
Abstract
Numerous oxidative modifications to proteins and amino acids have been identified with most susceptible, to varying degrees, of some form of oxidative modification. The consequence of oxidation on protein structure and function reveals that some of these modifications are functionally important. The discovery and accurate characterization/description of existing and new modifications requires modern instrumentation, great care, and attention to detail, especially if the modifications are present in low stoichiometric quantities or they only exist transiently. The focus of this brief review is on the use of mass spectrometry, protein chemistry, and proteomics methods and tools to identify oxidatively modified proteins and peptides along with the characterization of specific sites. Many of the specialized mass spectrometry technologies and techniques are becoming more widely available in research laboratories with mass spectrometry or proteomics facilities allowing even non-expert researchers in the field to accurately determine modifications. Illustrative examples of some approaches are provided from the author's work, collaborative research projects, and elsewhere.
Introduction
The determination of amino acid modifications caused by oxidation has gained greater importance over the last 10–15 years as these types of changes have been shown to regulate some important functions in vivo. These include signal transduction (OxyR) transcriptional regulation through disulphide bond formationCitation1,Citation2 and covalent addition of oxygen to Met, forming Met sulphoxide (Met(O)) that alters hydrophobicity, which may have functional consequences.Citation3 Dynamic changes in these modifications may be important mediators of cell signalling, protein removal, degradation, or contribute to some other cellular process or function.Citation4 These changes may be measured using quantitative mass spectrometry (MS). Protein oxidation may be caused by H2O2, HOCl, superoxide, or one of the other reactive oxygen species formed in vivo.Citation5 In vitro many possible reagents cause oxidative changes to amino acids and proteins.Citation6–Citation9 Fortunately, analytical techniques, approaches, and instrumentation also have developed allowing ever smaller quantities of material to be accurately analysed to determine the presence and identity of low sub-stoichiometric modifications. MS is a key technology for determining oxidative modifications of proteins, peptides, or amino acids. The techniques available to discover modifications are similar and do not really depend on where or how oxidations occur. However, more protein may be available with in vitro systems and this generally makes analysis easier. This review will focus on providing an overview of the determination of protein or peptide modification using MS.
Mass spectrometry
The two customary methods used to ionize proteins and peptides are matrix-assisted laser desorption ionization (MALDI) and electrospray (ESI). Both offer exceptional sensitivity, with the potential to analyse zeptomolar amounts with similar mass measurement accuracy expected from both.Citation10,Citation11
MALDI is performed by first co-crystallizing the sample with a large molar excess of low-molecular-weight UV-adsorbing matrix such as Sinapinic acid for proteins or α-cyano-4-hydroxycinnamic acid for peptides.Citation12 Other matrices or specialized sample preparation methods, that yield greater abundance of ion signal, may also be used depending on the analyte.Citation13 This crystalline mixture is generally irradiated by a UV-wavelength laser, and the ablated material containing positive or negative ions analysed using typically time-of-flight (Tof) MS. Tof instruments may be operated in high-resolution (called reflectron) or lower-resolution linear mode depending on the type of information required, with a single abundant protonated [M + H]+ ion normally observed in spectra of peptides and proteins (see C).
Figure 1. ESI and MALDI spectra of bovine CAn. (A) ESI spectrum recorded using static nanospray showing individual charge states of ions of native and oxidized forms (lower abundance series of satellite peaks). (B) Deconvoluted ESI spectrum showing native and two oxidized forms with m/z +16 and +32 greater mass. (C) MALDI spectrum shows abundant [M + H]+ and [M + 2H]2+ ions of native CAn (oxidized forms were not resolved). (D) Amino acid sequence of CAn (SwissProt; P00921, CAH2_BOVIN) showing no Cys, three Met, and seven Trp residues that are possible targets for oxidation. The calculated mass(av) of the native protein is identical (±error) to the values determined by ESI and MALDI.
![Figure 1. ESI and MALDI spectra of bovine CAn. (A) ESI spectrum recorded using static nanospray showing individual charge states of ions of native and oxidized forms (lower abundance series of satellite peaks). (B) Deconvoluted ESI spectrum showing native and two oxidized forms with m/z +16 and +32 greater mass. (C) MALDI spectrum shows abundant [M + H]+ and [M + 2H]2+ ions of native CAn (oxidized forms were not resolved). (D) Amino acid sequence of CAn (SwissProt; P00921, CAH2_BOVIN) showing no Cys, three Met, and seven Trp residues that are possible targets for oxidation. The calculated mass(av) of the native protein is identical (±error) to the values determined by ESI and MALDI.](/cms/asset/17d23dca-cc73-4580-9dc1-b9e299af9df2/yrer_a_11640818_f0001_b.jpg)
ESI requires the sample to be in solution. Desolvated ions form as solvent evaporates during the ESI process and transfer of the ions into the low vacuum region of the mass spectrometer.Citation14 Spectra of proteins and peptides have a unique appearance where instead of forming an abundant singly charged protonated ion, as observed after MALDI, ESI forms a series of multiply charged ions. For peptides, spectra may contain abundant ions corresponding to the charge states of +2, +3, +4, or +5 with one ion normally predominant, whereas proteins show a large variation in the observed charge state envelope, possibly +10 through +50 and with an approximate Gaussian shape (see A). The observed charge state is dependent on a number of factors including amino acid sequence, solvent composition, and ESI MS conditions.Citation15 Non-covalent interactions between proteins and membrane proteins may be observed and studied once solvent and MS conditions have been optimized.Citation16
ESI is ideally suited to interfacing with liquid chromatographic systems where greater sensitivity may be facilitated by the chromatographic clean-up that can remove salts etc. Many of the current proteomic workflows rely on coupling very-low-flow chromatography to an ESI mass spectrometer.
Both MALDI and ESI easily provide mass accuracies of 0.05% for proteins with low ppm mass measurements routinely obtained for peptides and proteins using ultra-high-resolution mass spectrometers. Mass measurement accuracy greatly depends on the type of mass spectrometer used for the measurement, with many current models, such as Orbitraps and Quadrupole Tof instruments, achieving this.
Modern mass spectrometers may be operated using a wide variety of scan methods or modes, with the terminology potentially confusing. The two major modes of operating mass spectrometers are termed MS and MS/MS. In MS, the mass spectrometer simply scans from low mass-to-charge ratio (m/z) to high m/z and a mass spectrum of all ions formed from the sample is obtained. The information recorded can then be interpreted to obtain the molecule mass of the peptide or protein (e.g. A and C). The other common scan mode is called MS/MS, MSCitation2, or tandem MS. In MS/MS, the mass spectrometer is programmed to select exclusively a single m/z that corresponds to one precursor m/z ion, and through a technique called collisional activation or collision-induced dissociation the selected precursor ion is fragmented and a spectrum containing fragment ions characteristic of the precursor ion is obtained.Citation17 For peptides, this characteristic fragmentation pattern may provide a very clear indication of the amino acid sequence since almost all amino acids have a different residue mass.Citation18 Where amino acids are modified, the expected amino acid m/z is altered in a predictable way. For example, addition of oxygen adds 15.9949 to the measured m/z and the residue mass of Met (m/z 131.0405) changes to m/z 147.0364 for Met(O).Citation6 This is shown in where the experimentally determined mass spectrum (by ESI) of carbonic anhydrase (CAn) contains peaks corresponding to the native (29 024) form and an additional two forms (29 041 and 29 057) corresponding to addition of one or two oxygens (see ).
Sample analysis and introduction methods/procedures
As described above, sample preparation is straightforward with MALDI MS, with the technique somewhat resistant to signal suppression by some common contaminates such as detergents and salts. After co-crystallization of the matrix and sample on a target surface, the sample plate is loaded into the mass spectrometer and the spectrum recorded within seconds. Typically, Tof or tandem Tof (Tof/Tof) instruments provide MS, or both MS and MS/MS, capabilities. Individual sample plates containing co-crystallized proteins/peptides may be carefully stored and analysed at a later date without major problems. Combining liquid chromatography (LC) with MALDI has shown some promise but the complexity of initial setup and performance has limited uptake. A great advantage of MALDI is the ability to analyse histological tissue sections, in a technique called MS imaging or MALDI imaging. This produces a molecular pattern or distribution of components across thin films such as brain, muscle, and other organ/tissue specimens.Citation19 Matrix is deposited uniformly on the surface, normally using a specialized piece of equipment or sprayer, and the entire sample analysed by rastering the laser across the surface, collecting individual spectra at each sampling point. Specialized software allows generation of molecular images that may distinguish control from diseased tissue.Citation20 These procedures are becoming more routinely available as methods develop and instrument and software capabilities improve.
A number of approaches may be used to analyse samples using ESI, and in all cases the same in principle applies; that is, the sample is dissolved in a solution suitable for ESI. The versatility of ESI allows samples to be introduced over wide solvent compositions and flow ranges; somewhat arbitrarily nanospray operates at 20–300 nl/min, microspray at 300–2000 nl/min, ESI 5–1000 µl/min with higher flow rates also possible. Lower detection limits are observed as the flow rate diminishes and this is predominantly where most protein/peptide research work occurs due to the desire to analyse/detect lower abundance proteins. Nanoflow rates with chromatographic separations may be difficult or inconsistent but many researchers have overcome these technical challenges and regularly achieve low attomole peptide detection, characterization, and quantification.Citation21 Typically, high-sensitivity ESI measurements are undertaken using quadrupole Tof or Orbitrap mass spectrometers.Citation22,Citation23 Both instruments are able to achieve low ppm mass accuracy and reliably collect 10–20 distinct MS or MS/MS spectra per second using data-dependent acquisitions.Citation24
General outline of identifying protein modifications
A great number of covalent modifications of amino acids have been described and along with sequence variations, splice variants, and other changes in the expected translated amino acid sequence, accounts for the large amount of diversity within the observed proteome (protein complement of a cell or system). Deciphering these diverse forms is complicated, and made more so by the high abundance of some proteins and the possible dynamic nature of some modifications. Despite this, many thousands of phosphopeptide modifications have been identified in a single experiment, together with the possibility of measuring changes in relative and absolute abundance using isotopic labelling techniques.Citation25
Amino acids modification by covalent attachment of oxygen or other elements results in a shift in mass, with these types of mass differences and modifications readily detected by MS or MS/MS experiments after data analysis. A great number of modifications have been identified and collated within UniMod (http://www.unimod.org). Of these, 22 have the key word ‘oxidation’ annotated in their description. Notable amino acids readily modified by oxidation include Cys, Met, and Trp, while most other amino acids generally require harsher conditions or prolonged exposure to oxidants to become modified. shows some of the Cys, Met, and Trp modifications so far described and similar information may be assembled for all amino acids. Some processes or modifications may cause or result in isomerization of amino acids (e.g. Asp to β-Asp) or even l–d racemization. These types of modifications may not be readily identified by MS alone because no change in m/z is observed and the MS/MS fragmentation pattern may also be very similar when compared. Changes in chromatographic retention time may be expected with these modifications and hint that further investigation is warranted.Citation26
Detection and characterization of oxidative modifications
With potentially all amino acids being susceptible to oxidative modification, the analytical techniques required to accurately elucidate these modifications must provide great specificity and, ideally, high sensitivity. Fortunately, MS provides these attributes in many cases. A general approach or workflow for the identification of oxidative modifications is shown in . Ideally, some preliminary information, by way of differences in electrophoretic migration or mass addition (A), may be useful in indicating that the protein is modified in some way (A). But even screening approaches, where no clear preliminary indication of the presence of oxidized amino acids is detected, are possible and these may find oxidative modifications (B).
Figure 2. Schematic of the workflow for the characterization of the oxidative modification of proteins or peptides. (A) Targeted approach where information or inkling of the nature of the modification is known from SDS/PAGE (for example). (B) A non-targeted approach may also be undertaken to simply look for possible oxidative modifications within a sample or specific protein/peptide. Database searches would normally be incorporated into these workflows.
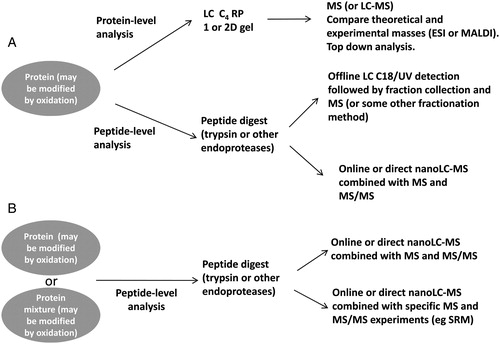
Targeted analysis
A targeted approach may be undertaken to determine the nature and location of an oxidative modification. Ideally, the protein mass is determined with high accuracy using MALDI or ESI and compared with the cDNA-derived theoretical mass (). These small mass errors (0.005%) allow mass differences of m/z 5 to be theoretically determined with a protein (m/z 100 000), but measurement errors of this size are generally only possible with m/z ∼<50 000. Thus, addition m/z 16 (addition of oxygen) would be readily detected, but the presence/absence of a disulphide bond would not be possible for larger proteins simply by measuring the m/z of a protein. In some instances ‘Top down’ MS sequencing, using electron transfer dissociation, may provide additional information allowing the identity and location of small m/z modifications to be determined. This includes disulphide bonds, because the smaller fragment ions are able to be more easily analysed with higher resolution and mass accuracy.Citation27 More routinely, proteins are digested with an endoprotease (e.g. trypsin) and the proteolytic peptides separated using C18 RP LC with either fractions collected and analysed or the eluate analysed immediately by MS. Peptide MS information may be used to identify individual peptide sequences based on comparisons with theoretical and experimentally determined peptide m/z.Citation28 The fragmentation spectrum (MS/MS) normally confirms the presence of the modification and may also allow the site of the modified amino acid to be specifically identified. As seen with protein MS, addition of a modification would normally cause a mass increase that would be identical to the experimental/theoretical mass difference observed for a peptide. Generally, mass accuracies for peptide measurements are very small (<4 ppm) enabling ready identification of oxidized peptides readily such as disulphide bond formation and even discrimination of S (31.9721) or O2 (31.9988) modified peptides. Interpretation of MS/MS spectra ‘de novo’ is specialized and spectra are generally interpreted automatically with software such as Mascot (http://www.matrixscience.com) or PEAKS (http://www.bioinfor.com). These types of software packages simplify the data analysis and generally work very well in correctly identifying and locating oxidative modifications.
As an example, in order to determine the locations of the modified amino acids in CAn (), a tryptic digestion was performed followed by LC-MS and LC-MS/MS. A shows the total ion chromatogram and contains a number of peaks that may be assigned to individual tryptic peptides based on their m/z. Three peptides contain Met (CAn59–76; MVNNGHSFNVEYDDSQDK, CAn213–224 EPISVSSQQMLK, and CAn227–251 TLNFNAEGEPELLMLANWRPAQPLK) and are potential targets for oxidation. Extracted ion chromatograms (XICs) performed using precursor masses of native and oxidized protonated peptides identified Met(O)-containing peptides for all candidates (B–D). The location of the modification was determined or confirmed by looking at the MS/MS pattern of ions and comparing individual sequencing ions from native and oxidized forms. The Met(O) is normally clearly visible because ions that contain this modification are shifted by m/z 16.9949 as shown in , which compares the fragmentation pattern of the [M + 2H]2+ ions of EPISVSSQQMLK and EPISVSSQQM(oxid)LK. Inspection of the other MS/MS spectra confirms the identity and location of Met(O) in the other two peptides (not shown).
Figure 3. Ion chromatograms from the LC-MS separation of tryptic peptides from a mixture of CAn and oxidized CAn. (A) Total ion chromatogram of tryptic peptides showing signals and retention times from all detected peptides. (B) XIC of m/z 673.86 and 681.85 (±0.1) corresponding to [M + 2H]2+ ions of native and oxidized EPISVSSQQMLK. (C) XIC of m/z 700.30 and 705.64 (±0.1) corresponding to [M + 3H]3+ ions of native and oxidized MVNNGHSFNVEYDDSQDK. (D) XIC of m/z 951.50 and 956.83 (±0.1) corresponding to [M + 3H]3+ ions of native and oxidized TLNFNAEGEPELLMLANWRPAQPLK. Specificity is gained by applying XIC mass filters and approximate quantities of each form may be estimated by comparing the relative peak heights or areas.
![Figure 3. Ion chromatograms from the LC-MS separation of tryptic peptides from a mixture of CAn and oxidized CAn. (A) Total ion chromatogram of tryptic peptides showing signals and retention times from all detected peptides. (B) XIC of m/z 673.86 and 681.85 (±0.1) corresponding to [M + 2H]2+ ions of native and oxidized EPISVSSQQMLK. (C) XIC of m/z 700.30 and 705.64 (±0.1) corresponding to [M + 3H]3+ ions of native and oxidized MVNNGHSFNVEYDDSQDK. (D) XIC of m/z 951.50 and 956.83 (±0.1) corresponding to [M + 3H]3+ ions of native and oxidized TLNFNAEGEPELLMLANWRPAQPLK. Specificity is gained by applying XIC mass filters and approximate quantities of each form may be estimated by comparing the relative peak heights or areas.](/cms/asset/84e6b3f6-6801-4678-b6e1-dbbaac42a71b/yrer_a_11640818_f0003_b.jpg)
Figure 4. Partial annotated MS/MS spectra of [M + 2H]2+ ions of (A) EPISVSSQQMLK and (B) EPISVSSQQM(oxid)LK (inset shows the peptide amino acid sequence and abundant sequence-specific ions). Individual sequence-specific y-type ions allow confirmation of the amino acid sequence and assignment of the oxidized Met because y-type ions up to y2 contain an additional m/z 16, indicating conversion to Met(oxid). Some ions containing Met(oxid) also show characteristic loss of m/z 64 (labelled with *), supporting the assignment and location of the modification.
![Figure 4. Partial annotated MS/MS spectra of [M + 2H]2+ ions of (A) EPISVSSQQMLK and (B) EPISVSSQQM(oxid)LK (inset shows the peptide amino acid sequence and abundant sequence-specific ions). Individual sequence-specific y-type ions allow confirmation of the amino acid sequence and assignment of the oxidized Met because y-type ions up to y2 contain an additional m/z 16, indicating conversion to Met(oxid). Some ions containing Met(oxid) also show characteristic loss of m/z 64 (labelled with *), supporting the assignment and location of the modification.](/cms/asset/e9bf6ff9-7577-428d-8ed2-f7aad32888bb/yrer_a_11640818_f0004_b.jpg)
Non-targeted search for modifications
In a non-targeted approach preliminary information about modifications may not be available, possibly due to the low quantity of protein that is readily obtainable. Or the protein of interest is part of a complex mixture making it difficult to isolate for sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS/PAGE) or MS analysis. In these cases, it is still possible to obtain information on the presence/absence of oxidized amino acids by creating a digest map of tryptic peptides. The information obtained from digesting a protein with trypsin, running LC-MS and LC-MS/MS, and extracting the expected m/z of individual tryptic peptides (±oxidation) from the protein of interest provides evidence for a modification (e.g. CAn, ). Typically all tryptic peptides from a protein are not detected, because some peptides are small, too large, highly hydrophilic, or simply do not provide a fragmentation pattern containing abundant sequence-specific b- and y-type ions. But, in most cases, digesting with additional endoproteases such as AspN, LysC, or GluC, and combining the results improves the experimentally determined sequence information, with sufficient overlap among the digests allowing 100% coverage. Once sufficient sequence coverage is obtained and the amino acids of interest are identified, based on MS and MS/MS, it is then possible to search for specific mass additions corresponding to known oxidative modifications using search software such as Mascot. Manual interpretation of each MS/MS may be needed to confirm the identity of the modification and may provide precise information on the location of the modified amino acid within the peptide/protein. Quantification of the relative amount of oxidized vs. unoxidized amino acids may be obtained by comparing XICs of each precursor m/z (e.g. B–D). This approach assumes similar ionization and transmission efficiency etc. during MS; in some cases these may be different and caution is warranted. Ideally, isotopic-labelled internal standards are incorporated if accurate absolute quantification is needed.Citation29 Additional oxidative-modification-specific MS methods such as selected reaction monitoring (SRM) may also be devised to identify or quantify specific modifications. These selective or filtering MS methodologies potentially increase sensitivity/specificity, but the number of modified peptide sequences identified is limited and information about what transitions to monitor needs to be known a priori. SRM methods have been adopted to quantify site-specific Cys oxidation in endogenous proteins.Citation30
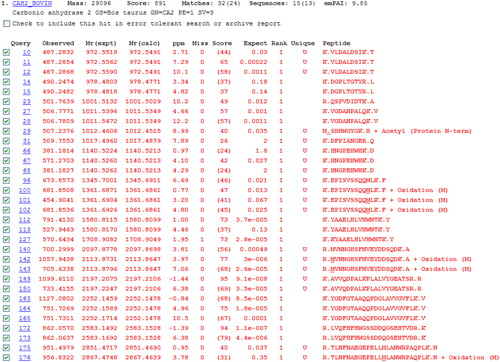
Both the targeted and non-targeted approaches rely on subsequent bioinformatic approaches/analysis; either manual interpretation or automated interrogation of the spectra is needed to retrieve the important and relevant information. Manual interpretation of spectra may be slow and requires experience to ensure correct labelling of ions. Automated approaches utilizing Mascot/SEQUEST searches or PEAKS software speed up and simplify analysis but manual validation of discoveries is warranted. Mascot searches interrogate individual MS/MS spectra and assign the most likely amino acid sequence within a protein database. If an oxidative modification is present, the software will assign the most likely amino acid that has the modification. shows the Mascot ‘Peptide Summary’ view after searching the SwissProt database with the spectral data obtained for the CAn LC-MS/MS experiment. Individual spectra are assigned to tryptic peptide sequences and some of these are modified by oxidation of Met. Additional information and search features, including additional modifications or ‘error tolerant’ searches may be obtained from hyperlinks within the software. Overall, combining automated and manual interpretations/validations generally leads to the best and most reliable information.
Figure 5. Mascot peptide summary report following nanoLC-MS/MS and Mascot database search of a tryptic digest of carbonic anhydrase. The top scoring protein identified corresponds to bovine carbonic anhydrase (75% sequence coverage was obtained). Individual peptides sequences identified, along with oxidized forms found, are shown.
Finally, various researchers have become interested in ‘Redox proteomics’. In these studies, efforts have been directed towards profiling ‘all’ redox active amino acids and their modifications, for example, determining proteome-wide differential disulphide bond analysis (between the control and treated samples)Citation31 or changes in protein expression due to oxidative stress. As with most proteomics experiments, maximizing reliable protein identifications ensures the best chance of understanding the functional changes and consequences induced by oxidation. Numerous approaches have been described and undertaken, with many requiring specific labelling with thiol active reagents like Isotope-coded affinity tag (ICAT) or enrichment strategies to allow detection of lower abundance peptides.Citation32 With potentially many thousands of individual MS/MS obtained from these types of experiments, only automated data analysis is realistically possible.
Conclusions
The susceptibility of proteins to oxidation and importance is irrefutable, with evidence that some of these modifications are required for biological activity. Many of the technical advances in MS and proteomics over the last 10 years may be used to accurately determine and quantify modifications using miniscule amounts of material. Method optimization is normally needed with these small quantities of material or if the protein of interest is part of a complex mixture. But, in many cases, the problems presented are not insurmountable. The approaches outlined are not only suitable for oxidative modification but can be applied for the analysis of any post-translational, chemical, or other processes leading to a sequence different from that described by transcription of DNA.
Disclaimer statements
Funding
None.
Conflicts of interest
None.
References
- Antelmann H, Helmann JD. Thiol-based redox switches and gene regulation. Antioxid Redox Signal 2011;14:1049–63.
- Christman MF, Storz G, Ames BN. OxyR, a positive regulator of hydrogen peroxide-inducible genes in Escherichia coli and Salmonella typhimurium, is homologous to a family of bacterial regulatory proteins. Proc Natl Acad Sci USA 1989;86:3484–8.
- Stadtman ER, Moskovitz J, Levine RL. Oxidation of methionine residues of proteins: biological consequences. Antioxid Redox Signal 2003;5:577–82.
- Lim SY, Raftery MJ, Geczy CL. Oxidative modifications of DAMPs suppress inflammation: the case for S100A8 and S100A9. Antioxid Redox Signal 2011;15:2235–48.
- Winterbourn CC, Kettle AJ. Biomarkers of myeloperoxidase-derived hypochlorous acid. Free Radic Biol Med 2000;29:403–9.
- Garner B, Witting PK, Waldeck AR, Christison JK, Raftery M, Stocker R. Oxidation of high density lipoproteins. I. Formation of methionine sulfoxide in apolipoproteins AI and AII is an early event that accompanies lipid peroxidation and can be enhanced by alpha-tocopherol. J Biol Chem 1998;273:6080–7.
- Nelson KJ, Klomsiri C, Codreanu SG, Soito L, Liebler DC, Rogers LC, et al. Use of dimedone-based chemical probes for sulfenic acid detection methods to visualize and identify labeled proteins. Methods Enzymol 2010;473:95–115.
- Perdivara I, Deterding LJ, Przybylski M, Tomer KB. Mass spectrometric identification of oxidative modifications of tryptophan residues in proteins: chemical artifact or post-translational modification? J Am Soc Mass Spectrom 2010;21:1114–7.
- Raftery MJ, Yang Z, Valenzuela SM, Geczy CL. Novel intra- and inter-molecular sulfinamide bonds in S100A8 produced by hypochlorite oxidation. J Biol Chem 2001;276:33393–401.
- Keller BO, Li L. Detection of 25,000 molecules of substance P by MALDI-TOF mass spectrometry and investigations into the fundamental limits of detection in MALDI. J Am Soc Mass Spectrom 2001;12:1055–63.
- Sun X, Kelly RT, Tang K, Smith RD. Membrane-based emitter for coupling microfluidics with ultrasensitive nanoelectrospray ionization-mass spectrometry. Anal Chem 2011;83:5797–803.
- Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal Chem 1988;60:2299–301.
- Dai Y, Whittal RM, Li L. Two-layer sample preparation: a method for MALDI-MS analysis of complex peptide and protein mixtures. Anal Chem 1999;71:1087–91.
- Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989;246:64–71.
- Li Y, Cole RB. Shifts in peptide and protein charge state distributions with varying spray tip orifice diameter in nanoelectrospray Fourier transform ion cyclotron resonance mass spectrometry. Anal Chem 2003;75:5739–46.
- Laganowsky A, Reading E, Hopper JT, Robinson CV. Mass spectrometry of intact membrane protein complexes. Nat Protoc 2013;8:639–51.
- Bordas-Nagy J, Jennings KR. Collision-Induced decomposition of ions. Int J Mass Spectrom Ion Process 1990;100:105–31.
- Hunt DF, Yates JR III, Shabanowitz J, Winston S, Hauer CR. Protein sequencing by tandem mass spectrometry. Proc Natl Acad Sci USA 1986;83:6233–7.
- Stoeckli M, Chaurand P, Hallahan DE, Caprioli RM. Imaging mass spectrometry: a new technology for the analysis of protein expression in mammalian tissues. Nat Med 2001;7:493–6.
- Norris JL, Caprioli RM. Analysis of tissue specimens by matrix-assisted laser desorption/ionization imaging mass spectrometry in biological and clinical research. Chem Rev 2013;113:2309–42.
- Hanke S, Besir H, Oesterhelt D, Mann M. Absolute SILAC for accurate quantitation of proteins in complex mixtures down to the attomole level. J Proteome Res 2008;7:1118–30.
- Andrews GL, Simons BL, Young JB, Hawkridge AM, Muddiman DC. Performance characteristics of a new hybrid quadrupole time-of-flight tandem mass spectrometer (TripleTOF 5600). Anal Chem 2011;83:5442–6.
- Olsen JV, Schwartz JC, Griep-Raming J, Nielsen ML, Damoc E, Denisov E, et al. A dual pressure linear ion trap Orbitrap instrument with very high sequencing speed. Mol Cell Proteomics 2009;8:2759–69.
- McCormack AL, Schieltz DM, Goode B, Yang S, Barnes G, Drubin D, et al.. Direct analysis and identification of proteins in mixtures by LC/MS/MS and database searching at the low-femtomole level. Anal Chem 1997;69:767–76.
- Dephoure N, Gould KL, Gygi SP, Kellogg DR. Mapping and analysis of phosphorylation sites: a quick guide for cell biologists. Mol Biol Cell 2013;24:535–42.
- Hooi MY, Raftery MJ, Truscott RJ. Interconversion of the peptide isoforms of aspartate: stability of isoaspartates. Mech Ageing Dev 2013;134:103–9.
- Cole SR, Ma X, Zhang X, Xia Y. Electron transfer dissociation (ETD) of peptides containing intrachain disulfide bonds. J Am Soc Mass Spectrom 2012;23:310–20.
- Sutton CW, Pemberton KS, Cottrell JS, Corbett JM, Wheeler CH, Dunn MJ, et al. Identification of myocardial proteins from two-dimensional gels by peptide mass fingerprinting. Electrophoresis 1995;16:308–16.
- Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci USA 2003;100:6940–5.
- Held JM, Danielson SR, Behring JB, Atsriku C, Britton DJ, Puckett RL, et al. Targeted quantitation of site-specific cysteine oxidation in endogenous proteins using a differential alkylation and multiple reaction monitoring mass spectrometry approach. Mol Cell Proteomics 2010;9:1400–10.
- Ghezzi P, Bonetto V. Redox proteomics: identification of oxidatively modified proteins. Proteomics 2003;3:1145–53.
- Gygi SP, Rist B, Griffin TJ, Eng J, Aebersold R. Proteome analysis of low-abundance proteins using multidimensional chromatography and isotope-coded affinity tags. J Proteome Res 2002;1:47–54.