Abstract
Imatinib is a chemotherapeutic drug used for the treatment of chronic myeloid leukemia (CML). Recent data showed imatinib‐induced cell death in various types of cancers. Autophagy is the physiological process in which cellular components are broken down by the lysosomal activation. In this study, we aimed to examine the effects of imatinib on autophagy in addition to apoptosis in CML cells. Results suggested that imatinib induces autophagy in CML cells through inducing over‐expression of BECLIN‐1 and ATG5 genes with the statistical significance. Our results demonstrated that autophagy might be involved in imatinib‐induced cell death.
Introduction
Chronic myeloid leukemia (CML) is a blood cancer caused by the replacement of healthy bone marrow cells with malignantly transformed leukemic cells and increased numbers of immature white blood cells in the bloodstream and blood forming tissues. The reciprocal translocation between the ninth and the twenty‐second chromosomes bringing the breakpoint cluster region (BCR) gene and V‐abl Abelson murine leukemia viral oncogene homolog (ABL) gene together is the main driving force of the malignant transformation. Resultant structure of this translocation is called Philadelphia chromosome. The BCR/ABL protein resulting from this translocation exerts constitutive tyrosine kinase activity which is important for signaling the cell to proliferate. Besides inducing cell proliferation, BCR/ABL is involved in differentiation, prevention of apoptosis, cell adhesion and migration.Citation1–Citation3 Therefore, this fusion protein is an important target for the treatment of CML.
Imatinib (STI‐571) is a chemotherapeutic agent that specifically binds to the adenosine triphosphate binding pocket of the BCR/ABL fusion protein. This binding inhibits the subsequent phosphorylation events of the target proteins and suppresses cell proliferation. Imatinib is highly selective for the transformed cells and it does not have cytotoxic effect up to the concentration of 10 μM on BCR/ABL‐negative cells.Citation4 Because of the selective nature of the drug, it is commonly used for the treatment of Philadelphia chromosome‐positive CML. In some of the patients, there are some mechanisms developed to gain resistance to imatinib. Resistance to chemotherapeutic agents such as imatinib is under active investigation and, with the increasing knowledge in this subject, we are able to delineate mechanisms underlying this phenomenon.Citation6,Citation6 We have also shown in our previous studies that the bioactive sphingolipids are also important in the development of imatinib resistance in CML.Citation7
Recycling the cellular components is as important as producing the new ones to maintain the homeostasis. Autophagy is the regulated pathway for cells to recycle their components. It is observed in basal levels in almost all types of cells in order to maintain the turnover of the organelles and long‐lived proteins.Citation8 Components of cells are degraded through activation of the lysosomal machinery upon induction of autophagy. Under starvation conditions, cells degrade some of their internal components via autophagy to derive monomers which will subsequently be used for the production of energy. There are three known types of autophagy: macroautophagy, microautophagy, and chaperone‐mediated autophagy. The first type is characterized by the encompassment of cellular components such as organelles and cytosolic protein aggregates by a double‐membrane structure called autophagosome. This event can be initiated through the activation of serine/threonine kinase target of rapamycin (TOR) upon nutrient starvation.Citation9 ATG family of genes having more than 15 members is important in the initiation, expansion, and completion of the autophagosome.Citation10 Especially ATG5 (or APG5L) is required for the vesicle expansion and completion.Citation11,Citation12 ATG6 (or BECLIN‐1) is responsible for vesicle nucleation by forming the phosphatidylinositol 3‐kinase complex along with Vps15 and Vps34 proteins.Citation14,Citation13 LC3 (or MAP1ALC3) is another key player required for the autophagosome expansion.Citation15 Completion of autophagosome harboring the cargo to be degraded is followed by the fusion with the lysosome. The fusion event requires some proteins other than ATG family members such as Vam3, Vam7, and Ypt7.Citation16 After fusion of autophagosome with lysosome, hydrolases degrade its constituents and resultant monomers can be further utilized by the cell. Unlike macroautophagy, in microautophagy, the cytoplasmic components are not surrounded by membrane that is to be fused with lysosome unlike the macroautophagy. Instead, lysosome engulfs targets to be degraded by invagination of the lysosomal membrane. This type of autophagy can also be initiated by mTOR signaling when nutrient in the medium is deprived and it is useful for decreasing the volume of the lysosome inside the cell since invaginated portion of the vacuolar membrane is degraded.Citation17 The last known mechanism of the autophagy, chaperone‐mediated autophagy, involves degradation of individual proteins. Autophagy plays roles in various physiological and pathophysiological processes in organisms. Activation of autophagy pathway may have positive or negative effects on the cell integrity. Tumor suppression and type‐II programmed death of cancer cells, defense against bacterial or viral infection, and prevention of neurodegeneration via degrading toxic protein aggregates can be given as examples of positive outcomes of autophagy. But in some other cases, autophagy might also contribute to tumor cell survival under nutrient‐deprived conditions or by protection against cancer treatments.Citation18–Citation20 Deregulation of autophagy may cause mortality of the cells due to excessive degradation of self‐materials in muscular diseases or such a scenario may also provide the pathogens with a suitable environment for their growth by supplying nutrients. Controversial as it sounds, autophagy might also cause the death of neuronal cells that harbor protein aggregates and thus contribute to neurodegenerative diseases. The role of autophagy in health and disease is currently under the intensive scope of scientific research as it was reviewed by numerous authors.Citation8,Citation22,Citation21
In the present study, we aimed to demonstrate the effects of imatinib in terms of autophagic induction on human CML cells. By this approach, we will shed light upon the link between autophagy and chemotherapy which might have important outcomes in the clinic.
Materials and Methods
Cell culture
Human K562 CML cells were obtained from the German Collection of Microorganisms and Cell Cultures and maintained in RPMI1640 (Roswell Park Memorial Institute) medium containing 10% fetal bovine serum and 1% penicillin‐streptomycin in 5% CO2 in a CO2 incubator at 37°C.
Gene expression analysis
Changes in the expression levels of autophagy related genes after the treatment with different concentrations of imatinib were quantified by SYBR Green‐based quantitative polymerase chain reaction (qPCR) technique. Cells were incubated in the presence of increasing concentrations of imatinib, and total RNA was isolated using NucleoSpin Total RNA Isolation kit (Macherey‐Nagel, Duren, Germany) according to manufacturer protocols. Isolated RNAs were converted to cDNA using First Strand cDNA Synthesis kit (Fermentas, Glen Burnie, MD, USA). Then, reverse transcriptase‐PCR (RT‐PCR) was performed with beta‐actin (F; CAGAGCAAGAGAGGCATCCT, R; TTGAAGGTCTCAAACATGAT), ATG5 (F; TGTCATTTTGCAATCCCATC, R; TTTGGCTTTGGTTGAAATAAG), and BECLIN‐1(F; CAAGATCCTGGACCGTGTCA, R; TGGCACTTTCTGTGGACATCA) primers. Bands observed in the agarose gel electrophoresis were quantified by the Quantity One 1D Gel Imaging software (BioRad, Hercules, CA, USA). The intensities of the bands were normalized to beta‐actin controls. Differences found by those calculations were correlated to the changes in the expression levels. In order to confirm densitometric analysis of PCR bands, qPCR was performed with the same primers by using LightCycler® FastStart DNA Master SYBR Green I kit (Roche, Basel, Switzerland). Finally, the data were analyzed with REST 2008 software (Corbett Research, Sydney, Australia).
Immunoblotting
In order to reveal the changes in protein levels, K562 cells were treated with increasing concentrations of imatinib for 72 hours. Following this treatment, cells were collected and lysed with lysis buffer [10 μl dithiothreitol (10 mM), 10 μl NP‐40, 50 μl NaCl (3M), 50 μl ethylenediaminetetraacetic acid (20 mM), and protease inhibitors aprotinin, leupeptin, pepstatin, phenylmethylsulfonyl fluoride, and 50 μl Tris‐buffered saline (1M, pH 7·5)] for 15 minutes on ice. Supernatant was harvested after centrifugation and protein concentration was measured via bicinchoninic acid protein assay kit. Equal amounts of lysates including the proteins were prepared and run on 12·5% sodium dodecyl sulfate polyacrylamide gel electrophoresis. Finally, immunoblotting was performed by using biotin labeled anti‐actin, anti‐atg6 and anti‐beclin‐1 antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA).
Results
Reverse transcriptase‐PCR
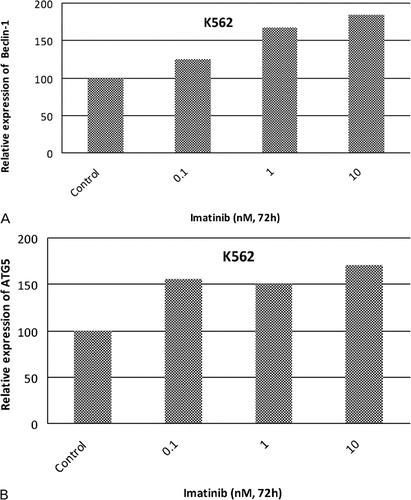
We previously determined the cytotoxic effects of imatinib on K562 cells.Citation23 In this study, IC50 value (the concentration of any chemical that inhibits cell proliferation 50% as compared to untreated control counterparts) of imatinib was found as 280 nM in K562 cells. To prevent a devastating effect of imatinib on K562 cells, we picked lower concentrations of imatinib for PCR experiments. Products of the PCR amplification appeared on agarose gel as seen in . Reverse transcriptase‐PCR (RT‐PCR) results demonstrated that there were significant increases in expression levels of Beclin‐1 and ATG5 as compared to untreated control counterparts and normalized to beta‐actin in response to 0·1, 1, and 10 nM imatinib. Changes in expression levels of Beclin‐1 and ATG5 genes were quantified by assessing the band intensities. Expression levels of Beclin‐1 were increased 1·25‐, 1·67‐, and 1·84‐fold in response to 0·1, 1, and 10 nM imatinib concentrations in K562 cells (). As shown in , there were 1·55‐, 1·50‐, and 1·71‐fold increases in expression levels of ATG5 in K562 cells exposed to 0·1, 1, and 10 nM imatinib, respectively. The results revealed that there were dose dependent increases in expression levels of ATG5 and Beclin‐1 genes in K562 cells.
Figure 1. mRNA levels of autophagy specific genes in response to imatinib in K562 cells. 0·1, 1, and 10 nM imatinib were applied to K562 cells for 72 hours and expression levels of autophagy specific genes were determined by RT‐PCR. Expression levels of beta‐actin were detected as an internal positive control.
Figure 2. Fold changes in expression levels of autophagy specific genes Beclin‐1 (A) and ATG5 (B) by densitometric analysis in response to imatinib in K562 cells. Intensities of RT‐PCR products were measured by software and normalized with beta‐actin gene expression as a house‐keeping gene.
qPCR analysis
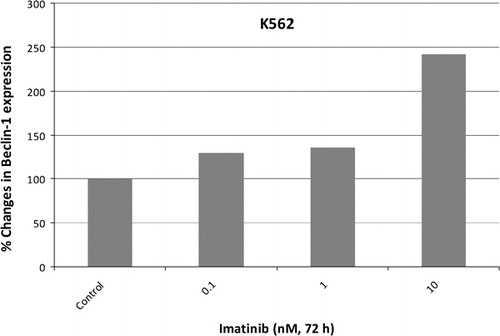
In order to validate densitometric analysis of RT‐PCR products, qPCR was performed to determine the relative expression levels of Beclin‐1. The results demonstrated that the same concentrations of imatinib increased expression levels of Beclin‐1 1·29‐, 1·36‐, and 2·25‐fold as compared to untreated controls and normalized to beta‐actin levels ().
Figure 3. Fold changes in expression levels of Beclin‐1 gene by real‐time PCR in response to imatinib in K562 cells. To confirm semi‐quantitative PCR results, expression levels of Beclin‐1 gene were assessed by real‐time PCR. Relative changes in expression levels of Beclin‐1 gene were found by normalizing with beta‐actin gene.
Immunoblotting
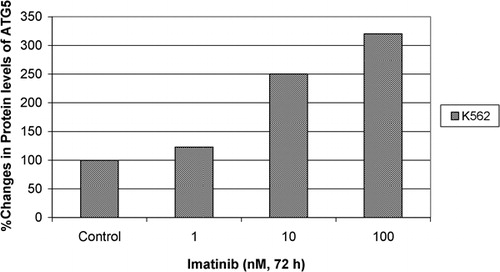
Protein levels of BECLIN‐1 gene under the different concentrations of imatinib were assessed via immunoblotting. Immunoblotting analyses demonstrated dose dependent increases in protein levels of BECLIN‐1 gene as compared to untreated controls (). Densitometric analysis was performed to quantify the protein levels and the results showed that there were 1·99‐, 2·42‐, 2·43‐, 2·61‐, and 2·93‐fold increases in BECLIN‐1 protein intensity in 0·1, 1, 10, 50, and 100 nM imatinib applied to K562 cells, respectively, compared to control group (). On the other hand, western blot analyses demonstrated that there were 1·23‐, 2·50‐, and 3·20‐fold increases in protein levels of ATG5 gene in response to 1, 10, and 100 nM imatinib in K562 cells, respectively ().
Figure 4. Protein levels of Beclin‐1 gene in response to imatinib in K562 cells. (A) Protein levels of Beclin‐1 gene in response to imatinib were determined by western blot. (B) Fold changes in protein levels were determined by densitometric analysis.
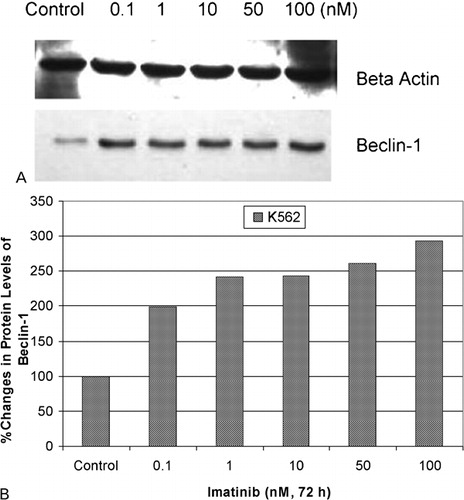
Figure 5. Protein levels of ATG5 gene in response to imatinib in K562 cells were determined by western blot and fold changes in protein levels were determined by densitometric analysis.
Discussion
In the current study, we showed that imatinib induces autophagy in a dose dependent manner in human K562 CML cells. This induction was shown by the increases in the expression and protein levels of autophagy‐specific genes which are considered as the key components of the formation of autophagosomes.
Autophagy is the physiological process in which cellular components are broken down. This event has importance for the maintenance of cellular homeostasis by partaking roles in organelle turnover, resistance to stress conditions and defense against infections. Besides these properties, the literature indicates that autophagy might contribute to cell death or cell survival depending on the circumstances. It was shown by different groups that autophagy is a tumor suppressing mechanism, and defects in the autophagy pathway were associated with the increased incidence of breast, ovarian and prostate cancers.Citation23–Citation26 Interestingly, some other groups demonstrated the significance of autophagy in cancer cell survival by providing nutrients especially in poorly vascularized areas and by contributing to the resistance to cellular stress induced by radiotherapy.Citation27–Citation29 Under the light of these findings, autophagy currently gains importance as a target pathway in cancer therapy.
Imatinib, being the first target specific drug, is a competitive inhibitor of adenosine triphosphate binding pocket of BCR/ABL oncogenic protein. Besides CML, imatinib was shown to be effective for the suppression of the growth of gastrointestinal stromal tumors, ovarian cancer cells, neuroendocrine tumor cells, and melanoma cells.Citation30–Citation33 It was shown that imatinib can suppress c‐KIT in addition to inhibition of BCR/ABL.Citation30 Inactivation of platelet‐derived growth factor receptor alpha and AKT proteins were documented to be the main mechanism of action of imatinib in the suppression of ovarian cancer cells.Citation31 Besides its targets counted so far, imatinib was also shown to inhibit telomerase activity in both BCR/ABL expressing and non‐expressing cells with the currently unknown intermediate agents.Citation34 Recently, ceramide metabolism was implicated to be related to the imatinib’s mechanism of action.Citation7 In another study, sphingosine kinase‐1 was shown as a downstream regulator of imatinib‐induced apoptosis.Citation35 Our group recently has shown that imatinib also induces apoptosis by inhibiting topoisomerase I and topoisomerase II enzyme activities in an enzyme specific manner. Since the efficacy of imatinib is not solely limited to CML, findings of the scientific studies involving imatinib might be exploited to other malignancies and diseases.
Autophagy induced by imatinib application might be either contributing cell death or it might protect the cell against the stress of this kind of chemotherapy. Currently ongoing projects aim to expand the scope of this research to reveal the cellular roles of autophagic induction by imatinib. Outcomes of this research may provide novel targets in CML therapy. If autophagy acts as a protective mechanism for cancer cell, in this case, drugs blocking autophagy pathway might be incorporated to the therapy along with conventional imatinib therapy. This approach might be useful in treating the imatinib‐resistant CML as well. In contrast, if autophagy is acting as a cell death mechanism besides the imatinib‐dependent apoptosis, this time, autophagy pathway might be promoted for more successful CML therapies. By conducting more research to understand the details of these scenarios, valuable information will be gained for developing more effective CML therapy.
This study was supported by the Turkish Academy of Sciences, Outstanding Young Investigator Programme to Y.B. We thank Dr Şermin Genç for her technical help and Biotechnology and Bioengineering Center staff of Izmir Institute of Technology for their help and technical support.
References
- Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood 2000;96:3343–56.
- Peled A, Hardan I, Trakhtenbrot L, Gur E, Magid M, Darash-Yahana M, et al.. Immature leukemic CD34+CXCR4+ cells from CML patients have lower integrin-dependent migration and adhesion in response to the chemokine SDF-1. Stem Cells 2002;20:259–66.
- Koca E, Haznedaroglu IC. Imatinib mesylate and the management of chronic myeloid leukemia (CML). Turk J Hematol 2005;2(4):161–72.
- Ertmer A, Huber V, Gilch S, Yoshimori T, Erfle V, Duyster J, et al.. The anticancer drug imatinib induces cellular autophagy. Leukemia 2007;21:936–42.
- Frame D. New strategies in controlling drug resistance. J Manag Care Pharm 2007;13:13–7.
- Baran Y, Ural AU, Gunduz U. Mechanisms of cellular resistance to imatinib in human chronic myeloid leukemia cells. Hematology 2007;12:497–503.
- Baran Y, Salas A, Senkal CE, Gunduz U, Bielawski J, Obeid LM, et al.. Alterations of ceramide/sphingosine 1-phosphate rheostat involved in the regulation of resistance to imatinib-induced apoptosis in K562 human chronic myeloid leukemia cells. J Biol Chem 2007;282:10922–34.
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004;306:990–5.
- Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie 2008;90:313–23.
- Kawamata T, Kamada Y, Kabeya Y, Sekito T, Ohsumi Y. Organization of the pre-autophagosomal structure responsible for autophagosome formation. Mol Biol Cell 2008;19:2039–50.
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al.. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001;152:657–68.
- Kametaka S, Matsuura A, Wada Y, Ohsumi Y. Structural and functional analyses of APG5, a gene involved in autophagy in yeast. Gene 1996;178:139–43.
- Kametaka S, Okano T, Ohsumi M, Ohsumi Y. Apg14p and Apg6/Vps30p form a protein complex essential for autophagy in the yeast, Saccharomyces cerevisiae. J Biol Chem 1998;273:22284–91.
- Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol 2001;152:519–30.
- He H, Dang Y, Dai F, Guo Z, Wu J, She X, et al.. Post-translational modifications of three members of the human MAP1LC3 family and detection of a novel type of modification for MAP1LC3B. J Biol Chem 2003;278:29278–87.
- Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol 2007;8:622–32.
- Mijaljica D, Prescott M, Klionsky DJ, Devenish RJ. Autophagy and vacuole homeostasis: a case for self-degradation? Autophagy 2007;3:417–21.
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen GH, et al.. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006;10:51–64.
- Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ 2007;14:500–10.
- Amaravadi RK, Yu DN, Lum JJ, Bui T, Christophorou MA, Evan GI, et al.. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007;117:326–36.
- Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol 2008;3:427–55.
- Todde V, Veenhuis M, van der Klei IJ. Autophagy: principles and significance in health and disease. Biochim Biophys Acta 2009;1792:3–13.
- Gucluler G, Baran Y. Docetaxel enhances the cytotoxic effects of imatinib on Philadelphia positive human chronic myeloid leukemia cells. Hematology, 2009;14(3):139–44.
- Ogier-Denis E, Codogno P. Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta 2003;1603:113–28.
- Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004;23:2891–906.
- Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, et al.. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 1999;59:59–65.
- Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, et al.. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem 2001;276:35243–6.
- Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, et al.. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res 2001;61:439–44.
- Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol 2004;14:70–7.
- Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al.. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347:472–80.
- Matei D, Chang DD, Jeng MH. Imatinib mesylate (Gleevec) inhibits ovarian cancer cell growth through a mechanism dependent on platelet-derived growth factor receptor alpha and Akt inactivation. Clin Cancer Res 2004;10:681–90.
- Fiorentini G, Rossi S, Lanzanova G, Biancalani M, Palomba A, Bernardeschi P, et al.. Tyrosine kinase inhibitor imatinib mesylate as anticancer agent for advanced ocular melanoma expressing immunoistochemical C-KIT (CD 117): preliminary results of a compassionate use clinical trial. J Exp Clin Cancer Res 2003;22:17–20.
- Lankat-Buttgereit B, Horsch D, Barth P, Arnold R, Blocker S, Goke R. Effects of the tyrosine kinase inhibitor imatinib on neuroendocrine tumor cell growth. Digestion 2005;71:131–40.
- Mor-Tzuntz R, Uziel O, Shpilberg O, Lahav J, Raanani P, Bakhanashvili M, et al.. Effect of imatinib on the signal transduction cascade regulating telomerase activity in K562 (BCR-ABL-positive) cells sensitive and resistant to imatinib. Exp Hematol 2010;38:27–37.
- Bonhoure E, Lauret A, Barnes DJ, Martin C, Malavaud B, Kohama T, et al.. Sphingosine kinase-1 is a downstream regulator of imatinib-induced apoptosis in chronic myeloid leukemia cells. Leukemia 2008;22:971–9.