Abstract
In recent years, therapeutic approaches for many tumors have broadened or even shifted entirely from cytotoxic chemotherapy to specific targeting of dysregulated proteins (predominately kinases), and more recently, harnessing of the anti-tumor immune response. The most prominent example of this shift is the management of metastatic melanoma, where BRAF and MEK inhibition and CLTA-4 blockade have established an entirely new standard of care in the last 3 years. Targeted kinase inhibition and immune checkpoint blockade have different strengths and weaknesses. Kinase inhibitors generally have rapid and impressive response rates but modest progression-free survival while immunotherapy can achieve durable tumor control, but is often associated with lower response rates and slower time to clinical benefit. These approaches would seem to be complementary however the results of early combination studies suggest that caution is advised when combining targeted kinase inhibition with immunotherapy. In this context, rigorous biomarker driven clinical trials are needed to further elucidate mechanisms of both benefit and toxicity. Depending on disease specific biology, it seems likely that both combination and sequential approaches of kinase inhibitors with immunotherapy will be required in order to harness the full potential of these approaches.
1. Introduction
Improvements have recently been made in the treatment of advanced cancer through the development of “targeted” cancer therapies, those that specifically inhibit a dysregulated oncogenic proteins, as well as advances in immunotherapy such as immune check-point blockade. This is a marked change from even a decade earlier when cytotoxic chemotherapy was still at the forefront of drug development. The drugs developed through these approaches have advanced patient care by improving overall survival, as well as quality of life, however have also generated many new questions regarding the future roles for combinations and sequences of targeted- and immunotherapies. Of all cancers, the treatment of BRAF mutant malignant melanoma has to date been impacted the greatest by these approaches and appears positioned to serve as the prototype whereby paradigms of combination or sequential kinase inhibitors with immunotherapy are evaluated and elucidated.
Over the past 3 years, four drugs have obtained Food and Drug Administration approval for the treatment of patients with BRAF mutant melanoma, and at least two further drugs are likely to be approved in the not distant future. These drugs include ipilimumab, the fully human monoclonal antibody against cytotoxic T-lymphocyte antigen 4 (CTLA-4), vemurafenib and dabrafenib, both serine-threonine kinase inhibitors targeting the mutant protein BRAFV600, as well as the MEK1/2 kinase inhibitor trametinib. All four of these drugs have been shown to improve the overall survival of patients with melanoma in Phase III clinical trials. Furthermore, dual BRAF and MEK inhibition in BRAFV600 mutant melanoma, by dabrafenib and trametinib, yields more durable tumor control and causes less toxicity than BRAF inhibition alone. Beyond these approved drugs, two anti-programmed death-1 (PD-1) antibodies, nivolumab and MK-3475, have shown impressive response rates and durable disease-free survival in early phase clinical trials. Importantly, the response rate of kinase inhibitors targeting mutant BRAF is high; however, response duration is limited in almost all patients. Ipillimumab has a more modest response rate and longer time to treatment benefit, but offers the potential for long term durable disease control, while PD-1 inhibition can achieve substantial response rates with relatively rapid onset and durability of responses.
Both targeted and immunotherapeutic treatments are important in the management of BRAF mutant melanoma; however, the optimal sequence and combination of the available treatment agents is not clear. The efficacy of immune check-point blockade may be improved by combinatorial approaches Citation[1]. Because of the substantial anti-tumor activity in BRAF mutant melanoma, inhibition of the mitogen-activated protein kinase (MAPK) pathway, through BRAF and MEK, may be particularly attractive to combine with immunotherapy. It has been hypothesized that antigen release through tumor cell death mediated by MAPK pathway inhibition may lead to increased antigen presentation or cross presentation to tumor specific T cells. Inhibition of the MAPK pathway was also found to increase the expression of melanoma differentiation antigens in vitro and in post treatment tumor samples. Additionally, inhibition of BRAF and MEK in melanoma cells leads to increased tumor specific T cell, as well as dendritic cell, function in vitro. Possible drawbacks of these kinase inhibitors when combined with CTLA-4 or PD-1/Programmed death ligand-1 (PD-L1) blockade include sometimes quickly emerging resistance and potential dampening of the immune response through inhibition of non-oncogenic target kinases.
Understanding the effects of kinase inhibitors on the immune response is thus essential for the development of rational combined targeted-immunotherapeutic drug regimens. Independent of immunotherapy, the combination of BRAF-MEK inhibition will likely displace single agent BRAF inhibitor treatment when results from Phase III clinical trials will be reported; it is therefore critical to delineate the impact of both drugs on the immune response.
2. Pre-clinical evidence for differential immune effects mediated by kinase inhibitors
There is increasing evidence that kinase inhibitors exert effects on the immune system in addition to the tumor cells they are designed to target. The specific mechanisms by which this occurs are variable, with examples including dysregulation of the T cell receptor through LCK Citation[2], reduction in cytokine production through SRC Citation[3], as well as reduction of immune tolerance induced by myeloid-derived suppressor cells through KIT Citation[4]. Kinase inhibitors such as sunitinib, imatinib or dasatinib have been shown to have both suppressive and stimulating effects on multiple sub-populations of immune cells, such as CD4+ and CD8+ T cells Citation[5], NK cells Citation[6], and dendritic cells Citation[7]. More importantly however, some of these effects have been associated with both improved and dampened preclinical Citation[5] and clinical outcomes with these agents Citation[6]. An example of such improved immune response after kinase inhibitor treatment has been described in gastrointestinal stromal tumors (GIST) Citation[8]. Using a spontaneous murine GIST tumor model, imatinib was shown to activate CD8+ T cells and induce regulatory T cell apoptosis by reducing tumor-cell expression of indoleamine 2,3-dioxygenase (IDO). This effect was augmented by CTLA-4 blockade.
BRAFV600 specific inhibitors, such as vemurafenib and dabrafenib, have relatively minimal effects on “off-target” kinases and may be good candidates for combination with immunotherapy (). Preclinical studies suggest that vemurafenib may improve recognition of melanoma antigens by T lymphocytes and this agent has been documented to induce increases in CD4+ and CD8+ tumor infiltrating lymphocytes into human melanoma tumors Citation[9]. Further, BRAF inhibition by vemurafenib has also been associated with reduced expression of the immunosuppresive cytokine interleukin-1 Citation[10] as well as increased trafficking and anti-tumor activity of adoptively transferred T cells in mouse melanoma xenografts Citation[11] and a BRAFV600E-driven murine model of melanoma Citation[12]. Similarly, dabrafenib has been described to have no detectable negative impact on existing systemic immunity or the de novo generation of tumor-specific T cells Citation[13]. Instead, in what may be a productive immunomodulatory role, dabrafenib treatment is associated with a slight increase in serum tumor necrosis factor-α and a modest increase in melanoma antigen specific CD8+ T cells.
Figure 1. Effects of MEK and BRAF inhibitors on dendritic cells and T cells. BRAF mutant melanoma cells release high levels of suppressive factors including cytokines such as interlukin (IL)-6, IL-10, vascular endothelial growth factor (VEGF) and other molecules. Treatment with MEK or BRAF inhibitors leads to increased release of melanoma differentiation antigens (MDA) which can increase presentation of these molecules by antigen presenting cells such as dendritic cells (DCs). BRAF inhibition is not associated with negative effects on T cell or DC function and has been shown to increase infiltration of tumor with CD4 and CD8 cells. MEK inhibition has possibly negative effects on T cell and DC function by decreasing the production of IL-12 and tumor necrosis factor-alpha as well as effect on T cell viability, decreased production of interferon-gamma and variable effects on FoxP3+ T regulatory cells.
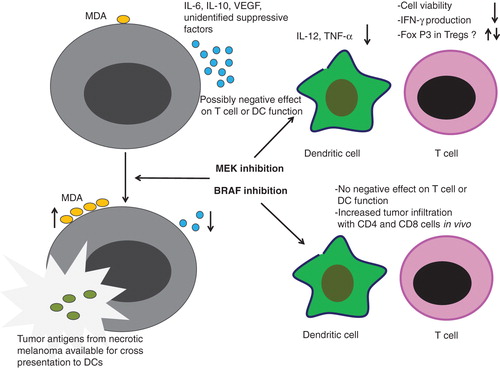
Whereas BRAFV600E specific inhibition does not appear to alter T-cell function, MEK inhibition has been shown to decrease proliferation and viability of T lymphocytes Citation[14] (). MEK inhibition significantly decreases T-cell release of interferon-γ, where a BRAF selective agent does not have this suppressive effect. MEK inhibition has also been shown to enhance the expression of forkhead box P3 (FoxP3)+ T cells Citation[15], thus, dampening the immune response and favoring T cell anergy. It also appears that RAS signaling is of importance in CD28-mediated co-stimulation of CD4+ T cells Citation[16]. This implies that downstream MEK inhibition could disrupt this process. Furthermore, MEK inhibition, but not BRAF inhibition, negatively impacts dendritic cell function as measured by IL-12 and TNF-α secretion and decreased expression of CD40, CD80, CD83, and MHC-I Citation[17].
In a potentially converse release of negative immune regulation, the use of MEK inhibitors to block ERK signaling has been shown to inhibit activation-dependent FoxP3 up-regulation in resting regulatory T cells, their transition to activated regulatory T cells and blockade of the resulting suppressive capacity Citation[18]. MEK inhibitors have also been shown to reverse the expression of PD-L1 that arises at the time of BRAF inhibitor resistance Citation[19]. Additionally, a recent small series described no significant difference in T cell infiltrates within human melanoma tumor specimens from patients receiving single agent BRAF inhibitor versus BRAF-MEK inhibitor combination Citation[20]. Thus, the role of MEK inhibitors on the immune response is unclear. While there is data suggesting a deleterious effect, other data suggests that rather than block immune activation, MEK inhibitors might have future therapeutic application in augmenting a cytotoxic T cell response.
3. Clinical experience
As preclinical data is rapidly evolving to describe the effects of kinase inhibitors on the immune system, clinical studies evaluating such combinations have already begun. Though the initial experience is small, a somewhat unanticipated high incidence of toxicity has been reported. In a Phase I study of the vascular-endothelial growth factor receptor (VEGFR) inhibitor, sunitinib, was combined with tremelimumab, a CTLA-4 monoclonal antibody Citation[21]. In this study, nine of 28 patients with renal cell cancer (in various dosing cohorts) experienced dose limiting toxicity, predominantly in the form of rapid onset acute renal failure.
In melanoma, an unexpected rise in toxicity with combination kinase inhibitor and immune check-point blockade has also been observed. In the Phase III trial of ipilimumab in combination with the chemotherapeutic agent dacarbazine, markedly more severe liver toxicity was observed than would be expected from each of the agents alone Citation[22]. Similarly, combinations of vemurafenib and immune check-point blockade have shown a pattern of increased toxicity, with immune-mediated hepatitis being of particular concern. A Phase I study of vemurafenib and ipilimumab was held after 6 of 10 patients experienced dose limiting immune-mediated hepatitis Citation[23]. Additionally, a Phase I study of vemurafenib in combination with the anti-PD-L1 antibody MPDL3280A had to be modified from continuous combination dosing to a staggered approach due to dose-limiting transaminitis Citation[24]. On-going studies of immune-check point blockade combinations are summarized in .
Table 1. Current immune check-point combinations in Clinical Trials.
The mechanisms of augmented liver toxicity seen with combination therapy are currently unknown. It is controversial whether these toxicities are mediated by the kinase inhibitor or an amplification of the known inflammatory toxicities observed with immune checkpoint blockade. Though some of these combinations have shown encouraging clinical benefit, care will have to be given in developing combinations of kinase inhibitors with inhibitors of immune check-points such as CTLA-4 and PD-1.
4. Expert opinion
The remarkable advances with selective small molecule kinase inhibitors and immune check-point blockade have ushered in a new era in cancer therapy. Despite well justified optimism, much work remains in elucidating maximally effective regimens with these new agents.
While BRAF and combined BRAF and MEK inhibition yields to rapid responses in a large proportion of advanced melanoma patients, the duration of response is generally modest. Conversely, immune checkpoint blockade has the potential for durable disease control, however the response rates, particularly with ipilimumab are lower. In this context, there is great hope that synergies between these two fundamentally different treatment approaches can be identified and translated into improved anti-tumor activity. However, caution and prudent evaluation will be necessary given the documented increased toxicity in early clinical studies with combined immune checkpoint blockade and MAPK inhibition and more broadly, with therapies such as CTLA-4 blockade in combination with other immunomodulators (cytokines or other immune check-point inhibitors), VEGF inhibition, and chemotherapy.
From a preclinical perspective, certain combinations of kinase inhibitors and immunotherapy would appear as more likely to be efficacious and tolerable. To date however, these findings have been difficult to translate into the clinic. Preclinical models strongly suggest a benefit in combining BRAF inhibitors with immune check-point blockade. It is quite notable therefore that the first in class BRAF inhibitor, vemurafenib, and the first in class immune checkpoint blockade agent, ipilimumab, were not tolerable when administered in combination. Some have argued that the toxicity may be drug-specific given that the BRAF inhibitor dabrafenib has a somewhat different side effect profile relative to vemurafenib (e.g., - arthralgia, fever etc.) and that other immune checkpoint inhibitors, such as anti-PD1/PD-L1 antibodies, appear to be less toxic than anti-CTLA-4. Vigilance must be employed when administering these new agents in combination and this should not be done outside the context of a clinical trial.
Moving forward, there are several studies evaluating concomitant administration of kinase inhibitors and immune checkpoint blockade. Examples include a study of dabrafenib, trametinib and ipilimumab in BRAF mutant melanoma (ClinicalTrials.gov Identifier: NCT01767454), a study of dasatinib, a KIT and SRC kinase inhibitor, with ipilimumab in GIST and other sarcomas (ClinicalTrials.gov Identifier: NCT01643278) as well as a study of sunitinib or pazopanib (a multi-kinase inhibitor with VEGFR activity), plus the anti-PD-1 antibody nivolumab in renal cell carcinoma (ClinicalTrials.gov Identifier: NCT01472081). Sequential approaches are also being evaluated in BRAFV600 mutant melanoma with studies launched, or being developed, including the combination of vemurafenib and ipilimumab (ClinicalTrials.gov Identifier: NCT01673854), vemurafenib and MPDL3280A (ClinicalTrials.gov Identifier: NCT01656642) as well as dabrafenib, trametinib and ipilimumab (ClinicalTrials.gov Identifier: NCT01940809).
Translational breakthroughs in molecular drug development and immunotherapy have set a new bar for the treatment of advanced melanoma and this is likely to become relevant to all cancers in the relatively near future. How to best use these therapeutic approaches in a synergistic manner is an open question that needs to be answered urgently. Optimal sequencing and dosing will need to be delineated in clinical trials and the development and rational incorporation of biomarkers will be critical in this endeavor. It seems likely that both combination and sequential approaches of kinase inhibitors with immunotherapy will be required depending on the disease biology of the malignancy under study.
Declaration of interest
The authors state no conflict of interest and have received no payment in preparation of this manuscript.
References
- Agarwala S. Novel immunotherapies as potential therapeutic partners for traditional or targeted agents: cytotoxic T-lymphocyte antigen-4 blockade in advanced melanoma. Melanoma Res 2010;20(1):1-10
- Schade AE, Schieven GL, Townsend R, et al. Dasatinib, a small-molecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood 2008;111:1366-77
- Weichsel R, Dix C, Wooldridge L, et al. Profound inhibition of antigen-specific T-cell effector functions by dasatinib. Clin Cancer Res 2008;14:2484-91
- Kao J, Ko EC, Eisenstein S, et al. Targeting immune suppressing myeloid-derived suppressor cells in oncology. Crit Rev Oncol Hematol 2011;77(1):12-9
- Ozao-Choy J, Ma G, Kao J, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res 2009;69:2514-22
- Kreutzman A, Juvonen V, Kairisto V, et al. Mono/oligoclonal T- and NK-cells are common in chronic myeloid leukemia patients at diagnosis and expand during dasatinib therapy. Blood 2010;116(5):772-82
- Ray P, Krishnamoorthy N, Oriss TB, et al. Signaling of c-kit in dendritic cells influences adaptive immunity. Ann NY Acad Sci 2010;1183:104-22
- Balachandran VP, Cavnar MJ, Zeng S, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med 2011;17:1094-100
- Wilmott JS, Long GV, Howle JR, et al. Selective BRAF inhibitors induce marked T cell infiltration into human metastatic melanoma. Clin Cancer Res 2011
- Khalili JS, Liu S, Rodriguez-Cruz TG, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res 2012;18:5329-40
- Liu C, Peng W, Xu C, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res 2013;19:393-403
- Koya RC, Mok S, Otte N, et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res 2012;72:3928-37
- Hong DS, Vence L, Falchook G, et al. BRAF(V600) inhibitor GSK2118436 targeted inhibition of mutant BRAF in cancer patients does not impair overall immune competency. Clin Cancer Res 2012;18:2326-35
- Boni A, Cogdill AP, Dang P, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res 2010;70:5213-19
- Gabrysova L, Christensen JR, Wu X, et al. Integrated T-cell receptor and costimulatory signals determine TGF-beta-dependent differentiation and maintenance of Foxp3+ regulatory T cells. Eur J Immunol 2011;41:1242-8
- Janardhan SV, Praveen K, Marks R, et al. Evidence implicating the Ras pathway in multiple CD28 costimulatory functions in CD4+ T cells. PLoS One 2011;6:e24931
- Ott PA, Henry T, Baranda SJ, et al. Inhibition of both BRAF and MEK in BRAF(V600E) mutant melanoma restores compromised dendritic cell (DC) function while having differential direct effects on DC properties. Cancer Immunol Immunother 2013;62:811-22
- Kalland ME, Oberprieler NG, Vang T, et al. T cell-signaling network analysis reveals distinct differences between CD28 and CD2 costimulation responses in various subsets and in the MAPK pathway between resting and activated regulatory T cells. J Immunol 2011;187:5233-45
- Jiang X, Zhou J, Giobbie-Hurder A, et al. The paradoxical activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res 2013;19:598-609
- Tompers Frederick D, Piris A, Cogdill AP, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res 2013;19:1225-31
- Rini BI, Stein M, Shannon P, et al. Phase 1 dose-escalation trial of tremelimumab plus sunitinib in patients with metastatic renal cell carcinoma. Cancer 2011;117:758-67
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517-26
- Ribas A, Hodi FS, Callahan M, et al. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med 2013;368:1365-6
- Hamid O, Sosman J, Lawrence D, et al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic melanoma (mM). J Clin Oncol 2013;31(Suppl):abstract 9010