Abstract
Background
Because of the insufficient number of cord blood hematopoietic stem cells (CB-HSC), expansion of these cells seems to be important for clinical application in adults. Cell cycle inhibitors are important regulators in normal hematopoietic regeneration. In this study, mRNA expression and promoter methylation status of p15INK4b were evaluated during CB-HSC ex vivo expansion using cytokines and in co-culture system with a mesenchymal stem cells (MSCs) feeder layer.
Methods
Ex vivo cultures of CB-HSCs were performed in three culture conditions for 14 days: cytokines with an MSCs feeder layer, cytokines without a MSCs feeder layer, and co-culture with MSCs without cytokines. After expansion, measuring the total number of cells, CD34+ cells, and CFU assay was performed. Methylation status of the p15INK4b gene promoter was analyzed using methylation-specific polymerase chain reaction and p15 mRNA expression was evaluated by real-time reverse transcriptase polymerase chain reaction.
Results
Maximum CB-HSC expansion was observed on day 10 of expansion. The data showed that after 10 days, p15 mRNA expression in the expanded cells in all the three culture conditions was higher than in CD34+ fresh cells (P < 0.01). p15 gene promoter of expanded CD34+ cells remained in an unmethylated form just like fresh CD34+ cells in all the three culture conditions at days 5, 10, and 14 of culture.
Conclusions
Expression of p15INK4b in HSCs was not decreased during ex vivo expansion. Also, no methylation of p15 promoter was observed, otherwise it would be capable of initiating some leukemic cell progression or disruption in hematopoietic regeneration.
Introduction
Umbilical cord blood (UCB) has been used as a source of hematopoietic stem cells (HSCs) for complete or partial human leukocyte antigen-matched allogenic transplantation in the treatment of hematologic diseases. Cryopreserved UCB has been used to reduce the risk of graft versus host diseases (GVHD) and treatment-related mortality and to improve survival.Citation1,Citation2 However, there are approximately 107 CD34+ cells in one unit of cord blood (CB) that is insufficient for clinical trials using CB. For a successful transplantation, 2.5 × 105/kg CD34+ cells are required; so one unit of CB can only be used for recipients <45 kg.3 Ex vivo expansion of UCB-HSCs is the best way to increase the number of CD34+ cells and improve the kinetics of UCB-HSCs engraftment.Citation1,Citation2 HSCs differentiation to precursor cells during ex vivo expansion leads to decreased long-term engraftment rate. So, HSCs proliferation and self-renewal capacity must be higher than the differentiation capacity, and some of the primitive stem cells should remain in the quiescence phase of cell cycle during expansion to be capable of long-term hematopoietic reconstitution.Citation4,Citation5 Cell cycle regulators including p21, p27, p15, p16, p18 as well as the D-type cyclins play critical roles in HSCs proliferation.Citation6 On the other hand, self-renewal capacity and multilineage differentiation of HSCs during a long-term survival period can be directly or indirectly involved in the formation of the ‘tumor stem cells’.Citation7 Disruption of the cell cycle inhibitors in normal stem cells may also contribute to abnormality in differentiation and apoptosis processes that may induce leukemic cells. When the self-renewal capacity of primitive stem cells coincides with a decrease in apoptosis and abnormality in cell differentiation, leukemic cells may be created.Citation8 In mammalian cells, cell cycle progression is regulated by alternative activation and inactivation of cyclin-dependent kinases (CDKs ) in G1 phase.Citation9,Citation10 INK4 constitutes a family of CDK inhibitors including p16INK4A, p15INK4B, p18INK4C, and p19INK4D, all of which inhibit the CDK4 and CDK6 in G1/S transient stage and unregulated proliferation.Citation8 Inactivation of these tumor suppressor genes via deletion, mutation and transcription suppression by promoter methylation induces tumor cells.Citation11 p16INK4a and p15INK4b gene deletion is seen in 25–60% of acute leukemias and 20–50% of lymphomas, but mRNA expression silencing by hypermethylation or mutation is more common.Citation12,Citation13
Several types of genetic changes including balanced translocations and alterations of proto-oncogenes and tumor suppressor genes may contribute to the leukemic phenotype. Transcriptional suppression of tumor suppressor genes by hypermethylation of promoter CpG islands has been identified as a significant event in acute myloid leukemia.Citation14
Because of the important role of p15INK4b tumor suppressor gene in normal hematopoiesis, in this study we analyzed the expression and methylation status of p15INK4b during ex vivo CB-HSC expansion using cytokines (FLT3L, SF, and TPO). On the other hand, mesenchymal stem cells (MSCs) that form a major part of the bone marrow microenvironment playing an important role in hematopoiesis regulation, homing, and HSCs maintenance in quiescence phaseCitation15 were used as the feeder layer. So, our results from expanded stem cells in a co-culture system were compared with those from stromal free cytokine cultures.
Materials and methods
Isolation of HSCs
HSCs were collected from fresh human UCB after written informed consent from normal full-term pregnant women according to guidelines approved by the Ethics Committee at the Iranian Blood Transfusion Organization. Mononuclear cells were isolated by density gradient centrifugation on Ficoll-Hypaque (Biosciences, Uppsala, Sweden). The mono nuclear cells (MNC) were incubated with antihuman CD34 conjugated with MicroBeads (Miltenyi Biotec, Auburn, CA, USA) for 30 minutes at 4°C. CD34+ cells were isolated using an affinity column.
Feeder layer cultivation
Mesenchymal stromal cells were isolated from fresh bone marrow after informed consent was received from healthy donors (at Taleghani hospital, Tehran, Iran). Briefly, MNCs were collected from a 10-ml bone marrow aspirate by density-gradient centrifugation on Ficoll-Hypaque (Biosciences), and were washed once in MSCs culture medium (low-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with penicillin and streptomycin). The MNCs were cultured in a 75-cm2 culture flask at 37°C. After 2–3 days, non-adherent cells were removed and adherent cells were left in MSCs culture medium until the cells achieved 80% confluence. MSCs were characterized by flow cytometric analysis using monoclonal antibodies against CD105, CD44, CD166, CD90, CD34, and CD45 (Dako, Copenhagen, Denmark). Differentiation potential of MSCs was investigated by an osteogenic differentiation kit (Chemicon, Billerica, MA, USA). To be used as a cell feeder layer, MSCs were harvested with 0 · 25% trypsin–EDTA solution (Stem Cell Technology, Copenhagen, Denmark), and 1 × 104 cells were plated in six-well plates. When the cells reached more than 90% confluence in DMEM, they were washed once with phosphate-buffered saline (PBS) and were replaced by serum-free medium (Stem Span, Copenhagen, Denmark) to co-culture with HSCs.
MSCs osteogenic differentiation assay
Osteogenic differentiation was performed on bonemarrow-derived MSCs. For this purpose, 5 × 104 cells were seeded in collagen-/Vitronectin-coated 24-well plates. After 100% confluence, DMEM enriched with FBS were replaced with differentiating medium containing 100 µm l-ascorbic acid-2-phosphate and 1 m β-glycerophosphate with 1 mm dexamethasone (Chemicon). On day 14, cells were fixed in 70% ethanol for 1 hour at 4°C and stained for 15 minutes with alizarin red-S (Chemicon, Billerica, MA, USA) at room temperature. Alkaline phosphatase staining was performed with an alkaline phosphatase kit (Sigma-Aldrich). Differentiated cells were fixed with acetone and Fast Blue RR salt in a naphthol AS-MX alkaline solution was used for staining. Mayer's hematoxylin was used as the counterstain.
Ex vivo expansion of CD34+-enriched cells
CD34+-enriched cells (1 × 105) were cultured in six-well plates (Nunc, Denmark) for 14 days in the serum-free medium (Stem Cell Technology, Copenhagen, Denmark) at 37°C under 5% CO2 humidified air in the three culture conditions: cytokines culture supplemented with SCF (100 ng/ml), TPO (100 ng/ml) and FLT3L (50 ng/ml) (Stem Cell Technology), co-culture with an MSCs feeder layer and above-mentioned cytokines and co-culture with MSCs without any cytokine.
Colony-forming cell assays
Fresh CD34+ cells and expanded cells at day 10 of culture (1–2 × 103) were seeded in semisolid culture (MethoCult GF H4434, StemCell Technologies, Copenhagen, Denmark) following the manufacturer's instructions. Methylcellulose-based media were aliquoted in 35-mm2 Petri dishes and incubated at 37°C in a 5% CO2 humidified air incubator. After 14 days of culture, the number of colonies was counted under an inverted microscope.
Proliferative and phenotypic analysis
Cell viability was determined at days 0, 5, 10, and 14 of culture by counting HSCs in each well using trypan blue stain (StemCell Technologies, USA), and analyzed for stem cell and lineage markers by flow cytometry (Partec, Görlitz, Germany) using monoclonal antibodies against CD2, CD19 (Dako) to evaluate lymphoid lineage differentiation; CD14, CD15, and CD13 to evaluate myeloid differentiation; Glycophorin A to evaluate erythroid differentiation; and CD34 and CD38 to assess the percentage of stem cells. Flow cytometric analysis was performed by incubating harvested cells with different fluorescent conjugated monoclonal antibodies at 4°C for 30 minutes. Then, the cells were washed in PBS and fixed with 2% paraformaldehyde (Sigma). Isotype controls were used in every experiment.
Samples and extraction of total RNA and DNA
DNA was extracted from fresh CB CD34+ cells and expanded cells in all the three culture conditions at days 5, 10, and 14 of culture, while total RNA was just extracted at days 0 and 10 of culture. They were immediately frozen and kept at −70°C until use. DNA was isolated by a QIAamp DNA Mini-kit (Qiagen, Valencia, CA, USA), and RNA was extracted using Trizol reagent (Roche, Mannheim, Germany).
Bisulfite modification and methylation-specific polymerase chain reaction
Genome DNA was subjected to bisulfite modification using an EpiTect Bisulfite kit (Qiagen). Modified DNA was purified, resuspended in distilled water, and stored at −20°C until use. Human genomic DNA was used to determine the bisulfate conversion efficiency.
Methylation-specific polymerase chain reaction (MSP): Modified DNA template was used to amplify the p15INK4b promoters with methylated and unmethylated-specific primer pairs, of which the sequences have been chosen for promoter regions containing frequent cytosines, and CpG pairs near the 3′ end of the primers to provide discrimination in the polymerase chain reaction (PCR) between methylated and unmethylated DNA.Citation16,Citation17 Primer sequences for the p15 gene, unmethylated reaction were as follows: 5′-TGTGATGTGTTTGTATTTTGTGGTT-3′ (sense) and 5′-CCATACAATAACCAAACAACCAA-3′ (antisense); and for methylated reaction as follows: 5′-GCGTTCGTATTTTGCGGTT-3′ (sense) and 5′-CGTACAATAACCGAACGACCGA-3′ (antisense).
The PCR mixture contained EpiTect Master Mix, 2× (Qiagen), 10 pmol of each primer and bisulfite-treated DNA (approximately 50 ng) in a final volume of 25 µl. PCR amplification of the modified DNA samples consisted of 1 cycle of 95°C for 10 minutes; 40 cycles of 95°C for 45 seconds, 56°C for 45 seconds, and 72°C for 30 seconds; and 1 cycle of 72°C for 10 minutes. Each modified DNA sample was amplified by unmethylated and methylated primers with 150-bp PCR product length band. Bisulfite-converted methylated and unmethylated EpiTect PCR DNA (Qiagen) were used as controls. Peripheral lymphocyte DNA from healthy donors was also used as the unmethylated control. The products were separated by electrophoresis on 2% agarose gel. Results were always confirmed by repeated MSP analyses.
Expression analysis and quantitative real-time PCR
Reverse transcription was performed on 1 µg total RNA, with an AccuPower CycleScript RT PreMix reverse transcription kit (Bioneer, Korea, Daejeon). The reaction was carried out in 12 cycles of 22°C for 30 seconds; 45°C for 4 minutes; 55°C for 30 seconds; and 1 cycle of 95°C for 5 minutes. Products were analyzed in 1.5% agarose gel under ultraviolet light. Quantitative reverse transcriptase (RT)-PCR was performed with the LightCycler technology, using 3 µl cDNA in 25 µl reaction volume with 0.4 µM of each primer and 12.5 µl of 2× Fast Start DNA Master SYBR Green I (Roche Molecular Biochemicals, Mannheim, Germany). The forward primer for p15INK4b was 5′-GATGTGCAAGCGACGACAGA-3′ and the reverse primer was 5′-GAGCAAAGGCCAGCATCCT-3′, and PCR product was 80 bp.Citation18 Thermal cycling was initiated at 95°C for 5 minutes followed by 40 cycles of PCR (95°C 30 seconds; 56°C, 30 seconds; and 72°C,20 seconds). Β2-microglobin was used as an endogenous control.
Statistical analysis
Results obtained from multiple experiments are expressed as the mean ± standard deviation (SD). The data were analyzed using the Student's t-test. Probability values <0.05 were defined as significant differences between test points.
Results
Mesenchymal stromal cells characterization
MSCs immunophenotyping
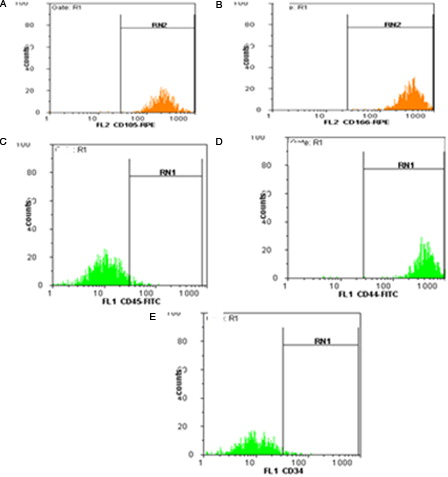
MSCs isolated from bone marrow were characterized by flow cytometric analysis of specific surface antigens. MSCs were found to be positive for the following adhesion molecules: CD44, CD166 and CD105, CD90 which together were considered as markers for MSCs. As shown in , the MSCs were negative for haematopoietic lineage markers, namely CD34 and CD45 ().
Figure 1. Flow cytometric analysis of mesenchymal stromal cells. (A) CD 105, 93%, (B) CD 166, 98.8%, (C) CD 45, 3.8%, (D) CD44, 99.5%, (E) CD 34, 2.8%.
Osteogenic differentiation assay
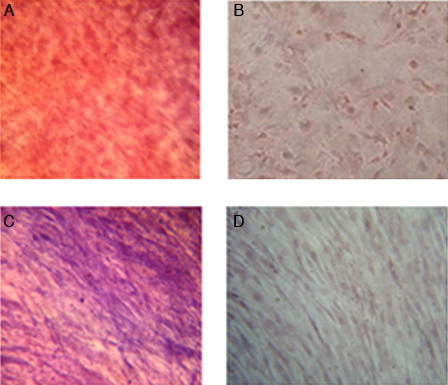
Osteogenic differentiation potential of bone marrow-derived MSCs was assayed by Alizarin red staining and evaluation of alkaline phosphatase activity. Both Alizarin red staining and alkaline phosphatase activity showed a positive reaction ().
Figure 2. Osteogenic differentiation of MSCs (×40). (A) Positive reaction in osteoblastic differentiated cells with Alizarin red staining. (B) Undifferentiated cells (negative control). (C) Increased alkaline phosphatase activity in osteoblastic differentiated cells. (D) Undifferentiated cells (negative control).
Ex vivo expansion of CD34+-enriched cells with selected cytokines, in the presence and absence of human mesenchymal stromal cells
Purity of separated CD34+ cells, determined by flow cytometric analysis, were 84 ± 14%. Moreover, 21.45 ± 7% of them were positive for the CD38 marker, and lineage-specific CD markers expression was low (n = 3) (A).
Figure 3. Flow cytometric analysis of fresh CD34+-enriched cells (A) and expanded cells in the co-culture system without cytokine (B) and cytokine culture without MSCs (C) and with MSCs (D). Specific staining was performed with anti-CD34-FITC and anti-CD38-PE antibodies. FL1: CD34 and FL2: CD38. (A) CD34: 70 % and CD38: 14.31%, (B) CD34: 70.90% and CD38: 58.31%, (C) CD34: 60.40% and CD38: 54.21%, (D) CD34: 79.54% and CD38: 66.68%.
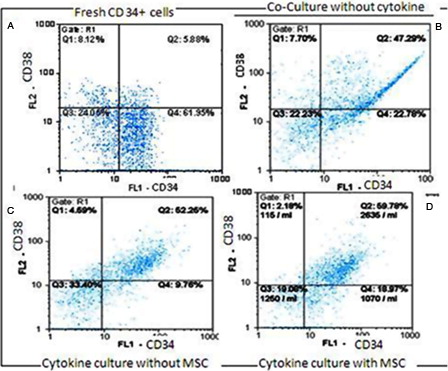
Ex vivo expansion of human CB CD34+-enriched cells in serum-free medium supplemented with SCF, TPO and FLT3L was evaluated either with or without feeder layer using flow cytometric analysis. B–D show the percentage of CD34+ and CD38+ cells of expanded cells in the co-culture system without cytokine, cytokine cultures without MSCs and with MSCs after 10 days, respectively.
A and B show the fold increase in the total number of cells (TNC) and the number of CD34+ cells during 14-day liquid cytokine culture in the presence and absence of human mesenchymal stromal cells and in the co-culture system without cytokine. Maximum expansion was observed at day 10 of culture in all the three culture conditions. The mean fold change of TNC was 12.66 ± 5.68 in cytokine culture without MSCs feeder layer and 69.56 ± 27.96 in the co-culture system with cytokines at day 10 of culture. The mean fold change of CD34+cells was 21 ± 2.3 in cytokine-supplemented culture and 109.56 ± 24.83 in the co-culture system with cytokines (n = 6). ()
Figure 4. (A) Mean fold change of TNC in cytokine-supplemented culture with and without MSCs feeder layer and co-culture with MSCs without cytokine. (B) Mean fold change of CD34 cells in cytokine supplemented culture with and without MSCs feeder layer and co-culture with MSCs without cytokine.
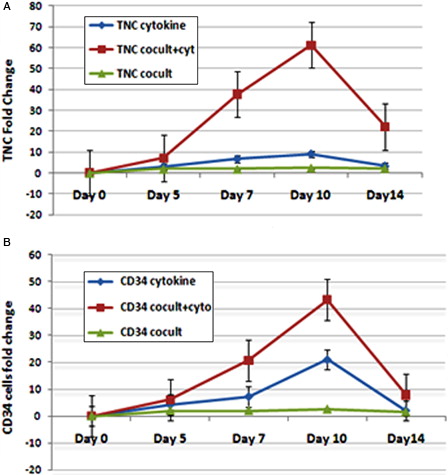
Expansion of CD34+-selected CB cells at day 14 of both cytokine culture and co-culture with cytokines was decreased, and a relative differentiation was also observed.
So the mean fold change of TNC and CD34+ cells at day 10 in the co-culture system with cytokines was higher than in the cytokine culture without MSCs feeder layer and the co-culture system without cytokines significantly (n = 6, P < 0.05). However, in the co-culture system without cytokines, TNC and CD34+ cell numbers were increased up to two folds, and the cell viability remained 80% after 14 days.
Colony-forming cell assays
The highest colony-forming unit (CFU) fold change was also observed in cytokine culture with MSCs at day 10 (90 ± 24) (). The CFU increase in cytokine culture without MSCs at day 10 was 87 ± 13, and in co-culture with MSC was 52 ± 9. So the mean fold change of CFU at day 10 in both cytokine cultures with and without MSCs feeder layer was higher than in the co-culture system without cytokines significantly (n = 3, P < 0.05), but there was no significant differences between two cytokine cultures with and without MSCs feeder layer ().
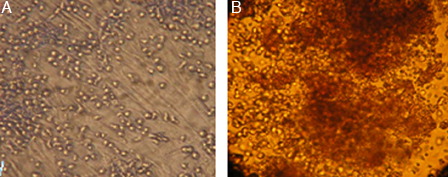
Figure 5. (A) Co-culture of HSCs with MSCs after 10 days expansion in serum-free media supplemented with cytokine cocktail. (B) CFU assay of expanded HSCs at day 10 of culture (×10).
mRNA expression of p15INK4b after 10 days ex vivo expansion of CB-HSCs
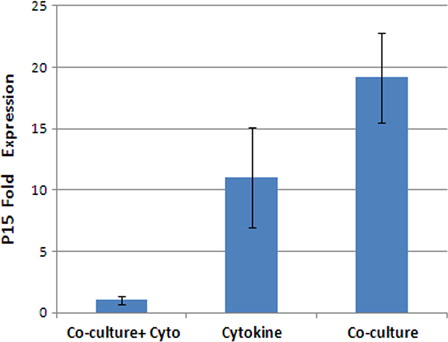
Expression of p15INK4b in fresh CD34+ cells and expanded cells after 10 days in all the three culture conditions was evaluated by real-time RT-PCR. The results showed that the fold change ratio of p15 mRNA expression in the expanded cells to the fresh CD34+cells was 1.05 ± 4.06 at the cytokine supplemented culture, 1.04 ± 0.34 in the co-culture system with cytokines and 19.16 ± 3.67 (n = 3) in the co-culture system without cytokines (). So, ex vivo expansion of CB-HSCs in the co-culture with MSCs feeder layer without cytokines compared to two other cytokines cultures resulted in significantly increased expression of p15INK4b (P < 0.001), and there was no significant difference between these two systems.
Figure 6. p15 mRNA expression fold change ratio in the expanded cells to the fresh CD34+ cells at the cytokine culture with and without MSCs and co-culture system without cytokine (P < 0.01).
Methylation status of the P15INK4b during ex vivo CB-HSCs expansion
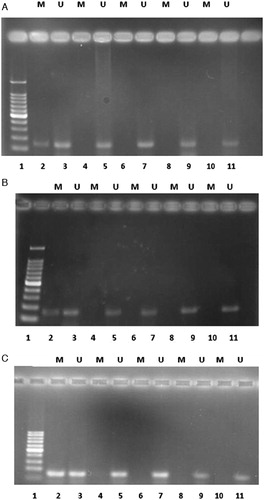
We examined the methylation status of p15INK4b gene promoter in fresh CD34+ cells and expanded cells in all the three culture conditions at days 5, 10, and 14 of culture with MSP analysis. Fresh CD34+ cell samples only showed an unmethylated band. Methylation of this gene did not occur in any of the eight samples taken from the cytokine-supplemented culture with and without MSCs feeder layer and co-culture with MSCs without cytokines at different days of expansion. In samples with methylated primer, no PCR products were detected. However, in samples with unmethylated primer, 150 bp bands were detected (). So, the methylation status of p15INK4b gene promoter during ex vivo expansion of CB-HSCs in all the three culture conditions was not changed.
Figure 7. Methylation-specific PCR of bisulfite-modified DNA using primer pairs specific for methylated and unmethylated p15INK4b promoter sequences in each case. (A) Expanded cells in the cytokines without MSCs feeder layer. (B) Expanded cells in the cytokine culture with MSCs feeder layer. (C) Expanded cells in the co-culture system without cytokine. M, primers specific for methylated DNA; U, primers specific for unmethylated DNA. Lane 1 is DNA ladder; 2 is methylated DNA control with the methylated primer pairs; 3 is unmethylated DNA control with the unmethylated primer pairs; 5, 7, 9, and 11 illustrate DNA amplification at days 0, 5, 10, and 14 of culture, respectively, with unmethylated p15 primers specific for unmethylated DNA; 4, 6, 8, and 10 have no band that illustrates lack of DNA amplification for expanded cells at days 0, 5, 10, and 14 of culture, respectively, with methylated p15 primers specific for methylated DNA.
Discussion
Ex vivo expansion of UCB-HSCs at different combinations of recombinant stimulatory cytokines is one way of increasing the number of CD34+ cells and the engraftment of UCB-HSCs.Citation19 Another way is by expansion of HSC in co-culture with MSCs that produce a more ‘natural’ haematopoietic microenvironment for proliferation, self-renewal and differentiation of HSCs.Citation20 However, it has been clearly shown that cytokines have a direct effect on cell cycle by affecting the expression levels of cell cycle regulators.Citation21 In this study, CB-CD34+ cells expansion with hematopoietic cytokines in the absence or presence of human bone marrow MSCs feeder layer and in co-culture system without cytokine was performed. mRNA expression and promoter methylation status of P15 gene were evaluated during expansion.
Maximum expansion was observed at day 10 of culture in the two cytokine-supplemented cultures.
Alvarado-Moreno et al.Citation21 also reported that UCB-derived CD34+ cells showed significant proliferation after 14 days in the cytokine culture and maximum expansion was observed at day 7 of culture.
The mean fold changes of TNC and CD34+ cells in the cytokine culture with MSCs were higher than in the cytokine culture without MSCs. However, in the co-culture system without cytokine TNC and CD34+ cell numbers were increased up to two folds, but cell viability was more than 80% and differentiation rate was low (data not shown).
In keeping with our results, Jang et al.Citation22 also showed that proliferation capacity of HSCs in the recombinant cytokine culture with UCB-MSCs feeder layer was higher than in the cytokine culture without MSCs, and in the co-culture system without cytokine, CD34+ cell numbers were increased up to two folds.
Da Silva et al.Citation23 cultured CB CD34+-enriched cells in serum-free medium supplemented with SCF, bFGF, LIF, and Flt-3, in the presence or absence of stroma, and observed that the stromal layers would support the process of expansion without exhaustion of the more primitive stem cells and differentiation induction.
Lewis et al.Citation24 suggested that INK4 proteins are growth suppressors in normal hematopoiesis, since INK4 deletion increases the differentiation rate of HSCs. In our study, p15INK4b mRNA expression after ex vivo expansion of CB CD34+cell with cytokines (FLt3L, SF, and TPO) in the absence or presence of mesenchymal stromal layer after 10 days has not decreased, and this constant expression of p15 might have supported the expansion of CD34+ cells and maintaining their clonogenic potential.
Lewis et al.Citation24 also indicated that the p16INK4a could modulate clonal expansion, as loss of p16INK4a increased progenitor cell expansion in vitro.
Alvarado-Moreno et al.Citation21 reported that the p16 and p21 expression level at CD34+-expanded cells in the co-culture without cytokine was increased, so in the co-culture system cell cycle regulators can directly control hematopoietic progenitor cells proliferation.
Furukawa et al.Citation25 reported that P16 Cdk inhibitor was highly expressed in fresh CD34+ progenitor cells and was down-regulated during expansion with different combinations of hematopoietic growth factors. This is consistent with our observations that expanded HSCs in the co-culture system without cytokine have the highest p15 mRNA expression, and their proliferation capacity is low. On the other hand, in the co-culture system with cytokines, expanded HSCs had less p15 mRNA expression with the highest proliferation capacity.
DNA methylation, an important mechanism for gene suppression, is one of the earliest processes in tumor cells initiation.Citation26 Belinsky et al.Citation27, McDermott et al.Citation28, and Reynolds et al.Citation29 reported that the tumor suppressor gene, p15INK4b, is one of the most common and earliest epigenetically mediated losses of tumor suppressor function events in human cancers.
Jones and BaylinCitation30 reported that epigenetic silencing of p15 gene in stem/precursor cells may serve to induce abnormal clonal expansion. This gene, like other similar genes, is termed ‘epigenetic gatekeeper’ because its normal epigenetic pattern of expression should allow it to be activated during stem/precursor cell differentiation.
After screening for methylation changes in expanded CB-HSCs at several time points, we have demonstrated that methylation of p15INK4b promoter in these cells during ex vivo expansion in all the three culture conditions has not occurred.
Sakashita et al.Citation31 showed that the percentage of the methylation of p15 CpG islands in HCB CD34+ cells treated with either granulocyte/macrophage colony-stimulating factor (GM-CSF) alone or in combination with stem cell factor increased to approximately 50–60% until 7 days after cytokine stimulation.
Zhao et al.Citation32 reported that GM-CSF may be able to induce de novo methylation of the p15 gene in hematopoetic cell line after a 96-hour treatment, and the cells exhibited p15 transcriptional silencing via the methylation. But in our study, FLT3L, SCF, and TPO showed no effects on methylation status of p15 gene in expanded CB-HSCs after 14 days of treatment. This is consistent with our real-time PCR results that showed no decrease in p15INK4b mRNA expression during 10 days of expansion.
Conclusion
Expression of p15INK4b in HSCs was not decreased during ex vivo expansion. Also, no methylation of p15INK4b promoter that may initiate some leukemic cell progression or disruption in hematopoietic regeneration was observed.
References
- Delaney C, Ratajczak MZ, Laughlin MJ. Strategies to enhance umbilical cord blood stem cell engraftment in adult patients. Expert Rev Hematol. 2010;3:273–83.
- Broxmeyer HE. Umbilical cord transplantation: epilogue. Semin Hematol. 2010;47:97–103.
- Wagner JE, Barker JN, DeFor TE, Baker KS, Blazar BR, Eide C, et al. Transplantation of unrelated donor umbilical cord blood in 102 patients with malignant and nonmalignant diseases: influence of CD34 cell dose and HLA disparity on treatment-related mortality and survival. Blood 2002;100:1611–8.
- Dahlberg A, Delaney C, Bernstein ID. Ex vivo expansion of human hematopoietic stem and progenitor cells. Blood 2011;23:234–45.
- Patricia D, Conrad SG. Ex vivo expansion of hematopoietic cells from umbilical cord blood for clinical transplantation. J Leukoc Biol. 1998;64:147–55
- Miyata Y, Liu Y, Jankovic V, Sashida G, Lee JM, Shieh JH, et al. Cyclin C regulates human hematopoietic stem/progenitor cell quiescence. Stem Cells. 2010;28(2):308–17.
- Tannishtha R, Sean J. Morrison. review article Stem cells, cancer and cancer stem cells. Nature 2001;414:105–11.
- Cheng T. Cell cycle inhibitors in normal and tumor stem cells. Oncogene 2004;23:7256–66.
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149–63.
- Quesnel B, Preudhomme C, Lepelley P, Hetuin D, Vanrumbeke M, Bauters F, et al. Transfer of p16inka/CDKN2 gene in leukaemic cell lines inhibits cell proliferation. Br J Haematol. 1996;95(2):291–8.
- Youichirou M, Toshiyuki S. INK4 Family – a promising target for ‘gene-regulating chemoprevention’ and ‘molecular-targeting prevention’ of cancer. Environ Health Prev Med. 2005;1:72–7.
- Toshiyasu H, Phillip K. Role of the cyclin-dependent kinase inhibitors in the development of cancer. Blood 1995;86:841–54.
- Nakamaki T, Kawamata N, Schwaller J, Tobler A, Fey M, Pakkala S, et al. Structural integrity of the cyclin-dependent kinase inhibitor genes, p15, p16 and p18 in myeloid leukaemias. Br J Haematol. 1995;91:139–49.
- Ogawa S, Hangaishi A, Miyawaki S, Hirosawa S, Miura Y, Takeyama K, et al. Loss of the cyclin-dependent kinase 4-inhibitor (p16; MTS1) gene is frequent in and highly specific to lymphoid tumors in primary human hematopoietic malignancies. Blood 1995;86:1548–56.
- Majumdar MK, Thiede MA, Haynesworth SE, Bruder SP, Gerson SL. Human marrow-derived mesenchymal stem cells (MSCs) express hematopoietic cytokines and support long-term hematopoiesis when differentiated toward stromal and osteogenic lineages. J Hematother Stem Cell Res. 2000;9:841–8.
- Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Med. Sciences 1996;93(18):9821–6.
- Xu XL, Yu J, Zhang HY, Sun MH, Gu J, Du X, et al. Methylation profile of the promoter CpG islands of 31 genes that may contribute to colorectal carcinogenesis. World J Gastroenterol. 2004;10:3441–54.
- Pattyn F, Speleman F, De Paepe A, Vandesompele J. The real-time PCR primer and probe database. Nucleic Acids Res. 2003;31:122–3.
- Tung SS, Parmar S, Robinson SN, De Lima M, Shpall EJ. Ex vivo expansion of umbilical cord blood for transplantation. Best Pract Res Clin Haematol. 2010;23:245–57.
- Yildirim S, Boehmler Am, Kanz L, Mohle R. Expansion of cord blood CD34+ hematopoietic progenitor cells in co-culture with autologous umbilical vein endothelial cells (HUVEC) is superior to cytokine-supplemented liquid culture. Bone Marrow Transplant. 2005;36:71–9.
- Alvarado-Moreno A, Chávez-González A, Cérbulo A, Arriaga L, Mayani H. In vitro cell cycle dynamics of primitive hematopoietic cells from human umbilical cord blood. Hematology 2010;15:11–20.
- Jang YK, Jung DH, Jung MH, Kim DH, Yoo KH, Sung KW, et al. Mesenchymal stem cells feeder layer from human umbilical cord blood for ex vivo expanded growth and proliferation of hematopoietic progenitor cells. Ann Hematol. 2006;85:212–25.
- Da Silva CL, Gonçalves R, Crapnell KB, Cabral JM, Zanjani ED, Almeida-Porada G. A human stromal-based serum-free culture system supports the ex vivo expansion/maintenance of bone marrow and cord blood hematopoietic stem/progenitor cells. Exp Hematol. 2005;33:828–35.
- Lewis JL, Chinswangwatanakul W, Zheng B, Marley SB, Nguyen DX, Cross NC, et al. The influence of INK4 proteins on growth and self-renewal kinetics of hematopoietic progenitor cells. Blood 2001;97:2604–10.
- Furukawa Y, Kikuchi J, Nakamura M, Iwase S, Yamada H, Matsuda M. Lineage-specific regulation of cell cycle control gene expression during haematopoietic cell differentiation. Br J Haematol. 2000;110:663–73.
- Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7:21–33.
- Belinsky SA, Nikula KJ, Palmisano WA, Michels R, Saccomanno G, Gabrielson E, et al. Aberrant methylation of p16 (INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc Natl Acad Sci USA 1998;95:11891–6.
- McDermott KM, Zhang J, Holst CR, Kozakiewicz BK, Singla V, Tlsty TD. p16 (INK4a) prevents centrosome dysfunction and genomic instability in primary cells. PLoS Biol. 2006;4:e51.
- Reynolds PA, Sigaroudinia M, Zardo G, Wilson MB, Benton GM, Miller CJ, et al. Tumor suppressor p16INK4A regulates polycomb-mediated DNA hypermethylation in human mammary epithelial cells. J Biol Chem. 2006;281:24790–802.
- Jones A, Baylin B. The epigenomics of cancer. Cell 2007;128:683–92.
- Sakashita K, Koike K, Kinoshita T, Shiohara M, Kamijo T, Taniguchi S, et al. Dynamic DNA methylation change in the CpG island region of p15 during human myeloid development. J Clin Invest. 2001;108:1195–204.
- Zhao XY, Sakashita K, Kamijo T, Hidaka E, Sugane K, Kubota T, et al. Granulocyte-macrophage colony-stimulating factor induces de novo methylation of the p15 CpG island in hematopoietic cells. Cytokine 2005;31(3):203–12.