linker DNA abrogated KDM2A binding but left CpG island chromatin intact, as verified by H3K4me3 ChIP. Together these observations are in keeping with our in vitro observations and demonstrate that KDM2A binds to CpG island chromatin through recognition of DNA between nucleosomes that encompasses linker DNA or more extended nucleosome-free regions.
Fig 5 KDM2A is targeted to CpG islands through recognition of linker DNA. (A) Formaldehyde-fixed chromatin was digested with micrococcal nuclease to produce polynucleosomes that contain linker DNA and mononucleosomes lacking linker DNA. (B) KDM2A binding to the Suv420h1 gene was analyzed by ChIP at tiled positions across the gene (A to F on the x axis) by quantitative PCR on both chromatin preparations. KDM2A binding (left panel) in the polynucleosomal fraction (blue line) corresponded to the CpG island region, and this signal was lost in the mononucleosomal preparation (red line). In contrast, the signal for H3K4me3 over the same regions remained constant for both samples (right panel). (C and D) The same analysis was extended to a series of four more CpG island genes, analyzing both the CpG island region and body region. In all instances, digestion to mononucleosomes released KDM2A from CpG island chromatin (C), while the H3K4me3 signal remained relatively constant (D). Together, these observations indicate that KDM2A interacts with linker DNA at CpG islands. In all cases, error bars correspond to standard errors of the means from biological triplicates. Numbers below the data for promoter (Prom) and body primer sets indicate the position of the primer set (kb from center of amplicon) with respect to the major transcription start site of the gene in question.
KDM2A is retained at CpG islands in the absence of DNA methylation.
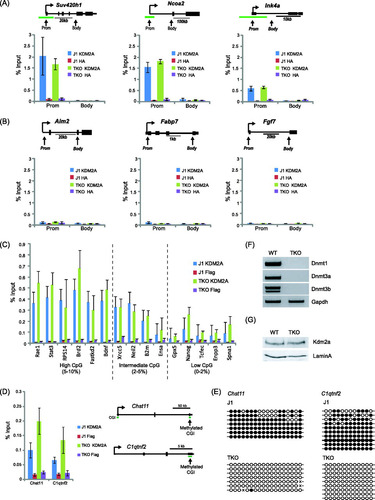
We have previously shown that KDM2A binds to nonmethylated CpG islands and that the acquisition of DNA methylation in a small fraction of CpG islands during differentiation is sufficient to block DNA binding (Citation4). CpG islands contain a much higher density of CpGs than the rest of the genome, and outside CpG islands, the remaining CpG dinucleotides are heavily methylated. Presumably these two features of the mammalian genome lead to efficient nucleation of KDM2A to CpG islands. To understand if DNA methylation in bulk chromatin is a determining factor for excluding KDM2A from these regions of the genome, mouse triple knockout (TKO) embryonic stem cells that lack both de novo methyltransferases DNMT3A and -B and the maintenance methyltransferase DNMT1 were obtained (F) (Citation48). These cells do not have any detectable CpG methylation. To examine whether the presence of nonmethylated DNA outside CpG islands in TKO cells alters KDM2A binding profiles, ChIP analysis was carried out on a panel of genes with CpG island promoters and genes with non-CpG island promoters, analyzing both promoter and body regions in wild-type (WT) and TKO cell lines (A and B). Interestingly, a loss of DNA methylation had no impact on the normal nucleation of KDM2A at the CpG island promoters examined (A) and did not create new binding sites at several non-CpG island promoters (B). Furthermore, the presence of nonmethylated DNA in the body of both classes of genes did not result in de novo binding of KDM2A over these regions of the genes tested. Importantly, KDM2A profiles were also maintained in the TKO cells when promoter regions of various CpG content were analyzed (C). However, loss of DNA methylation at some weak CpG islands that are subject to DNA hypermethylation in normal ES cells did lead to a modest renucleation of KDM2A in the TKO cells (D and E). This is in agreement with previous observations that CpG island methylation is a barrier to KDM2A binding (Citation4). Importantly, the maintenance of normal KDM2A binding profiles at the genes tested in the TKO cells was not simply due to increased KDM2A expression, as the levels of KDM2A were the same in the parental WT and TKO cell lines (G). From the regions analyzed here, it appears that in situations where a CpG island acquires methylation, KDM2A binding is inhibited (Citation4) (D), but loss of DNA methylation elsewhere in the genome does not significantly impact KDM2A binding (A, B, and C). Based on the observation that KMD2A is incapable of binding to nucleosome-bound DNA, it appears that low CpG dinucleotide frequency and occlusion of CpG binding sites by nucleosomes is a major barrier to KDM2A binding outside CpG islands. Therefore, KDM2A recognition of CpG island chromatin appears to rely on both nonmethylated CpG and accessibility of this binding site in extranucleosomal DNA.
Fig 6 KDM2A remains bound specifically to CpG islands in cells lacking DNA methylation. TKO mouse embryonic stem cells that lack all three known DNA methyltransferases (F) and have no detectable DNA methylation were obtained. ChIP was used to analyze KDM2A binding to three CpG island genes (A) and three non-CpG island genes (B) in both the WT parent line and the TKO cell line. In the three CpG island regions, KDM2A binding remained the same, indicating that loss of methylation elsewhere in the genome did not cause a loss of binding. In addition, the body of the CpG island genes (A) and non-CpG island genes (B) (both promoter and body) did not acquire KDM2A binding. (C) This analysis was then expanded to analyze a series of gene promoters that have high, intermediate, and low CpG content to demonstrate that KDM2A nucleation is largely unaffected by a loss of DNA methylation. (D) A similar ChIP analysis was performed at two CpG islands that are hypermethylated in wild-type J1 cells. Loss of DNA methylation in the TKO cells caused a modest increase in KDM2A nucleation at these weak CpG islands. (E) Bisulfite sequencing demonstrated that the Chst11 and C1qtnf2 CpG islands are hypermethylated in J1 but not TKO mouse ES cells. (G) Importantly, KDM2A protein levels remained the same in the WT and TKO cell lines. Error bars in all ChIP experiment data correspond to standard errors of the means from biological triplicates.
DISCUSSION
Despite over 25 years of active research on CpG islands, the mechanisms underpinning the function of these interesting elements remain poorly understood. The recent discovery that ZF-CXXC domain-containing proteins act as interpreters that translate the CpG island signal into a unique chromatin architecture has revealed that CpG islands are not simply passive genomic elements but that they function proactively in marking regulatory element chromatin (Citation4, Citation45). Based on these observations, there appears to be an intimate relationship between CpG island chromatin structure and function.
To better understand this relationship, we have taken advantage of a chromatin reconstitution system to define how KDM2A binding to CpG islands is achieved. Importantly, this work reveals that nucleosomal DNA is a major barrier to chromatin binding in vitro and that the ZF-CXXC domain specifically recognizes DNA between nucleosomes, including linker DNA or nucleosome-free regions. Despite the fact that KDM2A encodes a PHD domain, it does not appear to use this domain in recognition of chromatin. In vivo, recognition of CpG island promoters by KDM2A is achieved through interaction with linker DNA, revealing that a combination of nonmethylated CpG dinucleotide content and linker DNA accessibility may be utilized by ZF-CXXC proteins to recognize the CpG island signal and translate this into chromatin modification. As most ZF-CXXC domain-containing proteins function to modify chromatin, this would suggest that there is an intimate and potentially self-reinforcing cycle that defines CpG island functionality through chromatin modification and accessibility. For example, genome-wide studies analyzing nucleosome occupancy of transcription start sites suggest that CpG island-containing genes have high occupancy and more regularly phased nucleosomes (Citation47) than non-CpG island genes and other regions of the genome. It has been suggested that this could arise in part through the underlying GC-rich sequence (Citation46, Citation47). Together, elevated nonmethylated CpG content and regular nucleosome arrangement may create a situation at CpG islands where linker DNA is largely accessible and occupied by ZF-CXXC proteins. Indeed, both ZF-CXXC-containing proteins KDM2A and CFP1 occupy over 90% of CpG islands genome-wide and appear to be excluded from only a very small subset of CpG island genes that are highly repressed (Citation4, Citation40, Citation45). Nucleation of KDM2A at CpG islands removes H3K36me2, a modification that would normally recruit repressive histone deacetylase complexes and is inhibitory to transcriptional initiation (Citation6, Citation42). In contrast, CFP1 leads to deposition of H3K4me3, a modification that correlates positively with transcriptional potential (Citation12). Interestingly, H3K4me3 can act as a nucleation site for the NURF chromatin remodelling complex through the effector protein BPTF and the SAGA histone acetyltransferase complex through the effector protein SGF29 (Citation49, Citation50). Therefore, nucleation of these activities at CpG islands following CFP1-mediated deposition of H3K4me3 could result in a reinforcement of nucleosome phasing and histone acetylation, in keeping with genome-wide studies indicating that CpG island chromatin is often more accessible and acetylated than surrounding and bulk chromatin (Citation34, Citation35, Citation44, Citation51). Based on these properties, one can envisage that CpG islands may therefore function in a self-reinforcing manner whereby organized nucleosome structure and accessibility may permit access by ZF-CXXC DNA binding factors to nucleosome-free DNA, as described here, which then initiates a chromatin architecture through histone modification that allows effector protein-based reinforcement of accessibility. Importantly, this cascade could easily be reinitiated following cell division through binding of ZF-CXXC proteins to nonmethylated CpG islands in daughter cells and provide a simple mechanism by which CpG island chromatin architecture could be copied, based on a DNA template, to cellular progeny. Clearly, future studies are required to address these possibilities.
CpG islands are associated with the majority of genes, and ZF-CXXC proteins bind almost all CpG islands. This includes genes that cover the entire gene expression spectrum, including repressed, poised, and active genes. Together, these observations suggest that the presence of a CpG island does not directly impact the defined expression state of associated genes. Therefore, how do CpG islands impact gene regulatory element function and why are they so broadly associated with genes? Our work and the work of others indicate that chromatin accessibility (Citation35, Citation51) and defined chromatin modification architecture (Citation4, Citation45) are common features of CpG islands. Therefore, one possibility is that CpG islands use ZF-CXXC-dependent chromatin modification to set up a constitutively fluid and accessible chromatin structure that permits efficient recognition of gene regulatory elements by DNA binding transcription factors and perhaps eventually the core transcriptional machinery during gene activation. The fact that chromatin accessibility is a central feature of transcription factor nucleation is supported by recent work demonstrating that the binding of glucocorticoid receptor (GR) following hormone stimulation required not only the presence of the GR binding motif but also an accessible chromatin environment prior to stimulation for efficient GR nucleation (Citation17). This is in agreement with other genome-wide studies analyzing transcription factor binding to chromatin in vivo that indicate not all theoretical binding sites are recognized equally (Citation11, Citation13, Citation18, Citation24, Citation32). Therefore, the capacity of CpG islands to create permissive and accessible chromatin may be particularly important for highlighting gene regulatory elements in large and complex mammalian genomes. Given their association with both silent and active genes, this model places CpG islands as facilitators of gene regulatory processes as opposed to defining absolute transcriptional output. In keeping with this model, a recent study demonstrated that induction of CpG island-containing immediate early genes occurs rapidly without a requirement for the chromatin remodelling machinery (Citation33). In contrast, induction of non-CpG island-containing immediate early genes relies on the function of the ATP-dependent chromatin remodelling factor BRG-1 (Citation33). Therefore, it is tempting to speculate that chromatin modifications and accessibility that are intrinsic to CpG islands through the function of ZF-CXXC proteins contribute to this transcriptionally permissive and responsive state.
As we begin to uncover the factors that interpret CpG islands, the more apparent it becomes that chromatin structure and modification are important in defining CpG island function. Our observations that the CpG island signal is interpreted through recognition of linker DNA by the ZF-CXXC domain and an emerging trend from ChIP-seq studies analyzing transcription factor nucleation imply that nucleosome structure, occupancy, and modification may be important factors in gene regulatory element recognition and function in vivo. Therefore, a detailed mechanistic understanding of how DNA binding proteins interface with defined chromatin templates in vitro, and not simply their underlying naked DNA sequence motifs, in addition to genome-wide profiling endeavors will be central to understanding how selection and nucleation at target sites are achieved in vivo.
ACKNOWLEDGMENTS
We thank the late Jonathan Widom and Tom Owen-Hughes for the 601 nucleosome-positioning sequences; Yi Zhang, Timothy Richmond, and Matthew Simon for histone expression constructs; Sigrun Rumpel and Cheryl Arrowsmith for the histone peptide array analysis; James Parker for help with structural images; Masaki Okano for the TKO ES cells; and Neil Brockdorff for antibody reagents and valuable discussion during the course of this work.
This work is funded in the Klose laboratory by the Wellcome Trust, Cancer Research UK, EMBO, The Lister Institute of Preventive Medicine, and the Medical Research Council.
REFERENCES
- Bannister AJ, et al. 2001. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410:120–124.
- Bird A, Taggart M, Frommer M, Miller OJ, Macleod D. 1985. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell 40:91–99.
- Blackledge NP, Klose R. 2011. CpG island chromatin: a platform for gene regulation. Epigenetics 6:147–152.
- Blackledge NP, et al. 2010. CpG islands recruit a histone H3 lysine 36 demethylase. Mol. Cell 38:179–190.
- Capili AD, Schultz DC, Rauscher IF, Borden KL. 2001. Solution structure of the PHD domain from the KAP-1 corepressor: structural determinants for PHD, RING and LIM zinc-binding domains. EMBO J. 20:165–177.
- Carrozza MJ, et al. 2005. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123:581–592.
- Cierpicki T, et al. 2010. Structure of the MLL CXXC domain-DNA complex and its functional role in MLL-AF9 leukemia. Nat. Struct. Mol. Biol. 17:62–68.
- Dhalluin C, et al. 1999. Structure and ligand of a histone acetyltransferase bromodomain. Nature 399:491–496.
- Dyer PN, et al. 2004. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 375:23–44.
- Engeholm M, et al. 2009. Nucleosomes can invade DNA territories occupied by their neighbors. Nat. Struct. Mol. Biol. 16:151–158.
- Farnham PJ. 2009. Insights from genomic profiling of transcription factors. Nat. Rev. Genet. 10:605–616.
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. 2007. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130:77–88.
- Guertin MJ, Lis JT. 2010. Chromatin landscape dictates HSF binding to target DNA elements. PLoS Genet. 6:e1001114.
- Hodges C, Bintu L, Lubkowska L, Kashlev M, Bustamante C. 2009. Nucleosomal fluctuations govern the transcription dynamics of RNA polymerase II. Science 325:626–628.
- Izban MG, Luse DS. 1991. Transcription on nucleosomal templates by RNA polymerase II in vitro: inhibition of elongation with enhancement of sequence-specific pausing. Genes Dev. 5:683–696.
- Jeong S, et al. 2009. Selective anchoring of DNA methyltransferases 3A and 3B to nucleosomes containing methylated DNA. Mol. Cell. Biol. 29:5366–5376.
- John S, et al. 2011. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet. 43:264–268.
- Johnson DS, Mortazavi A, Myers RM, Wold B. 2007. Genome-wide mapping of in vivo protein-DNA interactions. Science 316:1497–1502.
- Kalkhoven E, Teunissen H, Houweling A, Verrijzer CP, Zantema A. 2002. The PHD type zinc finger is an integral part of the CBP acetyltransferase domain. Mol. Cell. Biol. 22:1961–1970.
- Klose RJ, Bird AP. 2006. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 31:89–97.
- Kouzarides T. 2007. Chromatin modifications and their function. Cell 128:693–705.
- Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. 2001. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410:116–120.
- Li G, et al. 2010. Highly compacted chromatin formed in vitro reflects the dynamics of transcription activation in vivo. Mol. Cell 38:41–53.
- Lidor Nili E, et al. 2010. p53 binds preferentially to genomic regions with high DNA-encoded nucleosome occupancy. Genome Res. 20:1361–1368.
- Lomvardas S, Thanos D. 2002. Modifying gene expression programs by altering core promoter chromatin architecture. Cell 110:261–271.
- Lomvardas S, Thanos D. 2001. Nucleosome sliding via TBP DNA binding in vivo. Cell 106:685–696.
- Lorch Y, LaPointe JW, Kornberg RD. 1987. Nucleosomes inhibit the initiation of transcription but allow chain elongation with the displacement of histones. Cell 49:203–210.
- Lowary PT, Widom J. 1998. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 276:19–42.
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389:251–260.
- Nady N, Min J, Kareta MS, Chedin F, Arrowsmith CH. 2008. A SPOT on the chromatin landscape? Histone peptide arrays as a tool for epigenetic research. Trends Biochem. Sci. 33:305–313.
- Orphanides G, Wu WH, Lane WS, Hampsey M, Reinberg D. 1999. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 400:284–288.
- Pique-Regi R, et al. 2011. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res. 21:447–455.
- Ramirez-Carrozzi VR, et al. 2009. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 138:114–128.
- Roh TY, Cuddapah S, Zhao K. 2005. Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes Dev. 19:542–552.
- Sabo PJ, et al. 2004. Genome-wide identification of DNaseI hypersensitive sites using active chromatin sequence libraries. Proc. Natl. Acad. Sci. U. S. A. 101:4537–4542.
- Saunders A, et al. 2003. Tracking FACT and the RNA polymerase II elongation complex through chromatin in vivo. Science 301:1094–1096.
- Saxonov S, Berg P, Brutlag DL. 2006. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. U. S. A. 103:1412–1417.
- Shogren-Knaak M, et al. 2006. Histone H4–K16 acetylation controls chromatin structure and protein interactions. Science 311:844–847.
- Simon MD, et al. 2007. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell 128:1003–1012.
- Smith E, Shilatifard A. 2010. The chromatin signaling pathway: diverse mechanisms of recruitment of histone-modifying enzymes and varied biological outcomes. Mol. Cell 40:689–701.
- Song J, Rechkoblit O, Bestor TH, Patel DJ. 2011. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 331:1036–1040.
- Strahl BD, et al. 2002. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol. Cell. Biol. 22:1298–1306.
- Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. 2007. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14:1025–1040.
- Tazi J, Bird A. 1990. Alternative chromatin structure at CpG islands. Cell 60:909–920.
- Thomson JP, et al. 2010. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature 464:1082–1086.
- Tillo D, Hughes TR. 2009. G+C content dominates intrinsic nucleosome occupancy. BMC Bioinformatics 10:442.
- Tillo D, et al. 2010. High nucleosome occupancy is encoded at human regulatory sequences. PLoS One 5:e9129.
- Tsumura A, et al. 2006. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells 11:805–814.
- Vermeulen M, et al. 2010. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 142:967–980.
- Wysocka J, et al. 2006. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 442:86–90.
- Xi H, et al. 2007. Identification and characterization of cell type-specific and ubiquitous chromatin regulatory structures in the human genome. PLoS Genet. 3:e136.