
Abstract
Objective and importance
Congenital dyserythropoietic anemia (CDA) represents a genotypically and phenotypically heterogeneous group of disorders. CDA type II, the most frequent variant, was recently shown to be caused by mutations in the gene encoding the secretory COPII component SEC23B. We report two siblings hailing from Punjab in northern India with classical CDA type II where this mutation was demonstrated.
Clinical presentation
A 7-year-old girl presented with transfusion-dependent anemia, splenomegaly, and progressive growth failure since 1 year of age. Her 5-year-old brother was similarly afflicted, but there was no other family history. Extensive prior work-up for hemolytic anemia, storage and metabolic disorders, and infectious diseases was negative. Hemoglobin was 71 g/l with normal leukocyte, platelet, and corrected reticulocyte counts. Bone marrow examination revealed marked normoblastic erythroid hyperplasia with dyserythropoiesis (36%) and the presence of bi- and multinucleated erythroblasts with equal-sized nuclei. Many pseudo-Gaucher cells were also seen. Iron stores were increased although ring sideroblasts were absent. Hereditary erythrocyte multinuclearity with positive acidified serum (HEMPAS) test revealed lysis of the red cells in four out of five control sera.
Technique
Genomic DNA sequencing of the SEC23B exon 12 revealed homozygosity for c.1385 A → G; Y462C mutations in both siblings.
Conclusion
CDA has traditionally been a difficult diagnosis to establish, since it requires exclusion of other causes of dyserythropoiesis and the performance of complex tests including HEMPAS and electron microscopy for confirmation. The availability of molecular genetic testing for SEC23B promises to streamline and hasten the diagnostic process for this rare and intriguing disease.
Introduction
The congenital dyserythropoietic anemias (CDA) are a group of genotypically and phenotypically heterogeneous disorders.Citation1 CDA type II, the commonest variant worldwide, has also been frequently reported from the Indian subcontinent.Citation2 Characterized by ineffective and dysplastic erythropoiesis, iron overload, and sub-optimal reticulocyte response to anemia, this autosomal recessive disorder was recently shown to be caused by a variety of mutations in SEC23B, the gene encoding the secretory COPII component.Citation1,Citation3,Citation4
We report two siblings hailing from Punjab in northern India with classical CDA type II, in whom the specific molecular defect described in south Asian kindreds could be demonstrated. The case study is pertinent for its likely implications on our diagnostic hematopathology practice.
Case presentation
A 7-year-old girl presented with transfusion-dependent anemia, abdominal distension, and progressive growth failure since 1 year of age. Her 5-year-old brother was similarly afflicted, but there was no other family history. They were born out of a non-consanguineous marriage. On examination, both siblings had hemolytic facies and splenomegaly (5.5 and 3 cm below left costal margins in the girl and the boy, respectively).
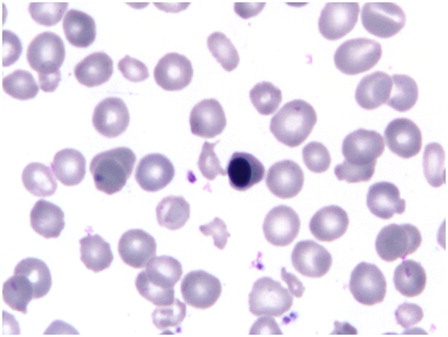
Extensive prior work-up for hemolytic anemia, storage and metabolic disorders, and infectious diseases was negative in both children. Specifically, hemoglobin high performance liquid chromatography (HPLC) (children and parents), antiglobulin tests, incubated osmotic fragility tests, glucose-6-phosphate dehydrogenase and pyruvate kinase deficiency screens, and urine for hemosiderin were normal or negative. Serum ferritin was >3000 ng/ml (>6741 pmol/l) in the girl. Her hemoglobin was 71 g/l, total leukocyte count (TLC) 8.8 × 109/l, platelet count 267 × 109/l, and corrected reticulocyte count was 2.6% with normal differential leukocyte count. Red cells showed moderate anisopoikilocytosis and were normocytic to microcytic, mildly hypochromic with irregularly shaped forms (). Three nucleated red blood cells (RBCs) were seen per 100 leukocytes.
Figure 1. Peripheral blood film shows normocytic to microcytic hypochromic red cells with circulating erythroblasts and irregularly shaped cells (May-Grunwald–Giemsa, ×1000).
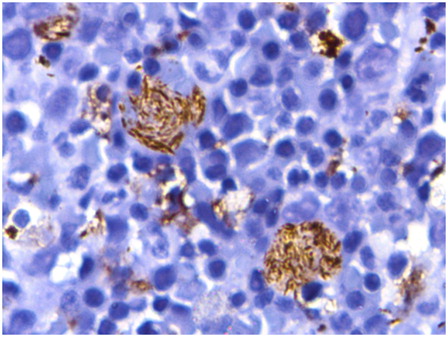
Bone marrow examination of the girl revealed marked normoblastic erythroid hyperplasia (myeloid to erythroid ratio 1:3.1) with a preponderance of intermediate and late erythroblasts. Dyserythropoiesis was seen in 36% of these cells, chiefly in the form of bi- and multinucleated erythroblasts with equal-sized nuclei (13% of all erythroid cells). Nuclear budding was also prominent (). Iron stores were increased (grade 5+) but ring sideroblasts were absent. Bone marrow biopsy was hypercellular with erythroid hyperplasia and conspicuous hemosiderin pigment deposition. Many sheets and large paratrabecular clusters of pseudo-Gaucher cells were also seen (). These cells were positive for periodic acid Schiff stain () and were positive for CD68 ().
Figure 2. Bone marrow aspirate shows binucleate erythroid precursors (May-Grunwald–Giemsa, ×1000).
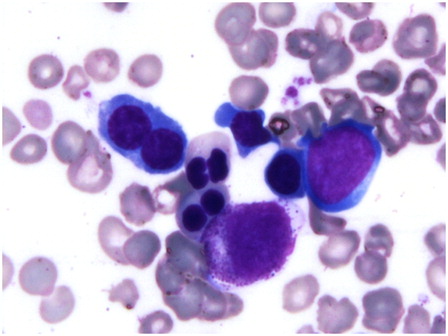
Figure 3. Hypercellular bone marrow biopsy shows erythroid hyperplasia with hemosiderin pigment. The paratrabecular area shows pale staining pseudo-Gaucher cells (hematoxylin and eosin, ×400).
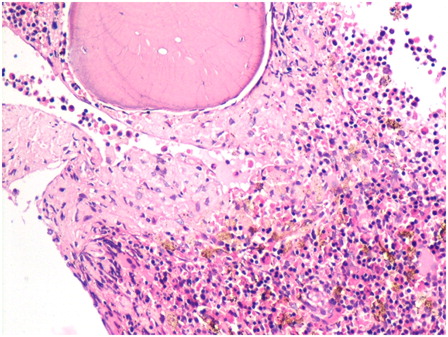
Figure 4. The pseudo-Gaucher cells are brilliantly positive with cytochemical stain for periodic acid Schiff (hematoxylin counterstain, ×400).
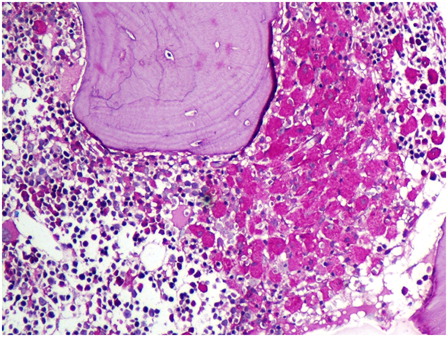
Figure 5. Immunohistochemistry for CD68 highlights the fibrillary cytoplasm of the pseudo-Gaucher cells (diaminobenzidine-based staining with hematoxylin counterstain, ×1000).
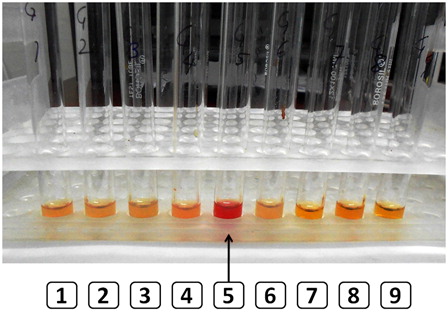
Hereditary erythrocyte multinuclearity with positive acidified serum (HEMPAS) test showed lysis of the girl's red cells in four out of five blood group-matched unrelated healthy control sera (). Flow cytometric eosin-5-maleimide (EMA) dye-binding test revealed borderline ratios of median fluorescent intensities (MFI) in both siblings. Test MFI to control MFI ratio was 0.85 and 0.89 in the two siblings. The reference cut-off for the laboratory is <0.80 for hereditary spherocytosis. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) of RBC membrane proteins was attempted for the children and the parents. Although sub-optimal, the gel suggested a contracted and slightly faster moving band 3 as compared to normal control.
Figure 6. The HEMPAS test shows lysis of the patient's red cells in four of five control sera (tube 5 shows lysis in one control, arrow) but not her own serum (tube 8).
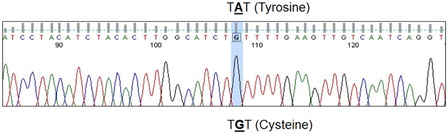
Since the clinical and laboratory findings were highly concordant with CDA type II, we resorted to genetic studies for a known Y462C point mutation in exon 12 of the SEC23B gene that was previously described in Pakistani and Indian cases.Citation3,Citation5 Genomic DNA was isolated from peripheral blood. The SEC23B exon 12 was amplified using the following primers: forward (SEC23B12F) 5′-TCATTAGCAATTAACCACAGGG-3′ (Tm 62.5°C, GC% = 40.9%); reverse (SEC23B12R) 5′-CAAAACAGGACACTAAGACATGAAG-3′ (Tm 62.7°C, GC% = 40.0%) and polymerase chain reaction (PCR) conditions described by Bianchi et al.Citation4 The PCR products were checked by agarose gel electrophoresis. Direct DNA sequencing performed on an ABI Prism 310 sequencer revealed the exon 12 c.1385 A → G; Y462C mutation in both siblings in the homozygous state ().
Figure 7. Sanger sequencing of exon 12 reveals the c.1385 A → G; Y462C mutation in homozygous state.
At 7 months follow-up, the siblings remain on folic acid, intermittent transfusion therapy, and iron chelation and are planned for splenectomy.
Discussion
CDA has traditionally been a difficult diagnosis to render for hematopathologists. Its rarity, coupled with the frequent presence of dyserythropoiesis in many hematological and non-hematological disorders make its distinctive morphology insufficient to clinch the diagnosis. Tests like the persistence of fetal i antigen are non-specific while confirmatory tests like HEMPAS, EMA binding, SDS–PAGE, and electron microscopy are more complex.Citation1
The genetics of CDA have been a focus of research in recent times. CDA type I results (in >80% cases) from mutations in the CDAN1 (codanin 1) gene on chromosome 15q15.2.Citation6 A minority of patients may harbor mutations in the C16ORF41 gene on chromosome 15q14.Citation7 CDA type III was recently attributed to a mutation in the KIF23 gene at 15q23 in a large Swedish family using haplotype analysis and targeted next-generation sequencing.Citation8
For CDA type II, the underlying defect was previously known to result in defective enzymatic glycosylation of erythrocyte membrane proteins due to incomplete processing of N-linked oligosaccharides. Bands 3 and 4.5 proteins in CDA-II red cells contain truncated short oligosaccharides instead of polylactosaminoglycans. The abnormal band 3 is particularly hydrophobic and clusters, causing cytoskeletal disruption. Unused polylactosamines (that are normally conjugated) also accumulate as glycolipids. These latter molecules may be responsible for formation of the neoantigen resulting in the positive acidified serum test.Citation1,Citation3–Citation5
Initially, abnormalities of two Golgi enzymes were suspected in the pathogenesis of CDA type II (N-acetylglucosaminyltransferase II and α-mannosidase II).Citation1,Citation5 However, further studies excluded linkage of CDA-II to these enzymes. Recently, gene expression profiling along with gene and epitope mapping of differentiating erythroid precursors led to the identification of the SEC23B gene as being responsible for the vast majority of CDA type II.Citation3,Citation4 The SEC23B gene product is a component of the coated vesicles transiting from the endoplasmic reticulum (ER) to the cis compartment of the Golgi apparatus (coat protein (COP) II complex). Frequent mutations include the R14W, E109K, R497C, and I318T substitutions, together accounting for >50% of all SEC23B gene mutations.Citation5 These molecular defects result in misglycosylation and impaired clearance of the ER cisternae past a erythroid differentiation stage. Accumulation of ER remnants at the cell periphery results. Molecular analysis also reveals a genotype–phenotype correlation: patients with a missense mutation and a nonsense mutation have a comparatively severe phenotype compared to those with two missense mutations.Citation5
Pseudo-Gaucher cells, as seen in our case, are long described in CDA, with one report finding them in 63% cases.Citation9,Citation10 Characterized by a functional rather than absolute lack of β-glucocerebrosidase enzyme, these morphological curiosities represent accelerated erythropoiesis as well as intramedullary death (ineffective erythropoiesis) in this condition. They are diagnostically unhelpful, being also seen in myriad conditions like thalassemia and hemoglobinopathies, myeloproliferative states, etc.Citation11 In fact, in a child with abdominal organomegaly and growth retardation, their presence in very large numbers may incorrectly suggest a diagnosis of storage disease. However, more often, as in our case, the secondary nature of such pseudo-storage cells is quite apparent.Citation12
Conclusion
The availability of molecular genetic testing for SEC23B promises to streamline and hasten the diagnostic process for CDA type II, a rare and intriguing disease. The molecular test result is also likely to be useful in the future for counseling the family and offering prenatal genetic diagnosis when indicated.
Disclaimer statements
Contributors RD and PS were the pathologists involved in diagnosis of the cases while DB and AT were the clinical hematologists responsible for their care. All authors contributed to writing of the report and provided critical intellectual inputs for the same.
Funding None.
Conflicts of interest None.
Ethics approval This case report does not require ethical approval by the institutional ethics committee. Consent was taken from the guardians of the children and their identity is not revealed in the report.
Prior presentation
The case was previously presented as a poster at the 19th AIPNA-ICP International CME 2014 at Histopathology Department, PGIMER, Chandigarh from 6 to 8 February 2014.
References
- Iolascon A, Heimpel H, Wahlin A, Tamary H. Congenital dyserythropoietic anemias: molecular insights and diagnostic approach. Blood 2013;122:2162–6.
- Marwaha RK, Bansal D, Trehan A, Garewal G, Marwaha N. Congenital dyserythropoietic anemia: clinical and hematological profile. Indian Pediatr. 2003;40:551–5.
- Schwarz K, Iolascon A, Verissimo F, Trede NS, Horsley W, Chen W, et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat Genet. 2009;41:936–40.
- Bianchi P, Fermo E, Vercellati C, Boschetti C, Barcellini W, Iurlo A, et al. Congenital dyserythropoietic anemia type II (CDAII) is caused by mutations in the SEC23B gene. Hum Mutat. 2009;30:1292–8.
- Iolascon A, Russo R, Esposito MR, Asci R, Piscopo C, Perrotta S, et al. Molecular analysis of 42 patients with congenital dyserythropoietic anemia type II: new mutations in the SEC23B gene and a search for a genotype-phenotype relationship. Haematologica 2010;95:708–15.
- Dgany O, Avidan N, Delaunay J, Krasnov T, Shalmon L, Shalev H, et al. Congenital dyserythropoietic anemia type I is caused by mutations in codanin-1. Am J Hum Genet. 2002;71:1467–74.
- Babbs C, Roberts NA, Sanchez-Pulido L, McGowan SJ, Ahmed MR, Brown JM, et al. Homozygous mutations in a predicted endonuclease are a novel cause of congenital dyserythropoietic anemia type I. Haematologica 2013;98:1383–7.
- Liljeholm M, Irvine AF, Vikberg AL, Norberg A, Month S, Sandström H, et al. Congenital dyserythropoietic anemia type III (CDA III) is caused by a mutation in kinesin family member, KIF23. Blood 2013;121:4791–9.
- Van Dorpe A, Broeckaert-van Orshoven A, Desmet V, Verwilghen RL. Gaucher-like cells and congenital dyserythropoietic anaemia, type II (HEMPAS). Br J Haematol. 1973;25:165–70
- Heimpel H, Kellermann K, Neuschwander N, Högel J, Schwarz K. The morphological diagnosis of congenital dyserythropoietic anemia: results of a quantitative analysis of peripheral blood and bone marrow cells. Haematologica 2010;95:1034–6.
- Sharma P, Khurana N, Singh T. Pseudo-Gaucher cells in Hb E disease and thalassemia intermedia. Hematology 2007;12:457–9.
- Sharma P, Kar R, Dutta S, Pati HP, Saxena R. Niemann-Pick disease, type B with TRAP-positive storage cells and secondary sea blue histiocytosis. Eur J Histochem. 2009;53:183–6.