Abstract
We describe the first case of prenatal diagnosis for pyruvate kinase (PK) deficiency in Chinese and emphasize that this disease is an important differential diagnosis in pediatric patients with non-spherocytic hemolytic anemia. A Han Chinese child with a history of severe transfusion-dependent hemolytic anemia was diagnosed to have PK deficiency. Prenatal diagnosis was performed on the second child based on the genetic findings from the family. The index patient was compound heterozygous for a missense mutation (c.1073G>A. p.Gly358Glu) from his father and a large deletion (c.283+1914_c.1434del5006) from his mother. The fetus was a simple heterozygote for the paternal mutation. Pregnancy was allowed to continue and a healthy baby was born. Severe PK deficiency warranting prenatal diagnosis is seen in Han Chinese. Genetic characterization and genotype–phenotype correlation studies on PKLR in different populations are indicated to better define the role of prenatal diagnosis in PK deficiency.
Introduction
Pyruvate kinase (PK) deficiency of red cells (EC: 2·7·1·40) is the commonest inherited enzyme deficiency in the glycolytic pathway, leading to hereditary non-spherocytic hemolytic anemia.Citation1Mutation in the PKLR gene follows an autosomal recessive inheritance. There are over 220 characterized mutations deposited in a public database (PKLR Mutation Database http://www.pklrmutationdatabase.com, accessed 8 January 2011). Heterozygous carriers are asymptomatic but homozygotes or compound heterozygotes can have significant anemia leading to transfusion dependence after birth, neonatal death, and hydrops fetalis.Citation2–Citation5All ethnic groups are affected, but data on Chinese are very scanty.
Case Report
The index patient was the first child in a Han Chinese family. He was born at full term by Caesarean section, with a birth weight of 2·9 kg. Antenatal history was uneventful. Hepatosplenomegaly, severe anemia (hemoglobin of 8·8 g/dl) and unconjugated hyperbilirubinemia (bilirubin 200 μmol/l) were detected at 2 hours after birth, necessitating exchange transfusion on day 1 and prolonged phototherapy until day 10 of life. He developed severe chronic non-spherocytic hemolytic anemia on follow-up, requiring regular red cell transfusion every 3–4 weeks to relieve symptoms and to maintain satisfactory growth. Iron chelation therapy was started at 2 and a half years old. He received splenectomy at the age of 4 years to reduce transfusion requirement. His PK enzyme levels varied from 4·96 to 7·0 IU/g Hb (reference range: 5·6–9·0 IU/g Hb) on multiple assays after he had been put on regular transfusion. Both parents had mildly reduced PK level (4·74 and 5·09 IU/g Hb). A diagnosis of PK deficiency was therefore suspected. Glucose-6-phosphate dehydrogenase level was normal.
His parents were unrelated and asymptomatic. They had normal hemoglobin and bilirubin level. There was no history of abortion or intrauterine death. A family history of anemia, gallstone, or splenectomy was not apparent. Because of the severe clinical course of their first child, the couple requested genetic testing and prenatal diagnosis in a second pregnancy to help obstetric decision-making. Chorionic villous biopsy was performed at 12 weeks of gestation. All procedures were carried out after an informed written consent had been obtained.
Methods and Results
DNA was extracted from EDTA blood sample collected from the proband and parents. The 11 exons of the PKLR gene and promoter were screened using a PCR-DHPLC followed by PCR-sequencing of fragments displaying an abnormal pattern as previously described.Citation6A QMP-SF method was employed to search for a deletion in the mother.Citation7Citation7,8Mapping of the deletion breakpoints was performed using gap-PCR primers flanking the deletion (PCR forward primer: PKLR_In3F4-2 5′-ATG GTG GCT CTT GCC TAT AAT C-3′, reverse primer: PKLR_I9145B 5′-GCC TCC AGC TGT CAT TGC TG-3′) followed by direct nucleotide sequencing of PCR products (forward primer: PKLR_3713F 5′-GAG ATC GAT ACC ATC CTG GC-3′).Citation9
Fetal DNA was extracted from micro-dissected chorionic villi as well as from cultured villous cells. PCR-sequencing and PCR-RFLP (Eco 47III Fermentas #ERO 321) was employed to detect the paternal mutation and QMP-SF was used to screen for the maternal deletion. The postnatal peripheral blood sample was analyzed by direct nucleotide sequencing of exon 8 of the PKLR gene using forward primer PKLR-ex8-F 5′-CAC CTT TCT TCT CCT GCC TG-3′ and reverse primer PKLR-ex8-R 5′-CCC TAA AAC CCA CAG AGT GC-3′.
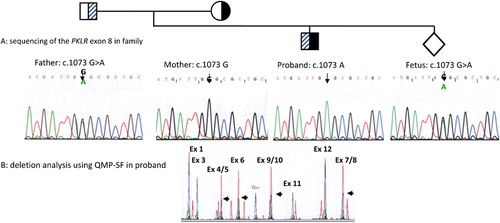
The father was found to carry a mutation in exon 8: PKLR: c.1073 G>A (p.Gly358Glu), while no point mutation could be detected in the mother. She was found to carry a large deletion removing exon 4 to exon 10 of the PKLR gene. The proband was found to carry the paternal point mutation and the maternal deletion (). The deletion extended from intron 3 to almost the 3′ end of exon 10 of PKLR gene. Sequence comparison revealed that this deletion was identical to that found in two other families from the Asia-Pacific region: a Vietnamese familyCitation6 and a family living in Australia.Citation9 The fetal sample was found to be heterozygous for the paternal mutation in exon 8 () with the absence of the maternal deletion. As a benign clinical state was predicted, the parents decided to carry on with the pregnancy. A healthy full-term baby boy was born. A postnatal peripheral blood sample was analyzed at 10 months of age, which confirmed inheritance of the paternal mutation but not the maternal deletion. A PK assay performed at the age of 9 months showed a normal level at 6·5 IU/g Hb. There was no evidence of hemolytic anemia after 2 years of follow-up.
Figure 1. Genetic analysis of proband, parents and prenatal sample. (A) PCR-sequencing of exon 8 shows that the father is heterozygous for the PKLR: c.1073G>A mutation. Mother is hemizygous for normal exon 8 sequence, while the proband is hemizygous for the PKLR: c.1073G>A mutation. The fetus is heterozygous for the PKLR: c.1073G>A mutation. (B) Detection of the c.283+1914_c.1434del5006 deletion. A semi-quantitative PCR is performed with eight sets of primers which cover the whole PKLR gene. A fragment located outside of chromosome 1 is used as internal control for normalization. Chromatogram of a normal sample (red) is superimposed on that of the test sample (blue). Comparison of peak heights between test and normal demonstrates a deletion removing exon 4 to exon 10 in the proband (black arrows).
Discussion
Compared with thalassemia, hereditary spherocytosis, and G6PD deficiency, PK deficiency is a relatively uncommon cause of hereditary anemia. The most recent epidemiological study in southern Chinese using standard quantitative assay reported an incidence of <0·1%.Citation10 Genetic data in Chinese are sparse. Only a handful of Chinese patients have been registered in public database or reported in English literature. The exact ethnic group is either not mentioned or is of non-Han southern Chinese minority group.Citation11Our family is the first Han Chinese who have been genetically characterized and with prenatal diagnosis performed.
Because of its perceived rarity and benignity in many ethnic groups, PK deficiency does not enter early into the differential diagnosis of anemia in pediatric patients. Yet its potential to cause severe disease is often overlooked and delay in diagnosis is common.Citation12This is exemplified in our patient where the diagnosis was suspected only after the start of empirical transfusion therapy. A definitive diagnosis by enzymatic assay was thus not possible. Genetic analysis was able to confirm the diagnosis in this case and also permitted accurate prenatal diagnosis. PK deficiency is uncommon among all indications for prenatal diagnosis. Although this largely reflects the low incidence of mutated PKLR alleles in most populations, failure to make a correct diagnosis and poorly defined indications for prenatal diagnosis could have limited the potential benefits of genotyping and prenatal diagnosis for PK deficiency. A better understanding of the spectrum of mutations in a particular population and their genotype–phenotype correlation will certainly improve the management of affected families.
Of all the mutation types in the PKLR Mutation Database, missense mutations are by far the commonest, accounting for over half of all mutations. Large deletions are very uncommon and only three well-characterized ones have been deposited. The two mutations detected in the current family have not been previously reported in Chinese. The missense mutation (c.1073G>A. p.Gly358Glu) was first found in a Syrian who was homozygous for the mutation.Citation13 He had severe transfusion-dependent hemolytic anemia, indicating that structural disruption at this site is detrimental to enzymatic stability or function. The severe phenotype of our index patient is also supportive of this. The other exon 4–11 deletion of PKLR detected in our family is the largest deletion so far reported. It involves a loss of 5006 bp and predictably results in no functional enzyme production. It was first reported in a Vietnamese,Citation6and later found and characterized in an Australian patient.Citation9 This finding indicates either that it occurred independently for several times in Asia or that the founder deletion occurred in Chinese, which was subsequently spread by Chinese migrants.
In conclusion, we reported the first case of prenatal diagnosis in a Han Chinese family with two PKLR mutations, both of which have not been previously found in Chinese. The importance of considering PK deficiency in the investigation of patients with hereditary hemolytic anemia is emphasized. Further genetic characterization and genotype–phenotype correlation studies in different populations should better define the role of prenatal diagnosis in PK deficiency.
Conflict of Interest and Ethical Approval
The authors have no conflict of interest to disclose. The study was approved by the Institutional Review Board of The Hospital Authority Hong Kong West Cluster/The University of Hong Kong.
References
- Hirono A, Kanno H, Miwa S, Beutler E. Pyruvate kinase deficiency and other enzymopathiesof the erythrocyte. In: , Scriver C R, Beaudet A L, Sly W S, Valle D, ed, editors. The metabolic and molecular bases ofinherited disease. New York: McGraw-Hill; 2001.p. 4637–64.
- Ferreira P, Morais L, Costa R, Resende C, Dias CP, Araújo F, et al.. Hydrops fetalis associated with erythrocytepyruvate kinase deficiency. Eur J Pediatr 2000;159:481–2.
- Gilsanz F, Vega MA, Gómez-Castillo E, Ruiz-Balda JA, Omenaca F. Fetal anaemia due to pyruvate kinase deficiency. ArchDis Child 1993;69:523–4.
- Hennekam RC, Beemer FA, Cats BP, Jansen G, Staal GE. Hydrops fetalis associated with red cell pyruvatekinase deficiency. Genet Couns 1990;1:75–9.
- Sedano IB, Röthlisberger B, Délèze G, Ottiger C, Panchard MA, Spahr A, et al.. PK Aarau: first homozygous nonsensemutation causing pyruvate kinase deficiency. Br JHaematol 2004;127:364–66.
- Costa C, Albuisson J, Le TH, Max-Audit I, Dinh KT, Tosi M, et al.. Severe hemolytic anemia in a Vietnamesefamily, associated with novel mutations in the gene encoding for pyruvatekinase. Haematologica 2005;90:25–30.
- Armour JA, Barton DE, Cockburn DJ, Taylor GR. The detection of large deletions or duplicationsin genomic DNA. Hum Mutat 2002;20:325–37.
- Casilli F, Di Rocco ZC, Gad S, Tournier I, Stoppa-Lyonnet D, Frebourg T, et al.. Rapid detection of novel BRCA1 rearrangementsin high-risk breast-ovarian cancer families using multiplex PCR of short fluorescentfragments. Hum Mutat 2002;20:218–26.
- Fermo E, Bianchi P, Chiarelli LR, Cotton F, Vercellati C, Writzl K, et al.. Red cell pyruvate kinase deficiency:17 new mutations of the PK-LR gene. BrJ Haematol 2005;129:839–46.
- Feng CS, Tsang SS, Mak YT. Prevalence of pyruvate kinase deficiency amongthe Chinese: determination by the quantitative assay. AmJ Hematol 1993;43:271–3.
- Kanno H, Wei DC, Chan LC, Mizoguchi H, Ando M, Nakahata T, et al.. Hereditary hemolytic anemia causedby diverse point mutations of pyruvate kinase gene found in Japan and HongKong. Blood 1994;84:3505–9.
- Pissard S, de Montalembert M, Bachir D, Max-Audit I, Goossens M, Wajcman H, et al.. Pyruvate kinase (PK) deficiency innewborns: the pitfalls of diagnosis. J Pediatr 2007;150:443–5.
- van Wijk R, Huizinga EG, van Wesel AC, van Oirschot BA, Hadders MA, van Solinge WW. Fifteen novel mutations in PKLRassociated with pyruvate kinase (PK) deficiency: structural implications ofamino acid substitutions in PK. Hum Mutat 2009;30:446–53.