
Abstract
Objectives: This multicenter, randomized, double-blind, placebo-controlled study with an enriched enrollment, randomized withdrawal design was conducted to evaluate the analgesic efficacy and safety of single-entity, once-daily hydrocodone 20 to 120 mg tablets (HYD) in opioid-naive and opioid-experienced patients with uncontrolled moderate to severe chronic low back pain (CLBP).
Research design and methods: The primary endpoint was week 12 pain intensity scores (11-point scale, 0 = no pain) using a mixed effect model with repeated measures incorporating a pattern mixture model framework. Responder analysis was a secondary endpoint. Safety was assessed.
Results: Out of 905 patients who were treated with HYD during the open-label titration period, 588 (65%) were randomized to continue to receive HYD (n = 296, 20 – 120 mg taken once daily, average daily dose 57 mg) or a matching placebo (n = 292). HYD demonstrated superior pain reduction (p = 0.0016); this result was supported by sensitivity analyses using different approaches to handling missing data. Proportions of patients achieving ≥ 30 and ≥ 50% improvement in pain from screening to week 12 also favored HYD (p = 0.0033 and 0.0225, respectively). HYD was generally well tolerated.
Conclusions: HYD was shown to be an efficacious treatment for CLBP in this study. There were no new or unexpected safety concerns detected.
1. Introduction
Immediate-release hydrocodone/acetaminophen (APAP) combination therapy has been the most prescribed medication for pain conditions, including chronic pain, in the US since 1997 Citation[1,2]. This medication is also the most frequently abused prescription opioid drug, with lifetime nonmedical use almost doubling between 2002 (13,093,000 persons) and 2012 (23,731,000 persons) Citation[3,4]. Hydrocodone combination products have recently been moved to a more restrictive Drug Enforcement Agency status (Schedule II vs Schedule III), in an effort to decrease inappropriate access to these drugs Citation[5].
In addition, the nonopioid component, APAP, may cause hepatotoxicity at high doses, and, especially when combined with hydrocodone, has been associated with a significant number of unintentional overdose cases which often lead to acute liver failure and death Citation[6-11]. As a result, the potential of APAP toxicity has created a dosage ceiling for those hydrocodone combination analgesics Citation[10].
HYD (Hysingla® ER, Purdue Pharma L.P., Stamford, CT) is a single-entity, once-daily, ER hydrocodone bitartrate tablet recently available in the US for the management of pain severe enough to require daily, around-the-clock, long-term opioid treatment for which other treatment options are inadequate. It is formulated with abuse-deterrent properties designed to deter abuse through manipulation and subsequent injecting or snorting. Clinical studies with HYD showing reduced abuse potential compared with immediate-release hydrocodone (solution or powder) have been previously reported and are not a part of the current study Citation[12,13]. Additionally, this single-entity hydrocodone tablet allows dose adjustment without the risks associated with higher doses of APAP.
This article presents the results of a pivotal study, as part of the clinical development program that evaluated the efficacy and safety of a new single-entity, once-daily, extended-release hydrocodone bitartrate tablet (HYD). The study employed an enriched enrollment, randomized withdrawal design, a standard approach to evaluate the efficacy of opioid analgesics for the chronic pain indication.
2. Patients and methods
2.1 Study design
This multicenter, randomized, double-blind, placebo-controlled study was conducted in 94 sites in the United States and enrolled patients with moderate to severe chronic low back pain (CLBP) that was uncontrolled by their current stable analgesic regimen. The primary objective was to evaluate the analgesic efficacy and safety of HYD (20 to 120 mg tablets once daily) for the treatment of moderate to severe LBP compared with placebo. The study consisted of a prerandomization phase (i.e., a screening period (up to 14 days) and an open-label run-in dose titration period (up to 45 days) that assessed patient qualification for randomization), a 12-week double-blind period, with a follow-up visit 5 – 7 days after the final double-blind visit (). Patients who discontinued study drug during the double-blind period were encouraged to remain in the study and complete all study visits, even though they were no longer receiving study drug (these patients were considered retrieved dropout patients). Retrieved dropout patients were allowed to resume their incoming pain medications and/or could take any medications recommended by the investigator.
Figure 1. Study design. Visits occurred at the start of the screening period, the start of the open-label period, at randomization, at weeks 1, 2, 4, 8, and 12 of the double-blind period, and at the end of the follow-up period.
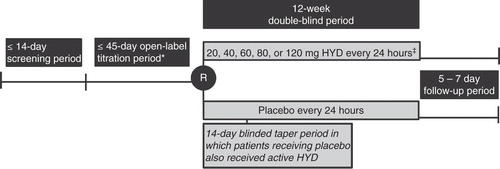
Beginning at screening, patients recorded pain scores on a daily basis on an electronic diary. At screening and during the open-label run-in and double-blind periods, patients recorded their “average pain over the last 24 h” scores on an 11-point numerical rating scale where 0 indicated “no pain” and 10 indicated “pain as bad as you can imagine.” This study and its informed consent form were reviewed and approved by the appropriate institutional review boards, and it was conducted according to current good clinical practice and all relevant parts of the United States Code of Federal Regulations Title 21. All patients provided written informed consent before any study-related procedures were performed.
2.2 Patients
Male and females aged 18 years or older with CLBP (i.e., LBP lasting for at least 3 months prior to the screening visit) who were either opioid-experienced or opioid-naive (defined as a patient receiving < 5 mg a day of oxycodone equivalent during the 14 days prior to screening) were eligible for participation in the study. Eligible patients must have met study inclusion/exclusion criteria, which included the following: must have been on a stable analgesic regimen; if receiving a stable opioid analgesic regimen, must have been receiving opioid medication equivalent to 100 mg/day oxycodone or less for 14 days prior to screening; must have had uncontrolled LBP (defined as an “average pain over the last 14 days” score of 5 or greater at screening, as well as 3 or more “average pain over the last 24 h” scores of 5 or greater during the screening period). The LBP was required to be nonradiating or radiating no further than above the knee (i.e., satisfying the criteria for Quebec Task Force Classification 1 or 2 only) Citation[14-16]. Female patients must have used adequate and reliable contraception. Oral corticosteroid use must have been stable, and adjunct therapy must have either been stable or stopped.
Pregnant or lactating women were not eligible for study entry. Patients were excluded if they had LBP with distal radiation (below the knee) with or without neurologic signs (i.e., Quebec Task Force Classification 3 to 6); inflammatory arthritis; surgical procedures directed toward the source of the CLBP within 6 months of the screening visit, or any major surgery scheduled during the study period; nerve/plexus block within 4 weeks; lumbar steroid injections within 6 weeks; or the presence of significant cardiac, pulmonary, neurologic, hepatic, renal, gastrointestinal, or psychiatric conditions.
2.3 Treatments
Upon entry into the open-label run-in period, patients discontinued all medications used for chronic pain and began treatment with HYD. Opioid naive patients began treatment with 20 mg HYD, and opioid-experienced patients were converted to an HYD dose that was 25 to 50% of their incoming opioid total daily dosage. Patients had their dosages titrated as needed, and were entered into the double-blind period if they met the qualification criteria (i.e., patient received the same dose for 7 ± 2 consecutive days; and if, for 3 consecutive days before randomization, patient had both an “average pain over the last 24 h” score that was ≤ 4 with a ≥ 2-point reduction in screening mean pain score, and also did not take more than the maximum daily dose of supplemental pain medication [see below]).
Upon entry into the double-blind period, patients were randomized in a 1:1 ratio to receive HYD (at the stable dose achieved during the open-label run-in period [20, 40, 60, 80, or 120 mg]) or matching placebo. Randomization was stratified by the patient’s HYD dose at the end of the open-label run-in period and by opioid status (i.e., naive or experienced). During the first 2 weeks of the double-blind period, patients randomized to placebo had their HYD doses tapered in a blinded manner, with a reduction in dose of approximately 25 – 50% every 3 – 4 days.
During the open-label run-in period, patients were permitted to take 5 mg immediate-release oxycodone tablets (10 mg maximum daily dose) as needed for their LBP. During the double-blind period, patients receiving either HYD 20, 40, 60, 80, and 120 mg or matching placebo were permitted to take maximum daily doses of immediate-release oxycodone of 10, 10, 15, 20, and 30 mg, respectively.
2.4 Efficacy assessments
The primary efficacy variable was the mean weekly pain intensity during the double-blind period. Secondary efficacy variables were proportions of patients achieving 30 and 50% reduction in pain intensity from screening to the end of double-blind period (responder analysis), the sleep disturbance subscale of the Medical Outcomes Study Sleep Scale – Revised (MOS Sleep – R) Citation[17] obtained during the double-blind period, the Patient Global Impression of Change (PGIC) Citation[18] obtained at the end of the double-blind period. Other (exploratory) efficacy variables included other subscales of MOS Sleep-R, Oswestry Disability Index (ODI) Citation[19], Brief Pain Inventory Short Form (BPI – SF) Citation[20-22], MOS 36-item short form (SF-36) Citation[23], “pain right now” scores collected at the time of supplemental pain medication use and measured on the same 11-point scale as the primary efficacy variable, and supplemental analgesic use.
2.5 Safety assessments
Safety assessments included AEs, clinical laboratory test results, vital sign measurements, physical examinations, and electrocardiogram (ECG) findings. Opioid withdrawal was defined as AEs of withdrawal as specified by the DSM-IV criteria and by the Standardized MedDRA Query (SMQ) subcategory of drug withdrawal. It was assessed during the conversion of incoming pain regimen to HYD (the first 2 weeks of the run-in period) and during the cessation of HYD (among patients receiving placebo during the first 2 weeks of the double-blind period or within 2 weeks of discontinuation of HYD). Additionally, the Clinical Opiate Withdrawal Scale (COWS) and the Modified Subjective Opiate Withdrawal Scale (SOWS) scores Citation[24] were used to assess opioid withdrawal for opioid-experienced patients during the open-label run-in period and for all patients during the double-blind period. The Screener and Opioid Assessment for Patients With Pain – Revised Citation[25], Addiction Behavior Checklist Citation[26], and Current Opioid Misuse Measure Citation[27,28] questionnaires were used to evaluate the risk and development of abuse, misuse, diversion, and other aberrant drug behaviors.
Cases of permanent hearing loss and hearing impairment had been reported in patients taking hydrocodone/acetaminophen combination analgesics, especially in overdose situations Citation[29-32]. In vitro investigations with hair cell culture Citation[33] and epidemiologic studies Citation[34,35] suggested that loss of hearing may be primarily attributable to APAP. Whether hydrocodone by itself caused hearing impairment had not been established. As a result, comprehensive audiologic assessments were conducted by licensed audiologists to evaluate the potential impact of HYD treatment on patients’ hearing functions in this trial. Assessments included bilateral pure-tone air conduction threshold audiometry, bilateral pure-tone bone conduction threshold audiometry Citation[36-38], speech reception threshold Citation[39], immittance audiometry (tympanometry), word recognition, and assessments using the Dizziness Handicap Inventory Citation[40-42] and the Tinnitus Handicap Inventory Citation[43,44]. Audiology assessments were evaluated using American Speech-Hearing Association (ASHA) criteria Citation[45].
2.6 Statistical analysis
All analyses were prespecified. Assuming a 2-sided significance level of 0.05, a desired detectable treatment difference of 0.70 between treatment means for the week 12 mean pain intensity score, a common within-treatment variance of 6.0 (an assumption that corresponded to a standard deviation of 2.45 and an effect size of 0.7/2.45 = 0.286), a 2-sided t-test had 90% power when the sample size was 259 patients per treatment group. This study was planned to randomize 300 patients per treatment arm, for an overall number of 600 patients to be randomized to double-blind treatment, which provided better than 80% power under different scenarios considered to account for early discontinuation of study treatment, and for treatment difference whether only on-study drug data were used (primary analysis) or retrieved dropout data were included (sensitivity analyses).
The enrolled population included all individuals who signed the informed consent form. The safety population was the group of patients who received ≥ 1 dose of study drug during the study. The randomized safety and full analysis populations were synonymous: the group of patients who were randomized and received ≥ 1 dose of double-blind study drug.
Efficacy analyses were based on the full analysis population according to the treatment assigned to each patient. Weekly mean pain intensity scores were calculated (from the daily diary “average pain over the last 24 h” scores) for each week of the double-blind period. Analysis of the weekly mean pain intensity scores was performed using a mixed effects model with repeated measures (MMRM) incorporating a pattern mixture model (PMM) framework to account for missing data, and using restricted maximum likelihood estimation. The current imputation approach assumed data not missing at random (NMAR) and employed different pre-specified imputation techniques for study withdrawals due to adverse events versus other reasons. This approach was an adaptation of a hybrid single imputation approach typically adopted for efficacy analysis of similar trials previously. The advantage of the current strategy is that it had more flexibility in handling different reasons for and timings of withdrawal and consequently the possible relationship between missing data and the outcome of interest.
The MMRM included treatment (2 levels: HYD or placebo), time (14 levels for weeks −7, 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, and 12), and opioid experience status (naive or experienced) as fixed effects. The screening and prerandomization mean pain scores were incorporated in the model as the weeks −7 and 0 values for the dependent variable. The primary efficacy comparison was based on the estimated treatment group means for week 12 based on the model using data collected while patients received study drug. Hypothesis tests were two-sided using α = 0.05 and CIs had 95% coverage probability. Responder analyses were conducted to assess patients who experienced improvements of pain scores of 30 and 50% from screening to week 12 Citation[46]. For all efficacy analyses, placebo values included patients who were randomized to placebo but who may have been exposed to HYD during the taper period in the double-blind period.
Four sensitivity analyses of the primary endpoint were conducted. The NMAR – All Observed Data analysis counted retrieved dropout patients as completers. The Missing at Random (MAR) – Observed Data on study drug analysis treated missing data as MAR, and used a standard MMRM on data observed while the patients were exposed to double-blind study drug. The Partial AE Penalty analysis replaced the screening mean pain value in the PMM formula specified in the primary endpoint analysis with the average of prerandomization and screening estimates for those patients in either treatment arm who withdrew due to AEs. The Hybrid BOCF/LOCF analysis used the MMRM model with all observed data on study drug and imputed data after study drug discontinuation using a hybrid imputation method: a baseline (i.e., screening) observation carried forward (BOCF) method was used for each patient who discontinued due to an AE or ASHA-related event, and the last observation carried forward (LOCF) was used for each patient who discontinued for any other reason than an AE. These analytical methods are consistent with recently published recommendations by the National Research Council Committee on National Statistics Citation[47].
Safety variables were summarized for the safety population and for the randomized safety population by treatment group.
3. Results
3.1 Patient characteristics
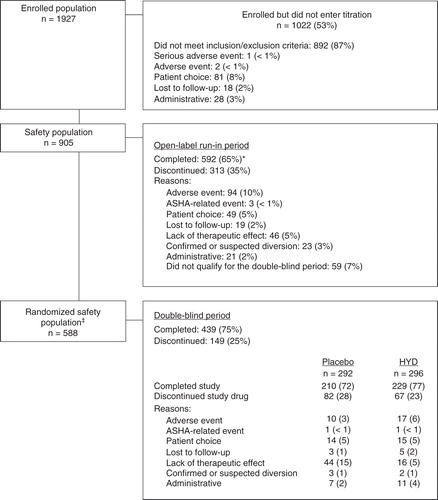
A total of 102 study sites in the United States were initiated for the study; 94 of these sites screened patients; 89 of these sites had patients entered into the open-label run-in period; and 76 of these sites had patients randomized into the double-blind period. Of 905 patients who entered the open-label run-in period, 592 (65%) completed it (). Of 588 patients who entered the double-blind period, 439 (75%) completed it (72% in the placebo group and 77% in the HYD group). The most common reasons for study drug discontinuation during the open-label run-in period were adverse events (10%), not qualifying for the double-blind period (7%), lack of therapeutic effect (5%), and patient choice (5%). During the double-blind period, the most common reasons for study drug discontinuation among patients receiving HYD were adverse events (6%), lack of therapeutic effect (5%), and patient choice (5%); among patients receiving placebo, the most common reasons were lack of therapeutic effects (15%), patient choice (5%), and adverse events (3%).
Figure 2. Patient disposition.
In general, patient characteristics, including demographics, screening pain intensity, medical history, and prior medication use were similar between the nonrandomized and randomized patients, and balanced between double-blind treatment groups (,, and ). Compared to the nonrandomized population, there was a slightly higher proportion of opioid-naive patients and a lower frequency of psychiatric disorders (e.g., depression, anxiety, and insomnia) in the randomized population.
Table 1. Demographic and screening characteristics, safety, and randomized safety populations.
Table 2. Incidence of medical history terms ≥ 15%, safety, and randomized safety populations.
Table 3. Prior opioid medications used for low back pain ≥ 3%, safety, and randomized safety populations.
Table 4. Prior nonopioid medications used for low back pain ≥ 3%, safety, and randomized safety populations.
3.2 Efficacy
3.2.1 Primary efficacy analysis
The primary efficacy analysis demonstrated analgesic efficacy in favor of HYD over placebo ( and ). Substantial pain reduction was achieved with open label HYD by the end of titration. After randomization, pain rebounded in the placebo-treated group, and to a lesser extent, in the HYD-treated group. The maximum treatment group separation was established by week 4 and maintained through week 12. At week 12 of the double-blind period, the difference in pain scores between treatment groups was statistically significant (–0.53, p = 0.0016). All sensitivity analyses were statistically significant and supported the primary analysis (), with the estimated treatment effects similar to that of the primary analysis.
Figure 3. Primary endpoint throughout the study.
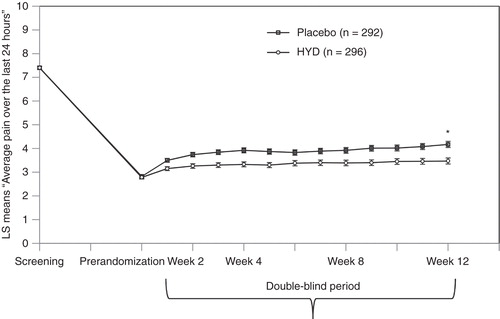
Figure 4. Primary analysis at week 12 of the double-blind period: LS mean difference was –0.53. Mixed model repeated measures with pattern mixture model.
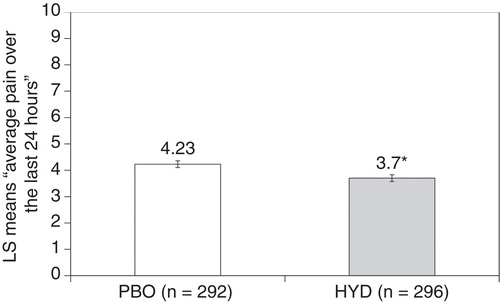
Table 5. Analysis of the primary efficacy variable at week 12.
3.2.2 Secondary efficacy analyses
Compared with patients receiving placebo, greater proportions of patients receiving HYD reported a reduction in pain intensity from screening to week 12 of ≥ 30% (65 vs 53%, p = 0.0033) and ≥ 50% (48 vs 39%, p = 0.0225; ). The proportion of patients reporting “very much improved” and “much improved” on the PGIC rating scale was significantly higher (61%) in the HYD treatment group compared to the placebo treatment group (49%, p = 0.0036; ). Improvements in sleep disturbance were observed during the open-label run-in period with HYD treatment, but a comparison between double-blind treatment groups found that the LS means for weeks 4, 8, and 12 of the double-blind period were not significant (p = 0.3462).
Figure 5. Plot of distribution of responders based on pain intensity at week 12.
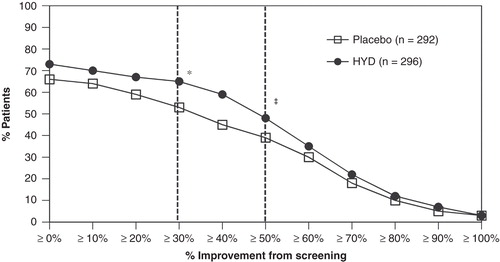
Table 6. Patient global impression of change (PGIC), randomized safety population.
3.2.3 Other (exploratory) efficacy analyses
The results of the analyses of the exploratory efficacy variables (including MOS Sleep-R, ODI, BPI-SF, and SF-36) showed that HYD treatment resulted in improvement from screening in these efficacy measures by the end of the open-label run-in period, but no statistically significant treatment differences between HYD and placebo were seen during the double-blind period (with exception of the BPI pain severity subscale, p = 0.0049). Mean “pain right now” scores during the double-blind period from week 1 through week 12 remained similar between treatment groups (mean scores > 5 for each visit and both treatment groups), indicating that supplemental pain medication was used appropriately when the patient was in pain. Supplemental pain medication use was low and below the protocol-permitted maximum daily doses (mean use of immediate-release oxycodone 5 mg tablets was 0.67 tablet and 0.90 tablet for HYD and placebo, respectively). The proportion of patients requiring no supplemental pain medication during the double-blind period was higher in the HYD group (22%) than that of placebo group (17%), although this difference was not statistically significant (p = 0.1677).
Treatment differences for weekly mean “average pain over the last 24 h” scores during the double-blind period were consistently in favor of HYD across subgroups when analyzed by age, sex, race, and BMI. There was no interaction seen between treatment and each subgroup.
3.3 Safety
Most treatment-emergent adverse events (TEAEs) were mild or moderate in severity. TEAEs that occurred at an incidence of ≥ 5% during the open-label run-in period included, by system organ class and preferred term, gastrointestinal disorders (nausea, vomiting, and constipation) and nervous system disorders (dizziness, headache, and somnolence; ). Among patients receiving HYD, TEAEs that occurred at an incidence of ≥ 5% during the double-blind period were, by system organ class and preferred term, gastrointestinal disorders (nausea [8%] and vomiting [6%]). The only TEAE that occurred with an incidence of ≥ 5% among patients receiving placebo during the double-blind period was nausea (5%). TEAEs for which the incidence was higher in the HYD group than that in the placebo group (difference of ≥ 2%) included nausea, vomiting, and influenza. No dose response relationship was observed between HYD dose levels and the incidence of TEAEs.
Table 7. Incidence of treatment-emergent adverse events ≥ 5%, safety, and randomized safety populations*.
Few patients experienced AEs associated with opioid withdrawal during opioid conversion and during cessation of HYD treatment. During the first 2 weeks of the open-label run-in period, no patient experienced AEs of opioid withdrawal as defined by the SMQ subcategory of drug withdrawal and by the DSM-IV criteria. During the first 2 weeks of the double-blind period, four patients (1%) in the placebo group experienced AEs of opioid withdrawal as defined by SMQ subcategory of drug withdrawal and three patients (1%) in the placebo group experienced AEs of opioid withdrawal as defined by DSM-IV. No patient had a total COWS score ≥ 13 during the study. During the open-label run-in period, 14 (3%) opioid-experienced patients had a total COWS score ≥ 5 and 108 (25%) opioid-experienced patients had a total SOWS score ≥ 10. During the first 2 weeks of the double-blind period, 8 (3%) patients in the placebo group reported a total COWS of ≥ 5 and 58 (20%) patients in the placebo group reported a total SOWS score of ≥ 10.
There were two deaths in the study; one occurred during the open-label run-in period (brain aneurysm) and one occurred in the placebo group during the double-blind period (squamous cell carcinoma of the lung). Both deaths were caused by concurrent medical events and not attributed to study drug by the investigators. A total of 10 patients developed treatment-emergent nonfatal SAEs during the study, 4 patients (7 events) in the open-label run-in period and 6 patients (10 events) during the double-blind period (4 placebo patients and 2 HYD patients). None of the SAEs were considered as possibly, probably, or definitely related to study drug by the investigators. The incidence of TEAEs that led to study drug discontinuation was 11% in the open-label run-in period and 4% in the double-blind period (4% in the HYD treatment group and 3% in the placebo treatment group).
Analysis of laboratory tests, vital signs, and ECG results revealed no new safety concerns during the study. Few patients (≤ 1%) experienced AEs associated with opioid withdrawal during opioid conversion or during cessation of HYD treatment. The comprehensive audiology evaluation found no signal of ototoxicity with HYD.
4. Discussion
CLBP is often challenging to treat, and this trial’s population likely represented an “undertreated” patient group whose baseline pain score averaged greater than 7 on the 0 – 10 NRS despite usage of a stable regimen of analgesic medicines. These patients had suffered from CLBP for an average of 10 years. The pain treatments at study baseline primarily consisted of NSAIDs and APAP for opioid-naive patients, and short-acting immediate-release opioids (such as hydrocodone/acetaminophen combination therapy, tramadol, and oxycodone/acetaminophen combination therapy) for opioid-experienced patients. Only a small number of patients received ER morphine, oxycodone, or fentanyl patch. The average overall daily dose of opioid was 27 mg at baseline. Many patients received concurrent muscle relaxants. A large portion of the trial patients came into this study with common comorbid conditions associated with chronic pain, including insomnia (20%), depression (17%), and anxiety (14%), suggestive of a challenging subgroup of the trial population that required multi-modal pain management approach. After conversion of all baseline pain regimens to HYD and gradual dose titration during the open-label period, a large portion of the patients (65%) were able to find a stable HYD dose which reduced their pain score substantially (> 4 points, from above 7 down to below 3). These were the patients who were randomized into the double-blind period where efficacy over placebo control was evaluated.
The study employed an enriched enrollment, randomized withdrawal design with a 12-week double-blind treatment period, a standard approach to evaluate the efficacy of a new analgesic or new formulation of an existing analgesic for the chronic pain indication. This type of design ensures that patient’s pain is well controlled prior to randomization, which may result in a relatively smaller treatment effect than non-enriched designs. The primary efficacy analysis demonstrated analgesic efficacy in favor of HYD over placebo. At week 12 of the double-blind period, the difference in pain scores between treatment groups was statistically significant (–0.53, p = 0.0016). This treatment difference was similar to the treatment differences observed with other extended-release mu opioid analgesics in studies of similar design, such as hydrocodone Citation[48], morphine Citation[49], and oxycodone Citation[50]. The robustness of the primary analysis results was supported by four sensitivity analyses that used different approaches for handling missing data (p < 0.01, differences ranged from p = 0.55 to 0.67), two responder analyses (p = 0.0033 and 0.0225), and an analysis of PGIC (p = 0.0036).
Supplemental pain medication allowances (use of immediate-release oxycodone up to one-fourth of the randomized dosage) were more generous in this trial than those seen in comparable trials of ER opioids Citation[48,49,51-53]. Its actual use during this trial was < 1 tablet on average across dose strengths and similar between treatment groups; greater use was observed among those randomized to higher dose strengths, as expected. This suggests that treatment with HYD during the double-blind period was adequate, and that supplemental pain medication usage was not likely to be a confounding factor in the estimated treatment effect for HYD. In contrast, dose-level-related supplemental pain medication use, though limited, may have contributed to the high completion rate for patients receiving placebo. Additionally, the combination of supplemental pain medication use and placebo effect is hypothesized to contribute to the observed effect sizes seen in the primary and responder analyses.
This study enrolled a patient population with a diverse background that was representative of that seen in pain practice in the community setting. The treatment groups were generally balanced with respect to sex, race, ethnicity, screening pain type and intensity, medical history, prior opioid and nonopioid medications, and concomitant medication use. The higher proportion of opioid-naive patients seen in the randomized population may have been an indication that, compared with opioid-experienced patients, disproportionally more opioid-naive patients presented with a deficit in their incoming pain regimen and responded well to opioid treatment. The lower proportion of patients with psychiatric disorders seen in the randomized population (mainly insomnia, depression, and anxiety) may have been a consequence of the stipulation that patients were asked to discontinue all medications used to treat pain (which also included treatments for these psychiatric conditions if they were used to treat pain) before entering the study. Of note, these differences in opioid experience and psychiatric disorders were not observed in a long-term study of HYD that employed a similar dose titration approach but permitted concomitant pain treatments with other medications Citation[54].
Patients in this study were converted to HYD during the open-label run-in period according to a prespecified conversion schedule, in which opioid-naive patients were initiated on the lowest dose (20 mg HYD) and opioid-experienced patients were converted to a reduced dose (∼ 25 – 50% reduction in total daily oxycodone equivalent). Subsequently, patients randomized to placebo had their dosages tapered in a blinded manner during the first 2 weeks of the double-blind period, with a reduction in dose of 25% approximately every 3 days. The appropriateness of both the conversion schema and the taper schema was supported by the low incidence of discontinuation due to lack of therapeutic effect in the HYD treatment group, low amounts of supplemental pain medication used, and the low incidence of opioid withdrawal within 2 weeks of changes to opioid treatment. During the first 2 weeks after conversion of incoming opioid regimen to HYD during the open-label period, no patients experienced AE of opioid withdrawal as defined by the DSM-IV criteria and by the SMQ subcategory of drug withdrawal; during the first 2 weeks of the double-blind period (when patients randomized to placebo were tapered off HYD treatment), three patients in the placebo group experienced AE of opioid withdrawal as defined by DSM-IV and four patients (1%) in the placebo group experienced AEs of opioid withdrawal as defined by SMQ subcategory of drug withdrawal.
HYD was generally well tolerated in this study. The TEAEs experienced by patients during the study were those typically associated with the use of central mu-opioid analgesics. Incidences of TEAEs and TEAEs leading to discontinuation were stable between the open-label run-in period for the overall randomized population (42 and 1%, respectively) and the double-blind period for the HYD treatment group (46 and 4%), indicating that once a stable HYD dose was achieved, tolerability to that dose was sustained. There were no new safety concerns detected during the study. Of note, a comprehensive audiology evaluation found no signal of ototoxicity with HYD.
HYD tablets are formulated with an abuse-deterrent technology (ResistecTM), a proprietary extended-release solid oral dosage platform that imparts hardness and hydrogelling properties to the tablets Citation[55]. These properties are anticipated to deter certain forms of product tampering for the purpose of abuse, such as crushing, chewing, snorting, or injecting the dissolved product. In addition, because HYD is difficult to crush and dissolve, it has the potential to reduce the risk of misuse and/or medication error by well-meaning patients or caregivers. Clinical abuse potential studies with HYD have shown reduced abuse potential compared with immediate-release hydrocodone (solution or powder) Citation[12,13]. The real-world impact of HYD’s abuse-deterrent technology is being assessed through epidemiologic studies.
5. Conclusion
This article represents the results of a double-blind 12-week study evaluating the efficacy and safety of a single-entity, once-daily hydrocodone product (HYD), which is formulated with an abuse-deterrent technology. HYD was shown to be an efficacious treatment for CLBP in this study. HYD was generally well tolerated. There were no new or unexpected safety concerns detected during the study.
Acknowledgments
Results presented in this manuscript have been previously presented in poster form at PAINWeek (Las Vegas, NV, September 3 – 6, 2014) and the American Academy of Pain Management (Phoenix, AZ, September 18 – 21, 2014). The authors wish to thank the principal investigators of this study: DD Altamirano, DO; FO Apantaku, MD; A Arslanian, MD; S Atkinson, MD; SJ Azzam, MD; FL Badar III, MD; JC Barlow, MD; R Beasley, MD; L Beltzer, MD; M Biunno, MD; K Blatt, MD; DG Bramlet, MD; C Breton, MD; E Bretton, MD, CPI; R Brownlow, MD; RJ Bunyak, MD, FACP; LB Chaykin, MD; F Chevres, MD; H Nelson Chipman III, MD; S Dar, MD, CCFP; G Dawson, MD; C DeBusk, MD; MJ Drass, MD; ME Duerden, MD; M Dunn, MD; R Eckert, MD; W Travis Ellison, MD; AW Farr, MD; D Figueroa, MD; DH Flaherty, DO; H Goldfarb, MD; H Goodman, MD; D Grant, MD; D Gruener, MD; ME Hale, MD; MW Harris, DO; DR Hassman, DO; C Herrera, MD, MPH; C Herring, MD; VJ Hershberger, MD; B Herskowitz, MD; E Heurich, DO; W Holloway Jr, MD; H Hong, MD; RE Jackson, MD, R Kalb, MD; JD Hudson, MD; JM Joyce, MD; A Khan, MD; JL Kirstein, MD; RM Kluge, MD; A Klymiuk, MD; JH Kopp, MD; D Krefetz, DO; P Kroll, MD; V Kumar, MD; D Levinsky, MD; LS Levinson, MD; LJ Levy Jr., MD; R Lipetz, DO; P Lodewick, MD; PA Lunseth, MD; S Lynd, MD; HG Mariano Jr., MD; G Martinovsky, MD; JP Maynard, MD; JW McGettigan Jr., MD; LJ McGill, MD; MR McGuire, MD, FACC, FAHA; R Michael, MD; SH Miller, MD; S Modugu, MD; RB Molpus, MD; S Nalamachu, MD; JM North, MD; T Nussdorfer, MD; IL Park, MD; KV Patel, MD; K Pollack, MD; GL Raad, MD; M Raikhel, MD, FAAFP; B Rankin, DO, CPI; LD Reed, PhD, MD; H Resnick, MD; TR Smith, MD; J Soufer, MD; ELH Spierings, MD; RZ Surowitz, DO; LA Taber, MD; GP Tarleton, MD; DR Taylor, MD; M Trevino, MD; BR Vaid, MD; LT Wadsworth III, MD; RJ Wagner, MD; AJ Weil, MD; PJ Winkle, MD, FACP, FACG, CPI; BA Wittmer, DVM, MD, CPI; A Yataco, MD; GD Yeoman, DO; and D Young, MD. The authors also wish to thank Henry Andrew Caporoso, MA, for his assistance in the preparation of this manuscript. The trial registration number for this study is NCT01452529.
Declaration of interest
This study was sponsored by Purdue Pharma L.P. W Wen, S Lynch, E He, S Ripa, and HA Caporoso are full-time employees of Purdue Pharma, L.P. S Sitar was an investigator for this study. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Notes
Bibliography
- Acetaminophen overdose and liver injury – background and options for reducing injury. Available from: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/DrugSafetyandRiskManagementAdvisoryCommittee/UCM164897.pdf
- Medicine use and shifting costs of healthcare: A review of the use of medicines in the united states in 2013 [Internet]. Available from: http://www.imshealth.com/portal/site/imshealth/menuitem.762a961826aad98f53c753c71ad8c22a/?vgnextoid=2684d47626745410VgnVCM10000076192ca2RCRD&vgnextchannel=736de5fda6370410VgnVCM10000076192ca2RCRD&vgnextfmt=default
- U.S. Department of Health and Human Services, Substance Abuse and Mental Health Services Administration. NHSDA Series H-22, DHHS Publication No SMA 03–3836. Office of Applied Studies; Rockville, MD: 2003. Results from the 2002 National Survey on Drug Use and Health: National Findings. Table 1.107 A & B
- Substance Abuse and Mental Health Services Administration, Center for Behavioral Health Services and Quality. 2013 Tables: Illicit Drug Use - 1.47 to 1.92 (PE). Table 1.89A - Nonmedical Use of Specific Pain Relievers in Lifetime, by Age Group: Numbers in Thousands, 2012 and 2013 Available from: http://www.samhsa.gov/data/sites/default/files/NSDUH-DetTabsPDFWHTML2013/Web/HTML/NSDUH-DetTabsSect1peTabs47to92-2013.htm [Last accessed 16 January 2015]
- Throckmorton DC; Re-scheduling prescription hydrocodone combination products. FDA Voice. Available from: http://blogs.fda.gov/fdavoice/index.php/2014/10/re-scheduling-prescription-hydrocodone-combination-drug-products-an-important-step-toward-controlling-misuse-and-abuse [Last accessed 6 October 2014]
- Duh MS, Vekeman F, Korves C, et al. Risk of hepatotoxicity-related hospitalizations among patients treated with opioid/acetaminophen combination prescription pain medications. Pain Med 2010;11(11):1718-25
- Summary minutes of the joint meeting of the drug safety and risk management advisory committee, nonprescription drugs advisory committee, and the anesthetic and life support drugs advisory committee of the U.S. food and drug administration to address the public health problem of liver injury related to the use of acetaminophen in both over-the-counter and prescription products. Held June 29 and 30, 2009 at Adelphi, MD. Available from: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/DrugSafetyandRiskManagementAdvisoryCommittee/UCM179888.pdf
- Acetaminophen overdose. Available from: http://www.nlm.nih.gov/medlineplus/ency/article/002598.htm
- Mort JR, Shiyanbola OO, Ndehi LN, et al. Opioid-paracetamol prescription patterns and liver dysfunction: A retrospective cohort study in a population served by a US health benefits organization. Drug Saf 2011;34(11):1079-88
- FDA drug safety communication: Prescription acetaminophen products to be limited to 325 mg per dosage unit; boxed warning will highlight potential for severe liver failure. Available from: http://www.fda.gov/drugs/drugsafety/ucm239821.htm
- Blieden M, Paramore LC, Shah D, Ben-Joseph R. A perspective on the epidemiology of acetaminophen exposure and toxicity in the United States. Expert Rev Clin Pharmacol 2014;7(3):341-8
- Harris SC, Colucci SV, Perrino P, et al. A single-center, randomized, double-blind, crossover study to evaluate the abuse potential, pharmacokinetics, and safety of oral milled and intact extended-release hydrocodone (HYD) in recreational opioid users [abstract 440]. J Pain 2014;15(4):S86
- Harris SC, Colucci SV, Perrino P, et al. A single-center, randomized, double-blind, crossover study to evaluate the abuse potential, pharmacokinetics, and safety of milled and intranasally administered extended-release hydrocodone (HYD) tablets in recreational opioid users [abstract 440]. J Pain 2014;15(4):S86
- Loisel P, Vachon B, Lemaire J, et al. Discriminative and predictive validity assessment of the Quebec task force classification. Spine (Phila Pa 1976) 2002;27(8):851-7
- Spitzer W. Scientific approach to the assessment and measurement of activity-related spinal disorders. A monograph for clinicians. Report of the Quebec task force on spinal disorders. Spine (Phila Pa 1976) 1987;12(7 Suppl):S1-9
- Werneke MW, Hart DL. Categorizing patients with occupational low back pain by use of the Quebec task force classification system versus pain pattern classification procedures: Discriminant and predictive validity. Phys Ther 2004;84(3):243-54
- Hays R, Stewart A. Sleep measures. In: Stewart A, Ware JJ, editors. Measuring functioning and well-being: the medical outcomes study approach. Duke University Press; Durham, NC: 1992. p. 235-59
- Farrar JT, Young JPJr, LaMoreaux L, et al. Clinical importance of changes in chronic pain intensity measured on an 11-point numerical pain rating scale. Pain 2001;94(2):149-58
- Fairbank JC, Pynsent PB. The oswestry disability index. Spine (Philadelphia PA 1976) 2000;25(22):2940-52; discussion 2952
- Cleeland CS, Ryan KM. Pain assessment: Global use of the brief pain inventory. Ann Acad Med Singapore 1994;23(2):129-38
- Daut RL, Cleeland CS, Flanery RC. Development of the Wisconsin brief pain questionnaire to assess pain in cancer and other diseases. Pain 1983;17(2):197-210
- Hoffman D, Friedman M, Colucci S, et al. Validation of the brief pain inventory for subjects with osteoarthritis. J Pain 2002;3:2 Supp 1
- Ware JEJr. SF-36 health survey update. Spine (Philadelphia, PA 1976) 2000;25(24):3130-9
- Wesson DR, Ling W. The clinical opiate withdrawal scale (COWS). J Psychoactive Drugs 2003;35(2):253-9
- Butler SF, Fernandez K, Benoit C, et al. Validation of the revised screener and opioid assessment for patients with pain (SOAPP-R). J Pain 2008;9(4):360-72
- Wu SM, Compton P, Bolus R, et al. The addiction behaviors checklist: Validation of a new clinician-based measure of inappropriate opioid use in chronic pain. J Pain Symptom Manage 2006;32(4):342-51
- Butler SF, Budman SH, Fanciullo GJ, Jamison RN. Cross validation of the current opioid misuse measure to monitor chronic pain patients on opioid therapy. Clin J Pain 2010;26(9):770-6
- Butler SF, Budman SH, Fernandez KC, et al. Development and validation of the current opioid misuse measure. Pain 2007;130(1-2):144-56
- Ho T, Vrabec JT, Burton AW. Hydrocodone use and sensorineural hearing loss. Pain Physician 2007;10(3):467-72
- Friedman RA, Adir Y, Crenshaw EB, et al. A transgenic insertional inner ear mutation on mouse chromosome 1. Laryngoscope 2000;110(4):489-96
- Oh AK, Ishiyama A, Baloh RW. Deafness associated with abuse of hydrocodone/acetaminophen. Neurology 2000;54(12):2345
- Schweitzer VG, Darrat I, Stach BA, Gray E. Sudden bilateral sensorineural hearing loss following polysubstance narcotic overdose. J Am Acad Audiol 2011;22(4):208-14
- Yorgason JG, Kalinec GM, Luxford WM, et al. Acetaminophen ototoxicity after acetaminophen/hydrocodone abuse: Evidence from two parallel in vitro mouse models. Otolaryngol Head Neck Surg 2010;142(6):814-19
- Curhan SG, Eavey R, Shargorodsky J, Curhan GC. Analgesic use and the risk of hearing loss in men. Am J Med 2010;123(3):231-7
- Curhan SG, Shargorodsky J, Eavey R, Curhan GC. Analgesic use and the risk of hearing loss in women. Am J Epidemiol 2012;176(6):544-54
- Calibration of speech signals delivered via earphones [Internet]. 1987. Available from: http://www.asha.org/policy/RP1987-00019.htm
- Carhart R, Jerger J. Preferred methods for clinical determination of pure-tone thresholds. J Speech Hear Res 1959;24:330-45
- Jerger J. Clinical experience with impendance audiometry. Arch Otolaryngol 1970;92(4):311-24
- Determining threshold level for speech [Internet]. 1988. Available from: www.asha.org/policy/GL1988-00008.htm
- Jacobson GP, Newman CW. The development of the dizziness handicap inventory. Arch Otolaryngol Head Neck Surg 1990;116(4):424-7
- Tamber AL, Wilhelmsen KT, Strand LI. Measurement properties of the dizziness handicap inventory by cross-sectional and longitudinal designs. Health Qual Life Outcomes 2009;7:101
- Whitney SL, Wrisley DM, Brown KE, Furman JM. Is perception of handicap related to functional performance in persons with vestibular dysfunction? Otol Neurotol 2004;25(2):139-43
- McCombe A, Baguley D, Coles R, et al. Guidelines for the grading of tinnitus severity: The results of a working group commissioned by the British association of otolaryngologists, head and neck surgeons, 1999. Clin Otolaryngol Allied Sci 2001;26(5):388-93
- Newman CW, Jacobson GP, Spitzer JB. Development of the tinnitus handicap inventory. Arch Otolaryngol Head Neck Surg 1996;122(2):143-8
- Clark JG. Uses and abuses of hearing loss classification. ASHA 1981;23(7):493-500
- Farrar JT, Dworkin RH, Max MB. Use of the cumulative proportion of responders analysis graph to present pain data over a range of cut-off points: Making clinical trial data more understandable. J Pain Symptom Manage 2006;31(4):369-77
- National Research Council. The prevention and treatment of missing data in clinical trials. Panel on handling missing data in clinical trials. Committee on national statistics, division of behavioral and social sciences and education. The National Academies Press; Washington, DC: 2010
- Rauck RL, Nalamachu S, Wild JE, et al. Single-entity hydrocodone extended-release capsules in opioid-tolerant subjects with moderate-to-severe chronic low back pain: A randomized double-blind, placebo-controlled study. Pain Med 2014;15(6):975-85
- Katz N, Hale M, Morris D, Stauffer J. Morphine sulfate and naltrexone hydrochloride extended release capsules in patients with chronic osteoarthritis pain. Postgrad Med 2010;122(4):112-28
- Friedmann N, Klutzaritz V, Webster L. Efficacy and safety of an extended-release oxycodone (Remoxy) formulation in patients with moderate to severe osteoarthritic pain. J Opioid Manag 2011;7(3):193-202
- Hale ME, Dvergsten C, Gimbel J. Efficacy and safety of oxymorphone extended release in chronic low back pain: Results of a randomized, double-blind, placebo- and active-controlled phase III study. J Pain 2005;6(1):21-8
- Hale ME, Ahdieh H, Ma T, et al. Efficacy and safety of OPANA ER (oxymorphone extended release) for relief of moderate to severe chronic low back pain in opioid-experienced patients: A 12-week, randomized, double-blind, placebo-controlled study. J Pain 2007;8(2):175-84
- Katz N, Rauck R, Ahdieh H, et al. A 12-week, randomized, placebo-controlled trial assessing the safety and efficacy of oxymorphone extended release for opioid-naive patients with chronic low back pain. Curr Med Res Opin 2007;23(1):117-28
- Wen W, Taber L, Lynch S, et al. 12-month safety and effectiveness of once-daily hydrocodone tables formulated with abuse-deterrent properties in patients with moderate to severe chronic pain. J Opioid Manag; In press
- Hysingla ER. [package insert]. Purdue Pharma L.P; Stamford, CT: 2014