
Abstract
The underlying mechanism of the central nervous system (CNS) injury after acute carbon monoxide (CO) poisoning is interlaced with multiple factors including apoptosis, abnormal inflammatory responses, hypoxia, and ischemia/reperfusion-like problems. One of the current hypotheses with regard to the molecular mechanism of CO poisoning is the oxidative injury induced by reactive oxygen species, free radicals, and neuronal nitric oxide. Up to now, the relevant mechanism of this injury remains poorly understood. The weakening of antioxidant systems and the increase of lipid peroxidation in the CNS have been implicated, however. Accordingly, in this review, we will highlight the relationship between oxidative stress and CO poisoning from the perspective of forensic toxicology and molecular toxicology.
Introduction
The radicals originating from molecular oxygen (O2) are generally named as reactive oxygen species (ROS). There has been much evidence in recent years that some of the symptoms and/or pathophysiology of carbon monoxide (CO) toxicity may be the result of increased free radical-mediated or ROS-mediated neuronal and/or cellular (e.g. erythrocytes) injury, as shown in both experimental animal studies and clinical studies.Citation1–Citation4 ROS-mediated pathology has also been supported by the amelioration of some pathologies, and by the reduced lipid peroxidation seen following treatment with antioxidants like melatonin,Citation5 atenolol,Citation5 some internal molecules like bilirubinCitation6 and hydrogen,Citation7 and some others such as hydrogen sulfide (H2S)Citation8 and other free radical blockers.Citation9 ROS-mediated neuronal injury occurs when oxidative stress exists. Oxidative stress is known as a state in which there is an imbalance between the oxidant and the antioxidant defense systems. It generally occurs as a consequence of elevated ROS production, or when the enzymatic or non-enzymatic antioxidant defense system is inefficient, or a combination of both. Oxidative stress, regardless of the primary cause, can result in the initiation of a number of pathophysiological processes leading to cellular injury and toxicity. ROS are generated in vivo during many of the normal biochemical reactions involving O2, including the mitochondrial electron transfer chain (oxygen is prematurely and incompletely reduced to give superoxide radical in Complexes I and III), microsomal electron transport system, and during phagocytic burst (NADPH oxidase and myeloperoxidase) ().
Figure 1. Schematic representation of the relationships among free oxygen radical formation, enzymatic antioxidant systems, and lipid peroxidation. O2−, superoxide anion radical; O2, molecular oxygen; H+, hydrogen ion, proton; H2O, water; SOD, superoxide dismutase; CAT, catalase; H2O2, hydrogen peroxide; GSH-Px, glutathione reductase; GSH, reduced glutathione; GSSG, oxidized glutathione; GSH-Red, glutathione reductase; NADPH + H+, reduced nicotinamide adenine dinucleotide phosphate; NADP+, oxidized nicotinamide adenine dinucleotide phosphate; Fe++, ferrous iron; OH−, hydroxyl ion; .OH, hydroxyl radical (the most potent free radical); tNOS, total nitric oxide synthases (neuronal NOS, endothelial NOS, and inducible NOS); NO., nitric oxide radical; ONOO−, peroxynitrite; MDA, malondialdehyde (the last product of lipid peroxidation of membrane phospholipids); NO2, nitrite; PUFA, polyunsaturated fatty acid; XO, xanthine oxidase.
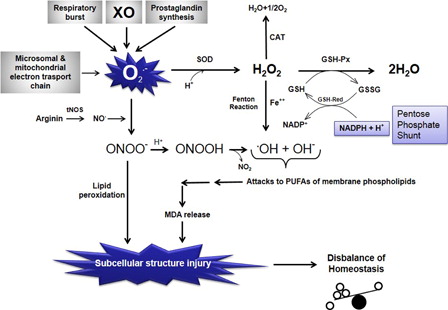
The pathophysiological effect of increased oxidative stress on whole organism and the tendency of central nervous system to toxic effects of ROS
Increased production of ROS and/or decreased detoxification ability of cells might cause increased oxidative stress in organs. Central nervous system (CNS) cells are more vulnerable to the toxic effects of ROS than other organs because they have a high rate of oxidative metabolic activity, a low level of protective antioxidant enzymes, a high ratio of membrane surface area to cytoplasmic volume, a neuronal anatomical network vulnerable to disruption, and high concentrations of readily oxidizable membrane polyunsaturated fatty acids (PUFAs).Citation10 The PUFAs located in cellular membranes of the brain can easily react with ROS and lead to lipid peroxidation. In case of lipid peroxidation of the membranes, it can markedly alter membrane transport mechanisms. Phospholipase A2 is the rate-limiting enzyme of membrane phospholipid metabolism and prostaglandin synthesis initiating the release of arachidonic acid from certain phospholipids. NOx interact with unsaturated lipids both in vitro and in vivo through a series of complicated mechanisms which lead to the initiation of oxidation and formation of nitrated lipid adducts. Nitric oxide (NO) and ONOO− also potently modulate the oxidative activity of enzymes which form the lipid signaling molecules, eicosanoids. Through this, NOx can have multiple and complicated effects on lipid-mediated signal transduction reactions. Studies using pure lipids (linoleic acid, phosphatidylcholine liposomes, cholesteryl linoleate, and free cholesterol) have shown that ONOO− can cause formation of several lipid oxidation products including conjugated dienes, malondialdehyde (MDA), lipid peroxides, lipid hydroxides, F2-isoprostanes, and oxysterols.Citation11 Increased production of ROS overwhelms the capacity of endogenous free radical scavengers. ROS attack the PUFAs in the membrane lipid and result in lipid peroxidation. Under these conditions, the defense system cannot prevent the escape of ROS, especially from the mitochondria, and their effects on the other intracellular compartments. Because of its high lipid content, the CNS is more sensitive to an ROS attack and lipid peroxidation than the other body compartments and organs ().Citation12
CO poisoning
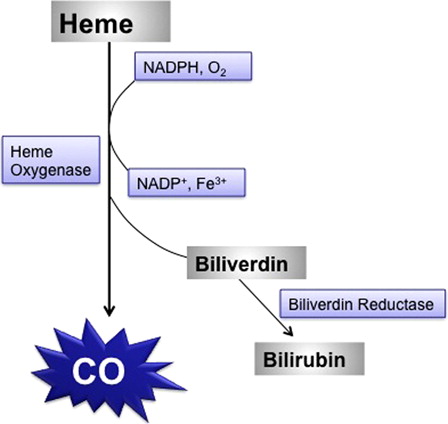
CO is a colorless, odorless, tasteless, and non-irritating toxic gas that is produced by incomplete combustion of organic compounds. Although everyone has CO in their blood (about <5%), heavy smokers and those in certain occupations, such as diesel engine operators, forklift operators, welders, police officers, industrial painters, firefighters, and warehouse workers, may reach 10% saturation. Torgny Sjöstrand in 1949 published his classic paper that the body generated CO intrinsically. Later, it was shown that it arises as part of the metabolism of the heme group, where for each heme split, one CO, one Fe, and one biliverdin molecule are generatedCitation13 (). Endogenous production of CO is unimportant because the values seldom exceed 3% COHb. The most important sources of CO poisoning are environmental and exogenous. Healthy individuals can survive with blood saturations of 40% for a minute or of 20% for a week. The most vulnerable organs to CO-induced hypoxia are the brain and heart because of their high metabolic rates. The amount of CO inhaled and/or its exposure time are the most critical factors that determine the severity of CO poisoning.Citation14
Figure 2. Intrinsic CO production during heme catabolism in the body.
The proposed basic mechanisms for CO toxicity
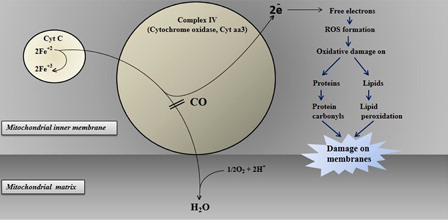
The mechanism of CO toxicity has been classified as hemoglobin, myoglobin, cytochrome oxidase, and cytochrome P450-dependent mechanisms. When inhaled, CO is readily absorbed from the lungs into the bloodstream, where it forms a tight but slowly reversible complex with hemoglobin. Therefore, this reversible combination with CO hemoglobin forms carboxyhemoglobin (HbCO), resulting in tissue hypoxia. Getting an individual away from the source of CO will lead to the eventual removal of CO. The affinity of CO to hemoglobin is about 250 times greater than O2. Oxygen bound to the HbCO produces a complex that does not give oxygen to peripheral tissues. The decreased O2 delivery to the CNS results in ventilator stimulation and increased CO uptake, elevation in HbCO, and respiratory alkalosis. Myoglobin has an affinity for CO 40 times greater than that for O2. Cardiac myoglobin is also a target for CO coupling. A reduction in the O2-carrying capacity because of the elevated HbCO level, exacerbated by impaired perfusion, which results from hypoxic cardiac dysfunctions, will trigger ischemia. The hypoxia and reduction in blood flow allow CO to bind to cytochrome c oxidase (), interfering with cellular respiration at the mitochondrial level (aerobic adenosine triphosphate synthesis).
Figure 3. The proposed toxic effect of CO in the electron transport chain of mitochondria connected with oxidative stress.
Autopsy studies revealed that CO poisoning affects several brain regions including the cerebral cortex, globus pallidus, caudate putamen, hippocampus, and striatum.Citation15 On the other hand, CO poisoning commonly results in acute and delayed neuropsychological sequelae including delirium, amnesia, urine and fecal incontinence, gait disturbance, Parkinson-like syndromes, depression, and anxiety, which persist 1–3 weeks after CO poisoning.Citation16 The basic manifestation of CO is dyspnea, nausea, vomiting, and dizziness. Tachycardia, tachypnea, weakness, and ataxia can also be seen after moderate exposure. Syncope, seizures, hypotension, coma, and death are the signs of more severe CO poisoning. One of the most affected organs is the heart, which may suffer premature ventricular contractions, atrial fibrillation, arrhythmia, heart block, and ischemic changes. CO poisoning causes brain hypoxia due to interference with the O2 supply to the brain (by binding hemoglobin), showing neuropsychological symptoms accompanied with pathological changes of brain tissues in humans. The delayed neuronal cell death following CO inhalation is characterized by the bilateral basal ganglia lesion.Citation17 Magnetic resonance images revealed multiple lesions in the subcortical white matter and basal ganglia in patients who had delayed encephalopathy after CO intoxication, showing that neurological manifestations correlated roughly with neuroimaging changes.Citation18
To characterize the mechanism of CO toxicity, an in vitro exposure system has been preferred for investigating the intracellular pathways leading to neural cell death. Studies showed that both hippocampal HT22 cells and glial D384 cells exposed to CO undergo apoptotic cell death.Citation19 Exposed cells have exhibited abnormal cell morphology with cell shrinkage and nuclear condensation; on the other hand, they exhibited a loss of mitochondrial membrane potential, release of cytochrome c into the cytosol, nuclei with chromatin condensation, and exposure of phosphatidylserine on the external leaflet of the plasma membrane. CO also triggers activation of caspase and calpain proteases.
A new approach for CO toxicity: ROS-induced damage
The direct effect of CO on intracellular targets remains poorly understood, although neurotoxicity and brain hypoxia are well known. Accumulated evidence from experimental studies implicates oxidative stress as a major reason for tissue injury in a variety of human diseases and experimental disease models. Oxidative stress plays a certain role in CO toxicity as well. It can be put forward that oxidative stress is the main element for CO-related neuronal injury. Even if there are plenty of proposed mechanisms, the basic mechanism of brain injury after CO poisoning is incompletely understood. The late changes associated with CO poisoning are mostly similar to post-ischemic reperfusion injuries. CO-induced tissue hypoxia may be followed by reoxygenation injury to the CNS. Hyperoxygenation facilitates the production of ROS, which in turn can oxidize nucleic acids and essential proteins, whereby resulting in typical reperfusion injury. In a study, 88 patients admitted to the emergency room for CO poisoning were tested in terms of total antioxidant capacity, total oxidative stress, and oxidative stress index of blood serum.Citation4 Total oxidative stress (17.14 versus 8.47 µmol H2O2 equivalent/l) and oxidative stress index (0.77 versus 0.40 arbitrary units) were found to be significantly increased compared to healthy controls. Hyperbaric O2 therapy or normal O2 therapy led to a significant decrease in these two parameters () .
Table 1. Proposed pathways for ROS in CO poisoning with detailed in vivo/in vitro studies and their outcomes
4-Hydroxy-2-nonenal in CO toxicity
ROS is normally believed to play an important role in neuronal death in degenerative diseases and cerebral ischemia. Studies demonstrated an increase in the immune reactivity of 4-hydroxy-2-nonenal (HNE), an aldehydic product of membrane lipid peroxidation, in the neurons and axons after ischemia.Citation30 Although there are no reports showing a direct relationship between CO and HNE formation, in light of the relevant literature, it is obvious that CO can cause HNE formation in neurons as well.Citation31,Citation32 The increase of HNE by CO inhalation suggests that lipid peroxidation is partly concerned in CO-induced cell injury. In a recent study whereby histological comparison of delayed neuronal cell death with HNE immune reactivity was detected, it was shown that CO inhalation injures neurons by ROS, independent of hypoxia.Citation20
Lipid peroxidation and glutathione in CO toxicity
CO-mediated delayed neuronal damage was investigated in Wistar male rats after CO poisoning had been performed by intraperitoneal injection (100 and 50 ml/kg).Citation1 After the step-down-type passive avoidance test, they were sacrificed on days 1, 3, 7, 14, and 21 and lipid peroxidation parameters together with antioxidant molecules were determined after brain samples were homogenized. Step-down latency was considerably shortened in the first 3 days, and maintenance of latency for the following 18 days and the numbers of errors in the first 3 days and the following days were significantly increased compared to day 0. MDA levels, the parameter of lipid peroxidation, in serum and nerve tissue were significantly increased, while glutathione reductase and glutathione peroxidase activities, the antioxidant enzymes, were significantly decreased. Glutathione (GSH) levels were also decreased the following days after a short increase in day 1. Authors suggested that CO-mediated deficits in learning and memory might be brought on by oxidative damage to the cerebral cortex, hippocampus, or both; therefore, the neuronal mitochondria and energy metabolites were damaged by free radicals generated through lipid peroxidation in the cells. Finally, the disruption contributed to the progressive memory dysfunctions induced by CO.
Mitochondrial dysfunction, oxidative stress, and lipid peroxidation are all expected after CO poisoning as a vicious circle (), in which the effect continues to feed the cause.Citation33 Oxidative damage on membranes from circulating lymphocytes of patients after acute CO poisoning was studiedCitation23 and a significant increase of lipid peroxidation was found compared to control individuals. A marked cytochrome c oxidase activity inhibition suggesting the involvement of similar mechanisms in the brain was also observed.
Blood plasma concentrations of thiobarbituric acid reactant substrates (TBARS, lipid peroxidation end product), oxidized proteins, GSH, and oxidized glutathione (GSSG) were found to be significantly increased in rats exposed to CO in a manner known to cause brain oxidative stress.Citation24 The elevation of GSH and GSSG was successfully inhibited in rats pretreated with the nitric oxide synthase (NOS) inhibitor, n-nitro l-arginine methyl ester hydrochloride (l-NAME). It can be alleged that NO may play a significant role with GSH elevations, because early perivascular oxidative changes during CO exposure are mediated by NO-derived ROS and these NO-mediated changes are required for the subsequent cascade of cellular and biochemical changes that lead to brain lipid peroxidation after CO poisoning.Citation22 Elevations in plasma TBARS, oxidized proteins, and GSH did not correlate with HbCO levels, but they occurred when rats were exposed to CO according to the pattern causing brain lipid peroxidation.Citation22,Citation34,Citation35 Plasma markers of oxidative stress were detected after the standard CO exposure model and not after the other patterns of CO poisoning. The reason would be that the standard model of exposure causes the most intense ROS-related changes.
Glutamate, an excitatory neurotransmitter amino acid, in CO toxicity
In an earlier study, delayed neurological damage after CO hypoxia was investigated in rats to determine whether programmed cell death, in addition to necrosis, is involved in neuronal death.Citation2 Male Sprague–Dawley rats were used to test this hypothesis by applying 2500 ppm of CO in air under mechanical ventilation for 30 minutes, followed by reoxygenation with air. Arterial blood pressure, blood gases, HbCO levels, brain temperature, cerebral blood flow, glutamate concentration, and ROS generation in the brain were measured and learning and memory (by using another group of animals) assessment in living animals as well as histopathological studies, ultrastructural analysis of dying cells, DNA fragmentation, and some other chemicals in death animals were performed. They found increases in glutamate release and OH generation after CO hypoxia. Rats showed learning and memory deficits after exposure associated with heterogeneous cell loss in the cortex, globus pallidus, and cerebellum. Ultrastructural features of both neuronal necrosis and apoptosis were also observed by electron microscopy. The mechanism by which CO hypoxia induces delayed cell death is not fully understood, although the increased excitotoxicity and ROS production after CO hypoxia may be involved.Citation36 The enhanced ROS generation could be mediated through glutamate receptor activation by increased excitatory amino acids including glutamate, based on stimulation of OH generation.Citation37
The mechanisms of enhancement of OH generation due to CO poisoning, including possible involvement of the glutamatergic system, have been studied in male Sprague–Dawley rats exposed to 3000 ppm of CO.Citation15 OH generation was determined by measuring the extracellular level of 2,3-dihydroxybenzoic acid (2,3-DHBA). The 2,3-DHBA levels in the striatum began to increase during the first 20-minutes period of CO exposure and a significant increase of 2,3-DHBA appeared during the following 20-minutes period. 2,3-DHBA levels were increased in the reoxygenation period shortly after the termination of CO exposure and then rapidly decreased to the control level. Exposure of rats to 3000 ppm of CO significantly increased extracellular glutamate in the striatum following reoxygenation. All of these findings show that CO poisoning stimulated OH generation in vivo. CO poisoning also causes an increase in extracellular dopamine, which could lead to ROS generation in the striatum together with glutamate increases.Citation29,Citation37 However, inhibiting of dopamine synthesis or glutamate receptors had little or no effect on CO-induced OH generation.Citation37 Another group reported an increase in extracellular glutamate, followed by enhancement of OH generation in the cortex and hippocampus due to CO poisoning in rats.Citation2 Glutamate seems to be a transmitter to participate in striatal OH generation in CO-poisoned rats.
NO and OH radical in CO toxicity
NO is synthesized from l-arginine (l-Arg) in the presence of NADPH, O2, tetrahydrobiopterin by three different forms of NOSs. It has a role in several physiological and pathological processes in the CNS. The effect of CO poisoning in the NO system in the brain was investigated by using in vivo brain microdialysis followed by an assay of the major oxidative NO products, nitrite and nitrate.Citation38 CO poisoning was found to reduce NO production in rat striatum, and this reduction might be due to suppression of the NOS activity by a lack of its substrate, arginine. On the other hand, some researchers found an NO level increase simply because of competitive binding with CO for intracellular heme protein-binding sites. CO has a stronger affinity than NO for heme protein-binding sites, and increased vascular NO promotes oxidative stress, leakage of various mediators, and triggers phagocyte adherence/activation.Citation39
In one study, rats were subjected to both a small volume of pure CO injection (1000 ppm) and 3000 ppm of CO in air for 20 minutes, followed by the determination of NO by electron paramagnetic resonance measurement spectroscopy and nitrotyrosine by immunohistochemistry using polyclonal antibody.Citation22 NO was found to be increased 9-fold immediately after CO poisoning. A burst of NO was detected in the brain at the termination of CO exposure, where blood flow was decreased to 50% and unconsciousness occurred.Citation34 In addition, nitrotyrosine, a product from the reaction of peroxynitrite with proteins,Citation40 was found to be deposited in vascular walls and also diffusely throughout the parenchyma with a 10-fold increase in the brain. Platelets were involved in the production of nitrotyrosine in the early phase of CO poisoning. When rats were subjected to the NOS inhibitor, l-NAME, formation of NO and nitrotyrosine in response to CO poisoning were abolished together with leukocyte sequestration in the microvasculature, endothelial xanthine dehydrogenase conversion to xanthine oxidase (XO), and lipid peroxidation of the cellular structure of the brain. This study may lead to a better understanding of CO poisoning as a reperfusion-like injury.Citation22 Exposure to 50 ppm or higher of CO for 1 hour increased the concentration of nitrotyrosine in the aorta.Citation26 Immunologically reactive nitrotyrosine was localized in a discrete fashion along the endothelial lining and this was inhibited by pretreatment with the NOS inhibitor, l-NAME.
Mitochondrial electron transport chain in CO toxicity
The mitochondrial electron transport chain is one of the targets of CO because of their heme groups. Among them, cytochrome c oxidase inhibition is crucial for some of the symptoms ascribed to CO toxicity, e.g. delayed neuronal injury (). Zhang and PiantadosiCitation31 demonstrated that cerebral oxidative injury is not the direct effect of hypoxia but it is due to ROS generated by brain mitochondria. Almost the same findings have been found in circulating lymphocytes from patients acutely intoxicated by CO.Citation23 Lipid peroxidations on membranes related to inhibition of mitochondrial enzymes could play a role in the pathophysiology of CO toxicity.
CO increases mitochondrial ROS production in vitro and in vivo, and mitochondria metabolize CO by oxygenation to CO2. CO also binds to reduced transition metals and other metalloenzymes including guanylate cyclase and cytochrome P450; therefore, CO–heme ligand formation tends to interfere with redox reactions involving O2 as well as NO. In some situations, CO can activate the expression of inducible nitric oxide synthase (iNOS), and in others, inhibits the expression of iNOS.Citation41
The impairment of energy metabolism after CO exposure of brain mitochondria is complex. The reason for continued toxicity after the period of reoxygenation attracted the attention of researchers. The first evidence was enhanced production of partially reduced oxygen species, ROS, after CO intoxicationCitation34 together with the finding of increased lipid peroxidation in the brain after experimental CO poisoning. It suggests a reoxygenation injury after CO hypoxia. The oxidative damage of proteins, lipids, or nucleic acids by ROS is a critical process in the pathogenesis of some of these injuries.Citation42 To test that hypothesis, Zhang and PiantadosiCitation31 planned a study in which the generation of ROS in the rat brains was subjected to 1% CO for 30 minutes and then reoxygenated on air for 0–180 minutes. They found that increased regional H2O2 generation probably arose from inactivation of the catalase enzyme. It indicates that the subcortical regions, which include the basal ganglia (known as highly vulnerable to neuropathological injury from CO intoxication),Citation43 may be a major source of ROS after CO exposure. They found that increased mitochondrial oxidized GSH shows a more deleterious effect than the other oxidation parameters because oxidized GSH can easily oxidize surrounding proteins in mitochondria.
Vitamin C and other antioxidants in CO toxicity
The results obtained from the related studies show that chemicals with the capacity to reduce free radical species can protect CNS function from the disruptive effects of CO. Vitamin C (ascorbic acid, vit C), a strong antioxidant vitamin in an aqueous environment in the body, administration leads to stimulation of ROS generation when administered parenterally, but not orally, suggesting the pro-oxidant action of vit C in vivo under normal physiological circumstances.Citation44 Hara et al.Citation45 found that pro-oxidant action of vit C was elicited in vivo by the increase in extracellular vit C in rat striatum following intraperitoneal administration of dehydroascorbate (the oxidized form of vit C). The same group demonstrated that both extracellular vit C and chelatable iron might play a role in OH generation in rat striatum due to CO poisoning, whereby the pro-oxidant nature of vit C might be elicited in the extracellular space.Citation21 On the other hand, authors suggested that the time course of changes in extracellular vit C could not be completely superimposed on that of the CO-induced OH generation. Moreover, the CO-induced OH generation was completely suppressed by an iron chelator, deferoxamine, suggesting that chelatable iron is the most likely candidate for CO-induced OH generation in rat striatum. In an in vitro study, when mouse hippocampal neurons and human glial cells were exposed to CO ranging from 300 to 1000 ppm in the presence of 20% oxygen, apoptosis and mitochondrial toxicity took place.Citation19 Pretreatment of cells with the antioxidant Mn (III) tetrakis (4-benzoic acid) porphyrin (MnTBAP) (a superoxide dismutase (SOD) mimetic) significantly reduced the number of apoptotic nuclei, pointing to a critical role of oxidative stress in CO toxicity.
Numerous strategies with regard to oxidative stress have been applied in the treatment of CO poisoning. Free radical scavengers, monoamine oxidase inhibitors, aggressive supportive care, and N-methyl-d-aspartate blockers as well as hyperbaric O2 therapy are the most known therapies that were studied extensively. In a review article, Mannaioni et al.Citation46 outlined that the addition of free radical scavengers such as GSH, acetylcysteine, and tempol to the standardized international protocols for the treatment of acute CO poisoning might be advisable.
H2S has been shown to have antioxidant and anti-apoptotic effects, which protected neurons against oxidative stress by scavenging ROS and reactive nitrogen species. It has been hypothesized that H2S might be an interesting potential strategy for treating acute CO poisoning via free radical scavenger properties.Citation8 Another hypothesis is that hydrogen may be a promising, effective, and specific treatment of acute CO poisoning. The hypothesis is based on the theory that molecular hydrogen can selectively decrease OH and ONOO−, given OH and ONOO− are much more reactive than other ROS.Citation7
CO exposure produces preferential high-frequency impairment in the auditory threshold. A study was designed to address the possibility that free radical generation occurs in the cochlea during CO hypoxia and that auditory impairment eventually occurs.Citation9 Inhibition of free radical production was accomplished by using two agents: phenyl-N-tert-butylnitrone (PBN), which is a spin trap agent scavenging free radicals, and allopurinol, which is a free radical inhibitor through the XO metabolic pathway. They found that both PBN and allopurinol might be protective in the cochlea. The results were consistent with those of other studies that have used these agents both in the cochlea (under different experimental protocols) and in other organ systems.Citation47
The effects of CO exposure on an isolated perfused rat heart concerning heart rate and perfusate flow were examined.Citation28 The data suggested that CO had two toxic effects: there was a dose-dependent decrease in perfusate flow post-exposure to CO, which was seen in all hearts exposed to CO and which is prevented by inclusion of the antioxidants ascorbate and Trolox C in the perfusion buffer. In the other possibility, there were CO-induced decreases in heart rate in some hearts either immediately after the start of the exposure or lagging 10–15 minutes after the end of the exposure that do not appear to be prevented by ascorbate and Trolox C in the perfusion buffer. They concluded that these effects were mediated by the CO-induced production of the oxidants, H2O2 and ONOO−. The administration of l-Arg during CO exposure, but not throughout the experimental period, was planned to examine its effect on the CO-induced OH generation.Citation25 The administration of l-Arg alone during CO exposure suppressed the CO-induced OH generation, seemingly supporting the authors' idea that reduction of l-Arg in CO poisoning facilitates production of ROS in preference to NO by NOS, resulting in stimulation of OH generation in rat striatum. However, d-Arg, which cannot be an NOS substrate, suppressed it as well. These findings suggest that the suppressive effect of l- and d-Arg on the CO-induced OH generation might be due to their ability to scavenge ROS. Studies were conducted with rats to investigate whether platelet-activating factor and NO-derived oxidants played a role in the initial adherence of neutrophils to vasculature in the brain after CO poisoning. Before CO poisoning, rats were treated with the competitive PAF receptor antagonist, WEB-2170 or with the ONOO− scavenger, selenomethionine.Citation27 Both agents caused significantly lower concentrations of myeloperoxidase in the brain after poisoning, indicating fewer sequestered neutrophils. On the other hand, the agents reduced the concentration of nitrotyrosine, indicating less oxidative stress due to NO-derived oxidants.
The effects of N-acetylcysteine and melatonin on histopathological and biochemical parameters after CO poisoning were studied in rats.Citation48 Both agents were found to be helpful in CO poisoning protecting brain and lung tissues from oxidative damage.
Antioxidant enzymes in CO toxicity
To test the hypothesis that chronic very mild prenatal CO exposure subverts the normal development of the cerebellar cortex, researchers exposed the rats to 25 ppm of CO.Citation3 Immunohistochemical studies together with immunolocalization of oxidative stress proteins, oxidative stress markers such as SOD, heme-oxygenase-1, iNOS, nNOS, eNOS, and ferritin were investigated. Chronic mild CO exposure was found to promote an increase in SOD-1, SOD-2, HO-1, iNOS, and nitrotyrosine in the cerebellum.
The potential harmful effect of reoxygenation by normobaric and/or hyperbaric oxygen therapy after CO poisoning
In clinical CO poisoning cases, hyperoxygenation can be achieved by breathing 100% O2 either at atmospheric pressure or by hyperbaric therapy. The goal is immediate saturation of blood with enough O2 to sustain life and to counteract tissue hypoxia in spite of high HbCO. O2 replacement causes rapid reduction of CO in the blood by mass action of O2. An increase in either O2 or CO results in a comparable increase in the corresponding compound with hemoglobin in this equation:
It assists in driving CO away from cytochrome oxidase and HbO2 reduces cerebral edema. These are the life-saving effects of O2 replacement. However, as we mentioned before, the late changes associated with CO poisoning are similar to post-ischemic reperfusion injuries. CO-induced tissue hypoxia may be followed by reoxygenation injury to the CNS. Hyperoxygenation facilitates the production of ROS, which in turn can oxidize macromolecules such as lipids, proteins, nucleic acids, and several others within the cell, thereby resulting in typical reperfusion injury. Oxidized products of lipids (e.g. MDA, HNE, lipid peroxides), proteins (e.g. protein carbonyl), and nucleic acids (e.g. 8-oxoguanine) are important evidence of oxidative reperfusion injury after hyperbaric CO poisoning treatment.Citation4,Citation49,Citation50 These cytotoxic products during rebound reperfusion insult to the sensitive organs, such as the CNS, promote the recruitment of inflammatory leukocytes and cause further injury and cell death secondary to both micronecrosis and apoptosis.Citation51
Conclusion
The above-mentioned findings add to the growing body of evidence for oxidative stress in CO intoxication. According to the findings in both oxidant and antioxidant systems, the oxidant/antioxidant imbalance may have a pathophysiological role in CO toxicity and it would be very interesting to test/include antioxidant drugs in the treatment of CO poisoning in addition to the classical treatments suggested by previous reports.Citation52,Citation53 Further investigations are needed to provide definitive information about the relationships between lipid peroxidation and membrane fatty acid composition in CO intoxication.
Disclaimer statements
Contributors
Conceived and designed the study: SA, SE, NI, SC, FU, OA. Performed the review methods and searches: SA, SE, MK, SD, OA. Wrote the paper: SA, SE, NI, SC, MK, FU, SD, OA.
Funding
None.
Conflicts of interest
None.
Ethics approval
None.
References
- Wang P, Zeng T, Zhang CL, Gao XC, Liu Z, Xie KQ, et al. Lipid peroxidation was involved in the memory impairment of carbon monoxide-induced delayed neuron damage. Neurochem Res 2009;34:1293–8.
- Piantadosi CA, Zhang J, Levin ED, Felz RJ, Schmechel DE. Apoptosis and delayed neuronal damage after carbon monoxide poisoning in the rat. Exp Neurol 1997;147:103–14.
- Lopez IA, Acuna D, Beltran-Parrazal L, Lopez IE, Amarnani A, Cortes M, et al. Evidence for oxidative stress in the developing cerebellum of the rat after chronic mild carbon monoxide exposure (0.0025% in air). BMC Neurosci 2009;10:53–71.
- Kavakli HS, Erel O, Delice O, Gormez G, Isikoglu S, Tanriverdi F. Oxidative stress increases in carbon monoxide poisoning patients. Hum Exp Toxicol 2011;30:160–4.
- Mizrak B, Celbis O, Parlakpinar H, Olmez E. Effect of melatonin and atenolol on carbon monoxide cardiotoxicity: an experimental study in rats. Basic Clin Pharmacol Toxicol 2006;98:565–8.
- Breimer LH, Mikhailidis DP. Could carbon monoxide and bilirubin be friends as well as foes of the body? Scand J Clin Lab Invest 2010;70:1–5.
- Shen MH, He JA, Cai JM, Sun QA, Sun XJ, Huo ZL. Hydrogen as a novel and effective treatment of acute carbon monoxide poisoning. Med Hypotheses 2010;75:235–7.
- Yu YP, Li ZG, Wang DZ, Zhan X, Shao JH. Hydrogen sulfide as an effective and specific novel therapy for acute carbon monoxide poisoning. Biochem Biophys Res Commun 2011;404:6–9.
- Fechter LD, Liu Y, Pearce TA. Cochlear protection from carbon monoxide exposure by free radical blockers in the guinea pig. Toxicol Appl Pharmacol 1997;142:47–55.
- Evans PH. Free radicals in brain metabolism and pathology. Br Med Bull 1993;49:577–87.
- Patel RP, Diczfalusy U, Dzeletovic S, Wilson MT, Darley-Usmar VM. Formation of oxysterols during oxidation of low density lipoprotein by peroxynitrite, myoglobin, and copper. J Lipid Res 1996;37:2361–71.
- Akyol O, Herken H, Uz E, Fadillioğlu E, Unal S, Söğüt S, et al. The indices of endogenous oxidative and antioxidative processes in plasma from schizophrenic patients. The possible role of oxidant/antioxidant imbalance. Prog Neuropsychopharmacol Biol Psychiatry 2002;26:995–1005.
- Sjöstrand T. Endogenous formation of carbon monoxide in man. Nature 1949;164:580.
- Bauer I, Pannen BHJ. Bench-to-bedside review: carbon monoxide – from mitochondrial poisoning to therapeutic use. Crit Care 2009;13:220–230.
- Hara S, Mukai T, Kurosaki K, Kuriiwa F, Endo T. Characterization of hydroxyl radical generation in the striatum of free-moving rats due to carbon monoxide poisoning, as determined by in vivo microdialysis. Brain Res 2004;1016:281–4.
- Chang KH, Han MH, Kim HS, Wie BA, Han MC. Delayed encephalopathy after acute carbon monoxide intoxication: MR imaging features and distribution of cerebral white matter lesions. Radiology 1992;184:117–22.
- Sawada Y, Sakamoto T, Nishide K, Sadamitsu D, Fusamoto H, Yoshioka T, et al. Correlation of pathological findings with computed tomographic findings after acute carbon monoxide poisoning. N Engl J Med 1983;308:1296.
- Hsiao CL, Kuo HC, Huang CC. Delayed encephalopathy after carbon monoxide intoxication – long-term prognosis and correlation of clinical manifestations and neuroimages. Acta Neurol Taiwan 2004;13:64–70.
- Tofighi R, Tillmark N, Dare E, Aberg AM, Larsson JE, Ceccatelli S. Hypoxia-independent apoptosis in neural cells exposed to carbon monoxide in vitro. Brain Res 2006;1098:1–8.
- Uemura K, Hoshino S, Uchida K, Tsuruta R, Maekawa T, Yoshida K. Hypothermia attenuates delayed cortical cell death and ROS generation following CO inhalation. Toxicol Lett 2003;145:101–6.
- Hara S, Mizukami H, Mukai T, Kurosaki K, Kuriiwa F, Endo T. Involvement of extracellular ascorbate and iron in hydroxyl radical generation in rat striatum in carbon monoxide poisoning. Toxicology 2009;264:69–73.
- Ischiropoulos H, Beers MF, Ohnishi ST, Fisher D, Garner SE, Thom SR. Nitric oxide production and perivascular nitration in brain after carbon monoxide poisoning in the rat. J Clin Invest 1996;97:2260–7.
- Miro O, Alonso JR, Casademont J, Jarreta D, Urbano-Marquez A, Cardellach F. Oxidative damage on lymphocyte membranes is increased in patients suffering from acute carbon monoxide poisoning. Toxicol Lett 1999;110:219–23.
- Thom SR, Kang M, Fisher D, Ischiropoulos H. Release of glutathione from erythrocytes and other markers of oxidative stress in carbon monoxide poisoning. J Appl Physiol 1997;82:1424–32.
- Hara S, Mukai T, Kurosaki K, Mizukami H, Kuriiwa F, Endo T. Role of nitric oxide system in hydroxyl radical generation in rat striatum due to carbon monoxide poisoning, as determined by microdialysis. Toxicology 2007;239:136–43.
- Thom SR, Fisher D, Xu YA, Garner S, Ischiropoulos H. Role of nitric oxide-derived oxidants in vascular injury from carbon monoxide in the rat. Am J Physiol 1999;276:H984–92.
- Thom SR, Fisher D, Manevich Y. Roles for platelet-activating factor and *NO-derived oxidants causing neutrophil adherence after CO poisoning. Am J Physiol Heart Circ Physiol 2001;281:H923–30.
- Patel AP, Moody AJ, Sneyd JR, Handy RD. Carbon monoxide exposure in rat heart: evidence for two modes of toxicity. Biochem Biophys Res Commun 2004;321:241–6.
- Hara S, Mukai T, Kurosaki K, Kuriiwa F, Endo T. Modification of the striatal dopaminergic neuron system by carbon monoxide exposure in free-moving rats, as determined by in vivo brain microdialysis. Arch Toxicol 2002;76:596–605.
- McCracken E, Valeriani V, Simpson C, Jover T, McCulloch J, Dewar D. The lipid peroxidation by-product 4-hydroxynonenal is toxic to axons and oligodendrocytes. J Cereb Blood Flow Metab 2000;20:1529–36.
- Zhang J, Piantadosi CA. Mitochondrial oxidative stress after carbon monoxide hypoxia in the rat brain. J Clin Invest 1992;90:1193–9.
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 1991;11:81–128.
- Ernst A, Zibrak JD. Carbon monoxide poisoning. N Engl J Med 1998;339:1603–8.
- Thom SR. Carbon monoxide-mediated brain lipid peroxidation in the rat. J Appl Physiol 1990;68:997–1003.
- Thom SR. Leukocytes in carbon monoxide-mediated brain oxidative injury. Toxicol Appl Pharmacol 1993;123:234–47.
- Piantadosi CA, Tatro L, Zhang J. Hydroxyl radical production in the brain after CO hypoxia in rats. Free Radic Biol Med 1995;18:603–9.
- Laplanche L, Kamenka JM, Barbanel G. The novel non-competitive N-methyl-D-aspartate antagonist gacyclidine blocks the glutamate-induced release of hydroxyl radicals in the striatum under conditions in which dizocilpine does not. A microdialysis study in rats. Neurosci Lett 2000;289:49–52.
- Hara S, Mukai T, Kurosaki K, Kuriiwa F, Endo T. Characterization of suppression of nitric oxide production by carbon monoxide poisoning in the striatum of free-moving rats, as determined by in vivo brain microdialysis. Brain Res 2003;979:27–36.
- Omaye ST. Metabolic modulation of carbon monoxide toxicity. Toxicology 2002;180:139–50.
- Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, et al. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys 1992;298:431–7.
- Calabrese V, Butterfield DA, Scapagnini G, Stella AM, Maines MD. Redox regulation of heat shock protein expression by signaling involving nitric oxide and carbon monoxide: relevance to brain aging, neurodegenerative disorders, and longevity. Antioxid Redox Signal 2006;8:444–77.
- Herken H, Uz E, Ozyurt H, Sogut S, Virit O, Akyol O. Evidence that the activities of erythrocyte free radical scavenging enzymes and the products of lipid peroxidation are increased in different forms of schizophrenia. Mol Psychiatry 2001;6:66–73.
- Sarsilmaz M, Songur A, Ozyurt H, Kuş I, Ozen OA, Ozyurt B, et al. Potential role of dietary omega-3 essential fatty acids on some oxidant/antioxidant parameters in rats' corpus striatum. Prostaglandins Leukot Essent Fatty Acids 2003;69:253–9.
- Chen Q, Espey MG, Sun AY, Pooput C, Kirk KL, Krishna MC, et al. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci USA 2008;105:11105–9.
- Hara S, Mizukami H, Kuriiwa F, Endo T. Hydroxyl radical generation dependent on extracellular ascorbate in rat striatum, as determined by microdialysis. Toxicology 2009;258:10–6.
- Mannaioni PF, Vannacci A, Masini E. Carbon monoxide: the bad and the good side of the coin, from neuronal death to anti-inflammatory activity. Inflamm Res 2006;55:261–73.
- Seidman MD, Shivapuja BG, Quirk WS. The protective effects of allopurinol and superoxide dismutase on noise-induced cochlear damage. Otolaryngol Head Neck Surg 1993;109:1052–6.
- Kekec Z, Seydaoglul G, Sever H, Ozturk F. The effect of antioxidants (N-acetylcysteine and melatonin) on hypoxia due to carbonmonoxide poisoning. Bratisl Lek Listy 2010;111:189–93.
- Uzun G, Eroglu M, Mutluoglu M, Senol MG. Comments on ‘oxidative stress increase in carbon monoxide poisoning’. Hum Exp Toxicol 2013;32:444–5.
- Sun Q, Cai J, Zhou J, Tao H, Zhang JH, Zhang W, et al. Hydrogen-rich saline reduces delayed neurologic sequelae in experimental carbon monoxide toxicity. Crit Care Med 2011;39:765–9.
- Knauert M, Vangala S, Haslip M, Lee PJ. Therapeutic applications of carbon monoxide. Oxid Med Cell Longev 2013;2013:360815.
- Weaver LK, Hopkins RO, Chan KJ, Churchill S, Elliott CG, Clemmer TP, et al. Hyperbaric oxygen for acute carbon monoxide poisoning. N Engl J Med 2002;347:1057–67.
- Gilmer B, Kilkenny J, Tomaszewski C, Watts JA. Hyperbaric oxygen does not prevent neurologic sequelae after carbon monoxide poisoning. Acad Emerg Med 2002;9:1–8.