Abstract
Almost half the world's population is infected by Helicobacter pylori (H. pylori). This bacterium increases the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) in human stomach, and this has been reported to impact upon gastric inflammation and carcinogenesis. However, the precise mechanism by which H. pylori induces gastric carcinogenesis is presently unclear. Although the main source of ROS/RNS production is possibly the host neutrophil, H. pylori itself produces O2•−. Furthermore, its cytotoxin induces ROS production by gastric epithelial cells, which might affect intracellular signal transduction, resulting in gastric carcinogenesis. Excessive ROS production in gastric epithelial cells can cause DNA damage and thus might be involved in gastric carcinogenesis. Understanding the molecular mechanism of H. pylori-induced carcinogenesis is important for developing new strategies against gastric cancer.
Introduction
No one had been able to imagine that a small infectious organism could live under acidic conditions in the human stomach and, still more, cause gastric cancer until 1983, when two researchers, Dr Barry J. Marshall and Dr J. Robin Warren, isolated Helicobacter pylori (H. pylori) from human gastric mucosa and unravelled the role of the pathogen in inflammation and ulceration of the stomach and duodenum.Citation1 The discovery of this small bacterium (3.5 mm × 0.5 mm) dramatically changed our perception of gastrointestinal pathogenesis. H. pylori is a Gram-negative, helical, microaerophilic bacterium that selectively colonizes the antrum of the human stomach and induces chronic gastritis with varying severity, which in around 10–15% of cases progresses to peptic ulcer and in 1–2% of cases ultimately results in mucosa associated lymphoid tissue lymphoma or gastric adenocarcinoma.Citation2–Citation4 Consequently, in 1994, H. pylori was defined as a group 1 (i.e. a definite) carcinogen by the International Agency for Research on Cancer, a part of the World Health Organization.Citation5 However, the mechanism through which H. pylori damages the gastric mucosa and leads to gastric carcinogenesis is not yet clear.
The most accepted hypothetical mechanism of H. pylori-induced gastric carcinogenesis is that of Correa et al.,Citation6 in which they proposed that gastritis could progress to gastric cancer through multiple ‘hits’, including oxidative stress and environmental toxins, that increase DNA mutation rates.
In this review, we summarize the reported mechanisms by which reactive oxygen species (ROS) and reactive nitrogen species (RNS) are produced in the H. pylori-infected stomach, and the possible mechanism by which H. pylori induces gastric carcinogenesis.
Redox biology of H. pylori
Among the factors that influence gastro-duodenal changes in H. pylori-infected patients are ROS and RNS. Excessive ROS/RNS production has been reported in H. pylori-infected human gastric mucosa,Citation7 and correlates well with histological mucosal damageCitation8 and with bacterial load.Citation9
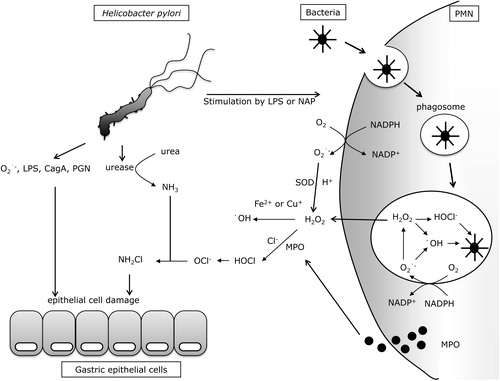
H. pylori itself has been reported to produce a large amount of superoxide anion (O2•−) in order to inhibit the bactericidal effects of nitric oxide (NO) synthesized by inflammatory cells.Citation10 On the other hand, O2•− might be passively produced by electrons leaking from the mitochondrial respiratory chain of H. pylori, since O2•− production by H. pylori can be suppressed by cyanide (CN−). The cytotoxicity of O2•− is moderate, but the cytotoxicity of hydroxyl radicals (•OH) produced via Fenton's reaction with metals and hydrogen peroxide (H2O2) is much higher, and therefore H. pylori-produced O2•− might indirectly induce gastric epithelial cell injury ().
Figure 1. H. pylori-induced oxidative stress in stomach. LPS: lipopolysaccharide; NAP: neutrophil-activating protein; PMN: polymorphonuclear neutrophil; CagA: cytotoxin-associated gene A; PGN: peptidoglycan; MPO: myeloperoxidase; H2O2: hydrogen peroxide; NH2Cl: monocloramine; OCl−: hypochlorite ion; NADP+/NADPH: nicotinamide adenine dinucleotide phosphate/reduced form of NADP+.
It is well known that H. pylori changes from its normal helical bacillary morphology to a coccoid morphology under unfavorable conditions.Citation11 Since the coccoid form cannot be cultured in vitro, it has been speculated that it is a dormant form involved in the transmission of H. pylori, and in H. pylori reinfection after antibiotic therapy.Citation12 Interestingly, the coccoid form of H. pylori produces more •OH than the helical form.Citation13 The precise role of •OH in the pathogenesis of the H. pylori-infected stomach is unknown.
Besides producing O2•− or •OH, H. pylori has various defense mechanisms against external ROS/RNS attack to protect it from elimination. Upon infection, H. pylori causes strong inflammation in the gastric mucosa. However, the human immune system cannot eliminate H. pylori, and the bacteria can survive in a neutrophil-rich environment in the stomach and induce chronic inflammation with a marked infiltration of polymorphonuclear neutrophils (PMN) that is unusual for chronic infectious diseases, although it is still unclear how these organisms evade phagocytic killing.Citation14 One of the reasons for this phenomenon is that H. pylori is rather non-invasive (although a very few H. pylori have been reported to invade the gastric mucosaCitation15), and therefore the host immune system-induced inflammatory reaction does not always recognize the bacterium. In addition, H. pylori itself causes immune tolerance by selecting H. pylori non-reactive T cells via induction of T-cell apoptosis.Citation16
Another characteristic defence strategy of H. pylori is its strong urease activity, which catalyzes the conversion of urea in the stomach to produce ammonia (NH3). By this mechanism, H. pylori neutralizes the surrounding gastric acid and protects itself from the strong acidity of the stomach. This NH3 in turn reacts with hypochlorite ions (OCl−) produced by activated neutrophils to form highly toxic monochloramine (NH2Cl) in the stomach, a hallmark of H. pylori infectionCitation17 and capable of eventually injuring gastric epithelial cells. By using urease activity, H. pylori indirectly quenches external NO attack. H. pylori induces the expression of inducible NO synthetase (iNOS) in gastric mucosal epithelial cells, vascular endothelial cells, or infiltrating inflammatory cells.Citation18 Among these cells, macrophages are the main source of NO induced by iNOS using l-arginine as a substrate.Citation19 Although NO produced by macrophages can kill H. pylori in vitro,Citation20 it cannot eliminate H. pylori in the human stomach and therefore chronic inflammation persists in the gastric mucosa. NO produced in the H. pylori-infected stomach rapidly reacts with O2•− to produce highly toxic peroxynitrite (ONOO−). Using carbon dioxide (CO2) produced by urease catalysis, H. pylori converts this ONOO− into non-toxic, short-lived ONOOCO2−. In addition, H. pylori synthesizes alkyl-hydroperoxide reductase, which can quench ONOO−.Citation21 H. pylori also suppresses NO production by inducing apoptosis of macrophages. Briefly, lipopolysaccharides (LPS) of H. pylori stimulates macrophages to produce polyamines by inducing arginase and ornithine decarboxylase, and the polyamines induce apoptosis of macrophagesCitation22 and suppress their iNOS expression.Citation23 H. pylori has other strategies for counteracting NO. Since the bacterium synthesizes arginase,Citation23,Citation24 it can produce urea using l-arginine as a substrate, and use urease to convert the urea to NH3. Since l-arginine is the substrate for both arginase and iNOS, it is possible that the production of arginase by H. pylori is a competitive strategy against NO production by host cells.
H. pylori also removes H2O2 produced by neutrophils by its catalase (KatA) and KatA-associated protein (KapA).Citation25 Some studies have reported that the amounts of catalase and superoxide dismutase (SOD) released by H. pylori are likely insufficient to clear excess extracellular oxidants,Citation26 as these enzymes primarily play a role in the elimination of ROS generated by the bacterium itself. This issue should be examined in more detail.
In summary, with various strategies, H. pylori quenches ROS/RNS that are potentially harmful to it, in order to survive the stressful conditions found in the stomach.
Redox biology of inflammatory cells
In H. pylori-infected gastric mucosa, neutrophils are believed to be the main source of ROS/RNS.Citation27 Many investigators have reported that in gastric mucosa with H. pylori infection, there is a remarkable infiltration of PMN and a significant increase in mucosal interleukin-8 (IL-8), a potent PMN chemotactic stimulant.Citation28 After successful H. pylori eradication therapy, the number of infiltrating PMN and level of mucosal IL-8 return to normal.Citation29 It is thus clear that PMN, transmigrated and localized into the gastric mucosa by IL-8 stimulation, plays an important role in the pathogenesis of H. pylori-induced gastric mucosal inflammation.Citation30
In neutrophils, nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase; Nox) on the cell membrane catalyzes ROS production.Citation31 Upon recognition of pathogenic bacteria, neutrophils immediately engulf the bacteria (phagocytosis) to form phagosomes, in which the invaginated pathogens are killed by actively produced ROS derived from Nox catalysis. During phagocytosis, gp91phox, the catalytic subunit of the phagocytic Nox, becomes activated and receives an electron from cytoplasmic NADPH. Subsequently, phagocytic Nox donates this electron to molecular oxygen in the phagosome or outside the phagocyte to produce O2•−, a precursor of microbicidal oxidants. This O2•− is converted to H2O2 by SOD catalysis, or by non-enzymatic dismutation in the phagosome. Since H2O2 can passively permeate cell membranes, it is converted to hypochlorous acid (HOCl), which is 100 times more toxic than H2O2. This conversion is mediated by myeloperoxidases released by Azure granules in phagocytes in the presence of chloride ions (Cl−). H2O2 also reacts non-enzymatically with O2•− to form •OH in the presence of ferrous (Fe2+) or cuprous (Cu+) ions. In general, the phagocytes use these highly reactive ROS (i.e. HOCl and •OH) to kill pathogenic bacteria. However, in the H. pylori-infected gastric mucosa, these ROS cannot eradicate the bacteria; therefore, the phagocytes produce more ROS. This excessive production of ROS is believed to be a major cause of gastric mucosal damage ().
Redox biology of gastric epithlial cells
Gastric epithelial cells are one source of ROS in the H. pylori-infected stomach. Upon stimulation by bacterial cytotoxic factors of H. pylori or cytokines,Citation32 gastric epithelial cells passively produce ROS as by-products of increased mitochondrial respiration rather than as active products. Recently, not only phagocytic cells but also non-phagocytic epithelial cells of the alimentary tract have been reported to express Nox.Citation33 H. pylori LPS not only activate neutrophils to produce O2•−, but also increase Nox on gastric epithetial cells and Toll-like receptor 4 expression on gastric epithelial cells leading to further production of O2•−.Citation34
Bacterial cytotoxic factors of H. pylori, such as vacuolating cytotoxin, cytotoxin associated gene product (CagA), and peptidoglycan, also stimulate gastric epithelial cells to cause oxidative stress. CagA and peptidoglycan have been reported to be injected into gastric epithelial cells via the needle-like structure of H. pylori, a type IV secretion system. By transfecting the cagA gene into gastric epithelial cells, we found that a fraction of the expressed CagA protein localizes to mitochondria and induces formation of a significant amount of ROS in the cells. In addition, increased ROS production might be involved in acceleration of the cell cycle and subsequent cell proliferation.Citation35 The mechanism by which CagA induces ROS production in gastric epithelial cells is not known. However, CagA localized to mitochondria may deregulate the function of the mitochondrial electron transport chain so that it produces primarily O2•−. To clarify the importance of ROS production in CagA-expressing gastric epithelial cells, an in vitro study using CagA-transfected normal rat gastric epithelial cells (RGM1) is currently under way in our laboratory.
Gastric epithelial cells protect themselves from oxidative stress by activating antioxidant defense mechanisms, including oxygen scavenger enzymes such as SOD, catalase and glutathione peroxidase,Citation36 as well as vitamins (Vit) E and C.Citation37 Those cells infected by H. pylori being predominantly on the antrum of the stomach, Vit E levels in the gastric body are known to decrease, which may reflect a mobilization of antioxidant defense mechanisms to maximally inflamed sites.Citation38 Another study in Mongolian gerbils concluded that Vit C or Vit E supplementation protects from H. pylori-induced gastritis in the short term, its effects seemingly declining during persistent infection.Citation39 Thus, although antioxidant systems seem to be involved in infection to counteract the increased ROS,Citation40 when H. pylori-induced excessive ROS generation on the epithelium is augmented by reduced effectiveness of antioxidant defenses, the risk of cytoxicity from oxidation, and DNA damage is potentially increased.Citation41 An impaired redox balance may then result in cell death, which would increase the proliferation rate of the remaining cells and thereby increase the chance of mutations leading to increased oncogene expression, which ultimately results in gastric cancer.Citation42
To summarize the findings described above, the role of H. pylori-induced oxidative stress on the gastric mucosa is multi-functional. For neutrophils, it is the result of excessive defense reactions of the human body against H. pylori intrusion, and for H. pylori, it might be a convenient tool for invading the human gastric mucosa. Although the role of H. pylori-induced oxidative stress in gastric epithelial cells is not clear, it might be involved in signal transduction in epithelial cells,Citation32,Citation43 or in gastric carcinogenesis. Further study is required to elucidate the effects of H. pylori-induced oxidative stress on gastric epithelial cells and its role in gastric carcinogenesis.
Redox biology of gastric carcinogenesis
In patients with H. pylori infection, chronic gastritis sometimes results in glandular loss, which triggers multifocal atrophic gastritis and intestinal metaplasia,Citation44 risk factors for the progression to gastric carcinoma.Citation45 Determinants and mechanisms involved in the occurrence of such divergent pathways are unknown and represent a primary focus of research.
One mechanism of H. pylori-induced carcinogenesis is now believed to be dependent on cumulative oxidative DNA damage.Citation46 This concept is supported by several reports of the production of 8-hydroxydeoxyguanosine (8-OHdG), the main oxidatively modified product of DNA. In gastric carcinoma patients, significantly higher levels of 8-OHdG were reported in tumor-adjacent tissues and tumor tissues than in normal tissues.Citation47 Although DNA damage induced by oxidative stress is made good by several repair genes, accumulated oxidative DNA damage such as 8-OHdG can be only partially repaired through enzyme pathways that may, in turn, cause further DNA damageCitation48 that induces DNA mutation and ultimately gastric carcinogenesis. Also, only a small proportion of the H. pylori-infected patients show clear reduction in the levels of the damaged DNA.Citation49 Moreover, repair genes damaged by oxidative stress may not able to repair damaged DNA and therefore may cause gastric carcinogenesis. Therefore, in younger patients with cagA-positive H. pylori, DNA oxidative damage in the gastric mucosa accumulates in earlier stages of their life, and may cause more extensive gastric mucosal derangement.Citation49,Citation50
In addition to 8-OHdG production, an accumulation of intracellular ROS/RNS can induce point mutation in the DNA, thus disrupting the expression and function of several tumor-suppressing genes such as p53, which might contribute to the pathogenesis of gastric cancer.Citation51 The importance of NO in H. pylori-induced gastric carcinogenesis has been shown in experiments using iNOS−/− mice, in which gastric carcinogenesis was induced by administration of N-methyl-N-nitrosourea. The overall incidence of gastric cancer after 50 weeks was significantly lower in iNOS−/− mice compared with their wild-type counterparts.Citation52 In gastric cancer, the most frequent gene mutation observed in the tumor suppressive gene, p53, is G:C to A:T conversion. Methylated cytosine (C) is deaminated in the presence of NO to form thymine (T) (G:methylated C to G:T). Since thymine is a usual nucleobase in DNA, the G:T mis-pair is rarely repaired, and therefore G:C to A:T transversion is fixed after DNA replication.Citation53,Citation54 This concept is supported by a report that human gastric cancer has a mutation with a high prevalence for G:C to A:T transversion.Citation55
As described above, CagA, a cytotoxin of H. pylori, induces ROS production in mitochondria. The mitochondrial DNA (mtDNA) is more susceptible to ROS damage than nuclear DNA because of its close proximity to the electron transport chain and its lack of protective histones or DNA-binding proteins.Citation56 In addition, mtDNA damage is not sufficiently repaired because of the low level of repair enzymes in some cells.Citation57 As a result, the respiratory enzymes containing the defective mtDNA-encoded protein subunits may exhibit impaired electron transport function and thereby increase the electron leak and ROS production, which in turn elevates oxidative stress and oxidative damage to mitochondria. Consequently, CagA might induce oxidative stress to the gastric mucosa, and may damage cellular components, including polyunsaturated fatty acids, proteins, and mtDNA, which may enhance nuclear DNA damage, and possibly result in the pathogenesis of gastric carcinogenesis.
Besides inducing DNA damage, H. pylori-induced oxidative stress has been shown to modify epithelial cell turnover in the stomach.Citation51 This notion is supported by studies describing an increase in both epithelial cell proliferation and cell death by apoptosis, in response to infection. Cell death, including apoptosis, is triggered by H. pylori as well as various inflammatory mediators. Firstly, activated T cells kill gastric epithelial cells directly, and the host response increases the expression of receptors for H. pylori and thus increases bacterial binding and the induction of apoptosis by the bacteria.Citation51 Next, H. pylori-induced epithelial cell death can stimulate the proliferative response of epithelial cell precursors.Citation51 Recently an interesting finding has been reported with regard to murine double minute 2 (MDM2), an inactivator of the tumor suppressor p53.Citation58 It was found that MDM2 expression is higher in biopsy specimens of intestinal metaplasia and gastric cancer than those from chronic gastritis in H. pylori-infected stomach and that interleukin-16 (IL-16), a T-cell chemoattractant that has been shown to be involved in H. pylori-induced gastric inflammation, increased the expression of MDM2 and cell proliferation of a gastric cancer cell line. Since the overexpression of MDM2 has been shown in several human tumors, and increased expression of MDM2 inactivates the apoptotic and cell cycle arrest function of p53, the expression of MDM2 in long-term H. pylori-infected gastric mucosa may indicate a risk for carcinogenesis, and IL-16 secretion in H. pylori-infected mucosa could well be one of the factors promoting gastric carcinogenesis.
Conclusion
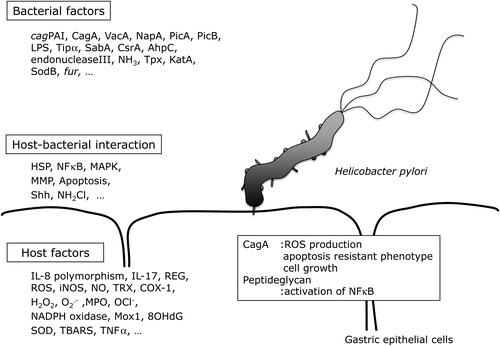
As well as host factors, bacterial factors, and host–bacterial interactions, there are many other factors that might be involved in the pathogenesis of gastric cancer (). For example, ingested food and tobacco smoke directly influence mucosal redox status,Citation41 as they expose the gastric epithelium to the ROS they generate within the gastric lumen in a sustained manner. This multiplicity of factors makes it difficult to fully understand the mechanism by which H. pylori-induces gastric carcinogenesis. Therefore, further research is needed to clarify this, and to identify useful strategies to combat the carcinogenic effects of H. pylori.
Figure 2. Factors in H. pylori-induced gastric inflammation. cagPAI: cag pathogenicity island; VacA: vacuolating cytotoxin A; PicA: permit the induction of cytokines A; PicB: permit the induction of cytokines B; Tipα: tumor necrosis factorα inducing protein; SabA: sialic acid-binding adhesin A; CsrA: carbon storage regulator A; AhpC: alkyl hydroperoxide reductase C; Tpx: thiol-peroxidases; KatA: catalase A; SodB: superoxide dismutase B; fur: ferric uptake regulator; HSP: heat shock protein; MAPK: mitogen-activated protein kinase; MMP: matrix metalloproteinase; Shh: sonic hedgehog; IL-8: interleukin-8; IL-17: interleukin-17; iNOS: inducible nitric oxide synthase; NO: nitric oxide; TRX: thioredoxin; COX-1: cyclooxygenase-1; NADPH: nicotinamide adenine dinucleotide phosphate; Mox1: mitogen oxidase 1; 8OHdG: 8-hydroxy deoxyguanosine; SOD: superoxide dismutase; TBARS: thiobarbituric acid reactive substances; TNFα: tumor necrosis factor alpha; ROS: reactive oxygen species; NFκB: nuclear factor kappa B; CagA: cytotoxim associated gene A; NapA: neutrophil-activating protein A; Lps: lipopolysacharide; COX1: cyclokxygenare; NH2Cl: monochloradine; ROS: reactive oxygen species; H2P2: hydrogen peroxide; OCl−: hypochloride ion.
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research (B) to T.Y. (no. 21390184) and (C) to Y.N. (no.22590705) from the Japan Society for the Promotion of Science, by a City Area Program to T.Y. and Y.N. from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and by an Adaptable and Seamless Technology Transfer Program through target-driven R&D to Y.N. from the Japan Science and Technology Agency.
References
- Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984;1:1311–5.
- Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 1991;325:1127–31.
- Moss S, Calam J. Helicobacter pylori and peptic ulcers: the present position. Gut 1992;33:289–92.
- Ernst PB, Gold BD. The disease spectrum of Helicobacter pylori: the immunopathogenesis of gastroduodenal ulcer and gastric cancer. Annu Rev Microbiol 2000;54:615–40.
- Collaboration AT. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Schistosomes, liver flukes and Helicobacter pylori. IARC Monogr Eval Carcinog Risks Hum 1994;61:177–240.
- Correa P, Cuello C, Duque E, Burbano LC, Garcia FT, Bolanos O, et al. Gastric cancer in Colombia. III. Natural history of precursor lesions. J Natl Cancer Inst 1976;57:1027–35.
- Suzuki H, Miura S, Imaeda H, Suzuki M, Han JY, Mori M, et al. Enhanced levels of chemiluminescence and platelet activating factor in urease-positive gastric ulcers. Free Radic Biol Med 1996;20:449–54.
- Davies GR, Simmonds NJ, Stevens TR, Sheaff MT, Banatvala N, Laurenson IF, et al. Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo. Gut 1994;35:179–85.
- Zhang Q, Dawodu JB, Etolhi G, Husain A, Gemmell CG, Russell RI. Relationship between the mucosal production of reactive oxygen radicals and density of Helicobacter pylori in patients with duodenal ulcer. Eur J Gastroenterol Hepatol 1997;9:261–5.
- Nagata K, Yu H, Nishikawa M, Kashiba M, Nakamura A, Sato EF, et al. Helicobacter pylori generates superoxide radicals and modulates nitric oxide metabolism. J Biol Chem 1998;273:14071–3.
- Benaissa M, Babin P, Quellard N, Pezennec L, Cenatiempo Y, Fauchere JL. Changes in Helicobacter pylori ultrastructure and antigens during conversion from the bacillary to the coccoid form. Infect Immun 1996;64:2331–5.
- Cellini L, Allocati N, Angelucci D, Iezzi T, Di Campli E, Marzio L, et al. Coccoid Helicobacter pylori not culturable in vitro reverts in mice. Microbiol Immunol 1994;38:843–50.
- Nakamura A, Park A, Nagata K, Sato EF, Kashiba M, Tamura T, et al. Oxidative cellular damage associated with transformation of Helicobacter pylori from a bacillary to a coccoid form. Free Radic Biol Med 2000;28:1611–8.
- Wang G, Alamuri P, Maier RJ. The diverse antioxidant systems of Helicobacter pylori. Mol Microbiol 2006;61:847–60.
- Oh JD, Karam SM, Gordon JI. Intracellular Helicobacter pylori in gastric epithelial progenitors. Proc Natl Acad Sci USA 2005;102:5186–91.
- Wang J, Brooks EG, Bamford KB, Denning TL, Pappo J, Ernst PB. Negative selection of T cells by Helicobacter pylori as a model for bacterial strain selection by immune evasion. J Immunol 2001;167:926–34.
- Naito Y, Yoshikawa T, Fujii T, Boku Y, Yagi N, Dao S, et al. Monochloramine-induced cell growth inhibition and apoptosis in a rat gastric mucosal cell line. J Clin Gastroenterol 1997;25 (Suppl 1):S179–85.
- Fu S, Ramanujam KS, Wong A, Fantry GT, Drachenberg CB, James SP, et al. Increased expression and cellular localization of inducible nitric oxide synthase and cyclooxygenase 2 in Helicobacter pylori gastritis. Gastroenterology 1999;116:1319–29.
- Tsuji S, Kawano S, Tsujii M, Takei Y, Tanaka M, Sawaoka H, et al. Helicobacter pylori extract stimulates inflammatory nitric oxide production. Cancer Lett 1996;108:195–200.
- Kuwahara H, Miyamoto Y, Akaike T, Kubota T, Sawa T, Okamoto S, et al. Helicobacter pylori urease suppresses bactericidal activity of peroxynitrite via carbon dioxide production. Infect Immun 2000;68:4378–83.
- Bryk R, Griffin P, Nathan C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature 2000;407:211–5.
- Bussiere FI, Chaturvedi R, Cheng Y, Gobert AP, Asim M, Blumberg DR, et al. Spermine causes loss of innate immune response to Helicobacter pylori by inhibition of inducible nitric-oxide synthase translation. J Biol Chem 2005;280:2409–12.
- Gobert AP, McGee DJ, Akhtar M, Mendz GL, Newton JC, Cheng Y, et al. Helicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: a strategy for bacterial survival. Proc Natl Acad Sci USA 2001;98:13844–9.
- McGee DJ, Zabaleta J, Viator RJ, Testerman TL, Ochoa AC, Mendz GL. Purification and characterization of Helicobacter pylori arginase, RocF: unique features among the arginase superfamily. Eur J Biochem 2004;271:1952–62.
- Harris AG, Wilson JE, Danon SJ, Dixon MF, Donegan K, Hazell SL. Catalase (KatA) and KatA-associated protein (KapA) are essential to persistent colonization in the Helicobacter pylori SS1 mouse model. Microbiology 2003;149:665–72.
- Annibale B, Capurso G, Delle Fave G. Consequences of Helicobacter pylori infection on the absorption of micronutrients. Dig Liver Dis 2002;34 (Suppl 2):S72–7.
- Naito Y, Yoshikawa T. Molecular and cellular mechanisms involved in Helicobacter pylori-induced inflammation and oxidative stress. Free Radic Biol Med 2002;33:323–36.
- Smith WB, Gamble JR, Clark-Lewis I, Vadas MA. Interleukin-8 induces neutrophil transendothelial migration. Immunology 1991;72:65–72.
- Ando T, Kusugami K, Ohsuga M, Ina K, Shinoda M, Konagaya T, et al. Differential normalization of mucosal interleukin-8 and interleukin-6 activity after Helicobacter pylori eradication. Infect Immun 1998;66:4742–7.
- Handa O, Yoshida N, Fujita N, Tanaka Y, Ueda M, Takagi T, et al. Molecular mechanisms involved in anti-inflammatory effects of proton pump inhibitors. Inflamm Res 2006;55:476–80.
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 2004;4:181–9.
- Handa O, Naito Y, Takagi T, Shimozawa M, Kokura S, Yoshida N, et al. Tumor necrosis factor-alpha-induced cytokine-induced neutrophil chemoattractant-1 (CINC-1) production by rat gastric epithelial cells: role of reactive oxygen species and nuclear factor-kappaB. J Pharmacol Exp Ther 2004;309:670–6.
- Sumimoto H, Miyano K, Takeya R. Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem Biophys Res Commun 2005;338:677–86.
- Kawahara T, Teshima S, Oka A, Sugiyama T, Kishi K, Rokutan K. Type I Helicobacter pylori lipopolysaccharide stimulates toll-like receptor 4 and activates mitogen oxidase 1 in gastric pit cells. Infect Immun 2001;69:4382–9.
- Handa O, Naito Y, Yoshikawa T. CagA protein of Helicobacter pylori: a hijacker of gastric epithelial cell signaling. Biochem Pharmacol 2007;73:1697–702.
- Mori M, Suzuki H, Suzuki M, Kai A, Miura S, Ishii H. Catalase and superoxide dismutase secreted from Helicobacter pylori. Helicobacter 1997;2:100–5.
- Calvino Fernandez M, Parra Cid T. H. pylori and mitochondrial changes in epithelial cells. The role of oxidative stress. Rev Esp Enferm Dig 2010;102:41–50.
- Phull PS, Price AB, Thorniley MS, Green CJ, Jacyna MR. Vitamin E concentrations in the human stomach and duodenum – correlation with Helicobacter pylori infection. Gut 1996;39:31–5.
- Sun YQ, Girgensone I, Leanderson P, Petersson F, Borch K. Effects of antioxidant vitamin supplements on Helicobacter pylori-induced gastritis in Mongolian gerbils. Helicobacter 2005;10:33–42.
- Bagchi D, Bhattacharya G, Stohs SJ. Production of reactive oxygen species by gastric cells in association with Helicobacter pylori. Free Radic Res 1996;24:439–50.
- Smoot DT, Elliott TB, Verspaget HW, Jones D, Allen CR, Vernon KG, et al. Influence of Helicobacter pylori on reactive oxygen-induced gastric epithelial cell injury. Carcinogenesis 2000;21:2091–5.
- Obst B, Wagner S, Sewing KF, Beil W. Helicobacter pylori causes DNA damage in gastric epithelial cells. Carcinogenesis 2000;21:1111–5.
- Handa O, Naito Y, Yoshikawa T. Rat Cytokine-Induced Neutrophil Chemoattractant-1 (CINC-1) in Inflammation. J Clin Biochem Nutr 2006;38:51–8.
- Fontham ET, Ruiz B, Perez A, Hunter F, Correa P. Determinants of Helicobacter pylori infection and chronic gastritis. Am J Gastroenterol 1995;90:1094–101.
- Filipe MI, Munoz N, Matko I, Kato I, Pompe-Kirn V, Jutersek A, et al. Intestinal metaplasia types and the risk of gastric cancer: a cohort study in Slovenia. Int J Cancer 1994;57:324–9.
- Nishibayashi H, Kanayama S, Kiyohara T, Yamamoto K, Miyazaki Y, Yasunaga Y, et al. Helicobacter pylori-induced enlarged-fold gastritis is associated with increased mutagenicity of gastric juice, increased oxidative DNA damage, and an increased risk of gastric carcinoma. J Gastroenterol Hepatol 2003;18:1384–91.
- Lee BM, Jang JJ, Kim HS. Benzo[a]pyrene diol-epoxide-I-DNA and oxidative DNA adducts associated with gastric adenocarcinoma. Cancer Lett 1998;125:61–8.
- Kuchino Y, Mori F, Kasai H, Nishimura S, Inoue H, Iwai S, et al. Misreading of 8-hydroxydeoxyguanosine-containing DNA in in vitro DNA replication. Nucleic Acids Symp Ser 1986:157–8.
- Farinati F, Cardin R, Russo VM, Busatto G, Franco M, Rugge M. Helicobacter pylori CagA status, mucosal oxidative damage and gastritis phenotype: a potential pathway to cancer? Helicobacter 2003;8:227–34.
- Maaroos HI, Vorobjova T, Sipponen P, Tammur R, Uibo R, Wadstrom T, et al. An 18-year follow-up study of chronic gastritis and Helicobacter pylori association of CagA positivity with development of atrophy and activity of gastritis. Scand J Gastroenterol 1999;34:864–9.
- Ernst P. Review article: the role of inflammation in the pathogenesis of gastric cancer. Aliment Pharmacol Ther 1999;13 (Suppl 1):13–8.
- Nam KT, Oh SY, Ahn B, Kim YB, Jang DD, Yang KH, et al. Decreased Helicobacter pylori associated gastric carcinogenesis in mice lacking inducible nitric oxide synthase. Gut 2004;53:1250–5.
- Shiao YH, Palli D, Buzard GS, Caporaso NE, Amorosi A, Saieva C, et al. Implications of p53 mutation spectrum for cancer etiology in gastric cancers of various histologic types from a high-risk area of central Italy. Carcinogenesis 1998;19:2145–9.
- Ranzani GN, Luinetti O, Padovan LS, Calistri D, Renault B, Burrel M, et al. p53 gene mutations and protein nuclear accumulation are early events in intestinal type gastric cancer but late events in diffuse type. Cancer Epidemiol Biomarkers Prev 1995;4:223–31.
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature 2007;446:153–8.
- Wei YH, Lu CY, Lee HC, Pang CY, Ma YS. Oxidative damage and mutation to mitochondrial DNA and age-dependent decline of mitochondrial respiratory function. Ann N Y Acad Sci 1998;854:155–70.
- Dobson AW, Xu Y, Kelley MR, LeDoux SP, Wilson GL. Enhanced mitochondrial DNA repair and cellular survival after oxidative stress by targeting the human 8-oxoguanine glycosylase repair enzyme to mitochondria. J Biol Chem 2000;275:37518–23.
- Nakajima N, Ito Y, Yokoyama K, Uno A, Kinukawa N, Nemoto N, et al. The Expression of Murine Double Minute 2 (MDM2) on Helicobacter pylori-Infected Intestinal Metaplasia and Gastric Cancer. J Clin Biochem Nutr 2009;44:196–202.