
Abstract
Technology often serves as a handmaiden and catalyst of invention. The discovery of safe, effective medications depends critically upon experimental approaches capable of providing high-impact information on the biological effects of drug candidates early in the discovery pipeline. This information can enable reliable lead identification, pharmacological compound differentiation and successful translation of research output into clinically useful therapeutics. The shallow preclinical profiling of candidate compounds promulgates a minimalistic understanding of their biological effects and undermines the level of value creation necessary for finding quality leads worth moving forward within the development pipeline with efficiency and prognostic reliability sufficient to help remediate the current pharma-industry productivity drought. Three specific technologies discussed herein, in addition to experimental areas intimately associated with contemporary drug discovery, appear to hold particular promise for strengthening the preclinical valuation of drug candidates by deepening lead characterization. These are: i) hydrogen–deuterium exchange mass spectrometry for characterizing structural and ligand-interaction dynamics of disease-relevant proteins; ii) activity-based chemoproteomics for profiling the functional diversity of mammalian proteomes; and iii) nuclease-mediated precision gene editing for developing more translatable cellular and in vivo models of human diseases. When applied in an informed manner congruent with the clinical understanding of disease processes, technologies such as these that span levels of biological organization can serve as valuable enablers of drug discovery and potentially contribute to reducing the current, unacceptably high rates of compound clinical failure.
1. Technology informs and enables translational drug innovation
Multiple challenges blanket the pharmaceutical industry under darkening clouds of inefficiency and instability as staggering development costs and moribund drug-approval rates erode returns on the immense up-front time and capital investments required for therapeutics invention Citation[1-3]. Many unsolved medical problems persist for which safe, effective, population-based therapies are lacking, particularly with regard to complex, multifactorial diseases whose etiologies and therapeutic outcomes involve dynamic interplay among myriad environmental and genetic factors and biological signaling pathways, often across multiple organs/organ systems Citation[4]. Pharma-industry’s challenges and opportunities have made the identification and characterization of agents that produce a desired salutary effect on a disease process a central focus of preclinical drug discovery in order to triage and optimize those very rare advanced leads worthy of human testing. The view has been espoused that shallow preclinical profiling of drug leads has decisively undermined sustainable growth potential in the pharmaceutical industry across therapeutic areas and indications. Myopic appreciation of the biological properties of lead drug candidates, in turn, can jeopardize product innovation and limit value creation by sanctioning leads of insufficient quality, validity and translational promise for pipeline advancement and entry into clinical trials Citation[5]. Inadequate preclinical lead characterization is, therefore, a likely contributor to the unprecedentedly high failure rates of investigational drugs despite the increasing numbers of compounds in development and burgeoning total research and development (R&D) expenditures Citation[6].
These circumstances suggest the need to raise the quality standards for development leads and strengthen lead-validation packages through more comprehensive and disease-relevant biological profiling. Although this view may appear to position medicinal chemistry ancillary to pharmacologial drug-discovery endeavors, the author considers that synthetic chemistry is operationally better defined and standardized than is biological profiling, the discovery ideal represented by coherence between medicinal chemistry and biological/pharmacological research as informed by the clinic. Arguably, then, a major – if not decisive – value-added proposition in preclinical drug discovery comes from thorough and rigorous compound characterization in disease-relevant biological systems, information from which should iteratively inform allied medicinal chemistry efforts. Toward this end, technological advances affording more comprehensive, in-depth candidate characterization have been demonstrated to empower preclinical discovery efforts Citation[7].
From this vantage point and the author’s R&D experience of over three decades in discovery-related activities within both public and private sectors that generated numerous development leads and clinical candidates as well as approved therapeutics for diverse indications, this commentary will discuss three specific technologies that, in the author’s opinion, have demonstrated promise for enriching the innovation economy of drug development by improving compound profiling and enhancing our understanding of disease mechanisms. Since the technologies considered are themselves the subjects of refinement and scope and application extensions, their ultimate impact on therapeutics innovation cannot be definitively gauged at present. The primary purpose of this presentation, therefore, is to exemplify and promote awareness of emerging technologies for their potential to enrich the preclinical profiling of drug leads and strengthen lead validation in ways directly relevant to evaluating therapeutically attractive drug candidates. Small-molecule discovery aimed at modulating protein (enzyme, receptor) function will be emphasized, since research along this line has generated the majority of known therapeutics and is foundational to most contemporary discovery efforts Citation[1,3].
2. Dynamic structural biology of disease-related proteins: hydrogen–deuterium exchange mass spectrometry
The 2012 Chemistry Nobel Prizes well illustrate the appreciable research and translational significance of ‘chemical pharmacology,’ especially structure–function correlates of protein modulation by therapeutic ligands Citation[8]. Three-dimensional crystal structures of disease-relevant proteins have aided understanding of their interaction profiles with small-molecule drugs, thereby informing structure-led drug design Citation[9]. Recent methodological advances in (especially membrane) protein isolation and crystallization, X-ray sources and allied instrumentation, and data handling auger well for future crystallographic protein analyses Citation[10]. Nonetheless, unease exists as to the fidelity with which the conformation of an isolated, crystalline protein is able to represent that protein’s native structure and the degree to which even a suite of non-contradictory static X-ray maps per se can reflect the diversity of a protein’s conformational repertoire Citation[11]. The concern is underscored by the important roles that protein structural transitions play in the therapeutic action of drugs on receptor-mediated information transmission and enzyme catalysis Citation[12,13].
During the past two decades, the technique of peptide-level hydrogen–deuterium exchange (HDX) mass spectrometry (MS) (HDX-MS) has met with increasing application in the experimental interrogation of higher-order protein conformation and structural dynamics. As detailed in comprehensive historical and technical reviews Citation[14-16] and schematized (), HDX-MS leverages two phenomena: i) the unique, fundamental property of protein backbone amide hydrogen atoms to exchange with solvent deuterons from ‘heavy’ water (i.e., D2O) in real time under physiological conditions; and ii) the ability of MS to quantify the protein mass so increased. Under set conditions of pH and temperature, the pattern of hydrogen exchange reflects the degree of solvent accessibility and intramolecular hydrogen bonding in a conformationally and structurally sensitive manner. Generally, solvent-exposed and highly dynamic protein regions (e.g., unstructured domains interposed between helices or loops) will exchange rapidly. Domains involved in hydrogen-bonding networks or within the protein’s interior (e.g., buried α-helices or β-sheets) tend to be less dynamic and exchange more slowly. Alteration in the location and/or rate of deuterium incorporation signals a motional change in solvent accessibility and/or hydrogen bonding in the protein. The basic HDX-MS technology may be augmented with sophisticated MS techniques to enhance spatial (i.e., from short peptide regions of the protein [] to the single amino acid level) and temporal (i.e., in the sub-second time frame) resolution of the deuteration profile Citation[17-19].
Figure 1. Diagrammatic illustration of prototypical HDX-MS analyses. Under physiological conditions (temperature, pH), a protein sample in compatible buffer is diluted with the same buffer containing 99.9% ‘heavy’ water (D2O) in place of H2O. Deuteration of the intact protein is allowed to proceed for various durations prior to quenching by rapid acidification to pH 2.5 and cooling to 0°C. The intact deuterated protein may either be directly injected into the mass spectrometer or digested under quench conditions with an acid protease prior to UPLC of the resulting peptide hydrolysate at 0°C (to restrict deuterium back exchange) followed by peptide-level mass analysis by MS. The mass spectra allow determination of the kinetics of deuterium uptake by the protein and/or its peptic peptides, the latter identifiable by MS/MS of the protein itself.
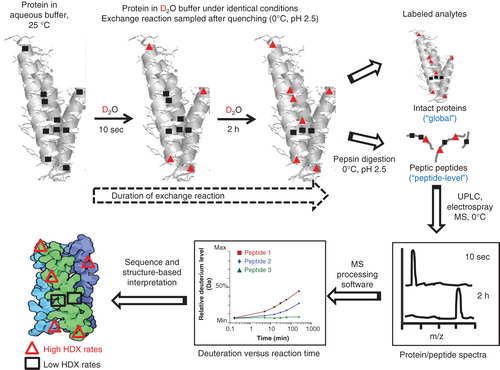
HDX-MS has enabled experimenter access to details on the intrinsic structures and conformational dynamics of a range of proteins and the functional implications of their structural properties/dynamics Citation[16,20,21]. Recent specific examples of discovery-related HDX-MS demonstrate that HDX-MS can afford unique insights into the function and dynamics of disease-relevant protein targets and protein therapeutics unobtainable by other structural biology techniques () Citation[22-31]. In these regards, HDX-MS was key to obtaining the first definitive experimental corroboration of implications from molecular-dynamics simulations and energy landscape theory that local unfolding guides intermediate structural trajectories among active and inactive receptor conformations. Existing crystal structures of the druggable protein studied (the EGFR) did not correlate with the simulated intermediate protein conformations Citation[21]. HDX-MS enabled definition of structural elements essential to selective therapeutic-ligand recognition by pathologically relevant proteins and informed thereby the design and optimization of novel enzyme and receptor modulators with improved potency, selectivity and/or functional specificity – properties often decisive to the pipeline viability of new chemical entities as leads toward clinical candidacy. Regional changes in DOT1-like, histone H3 methyltransferase upon binding of a potent, highly selective inhibitor apparent by HDX-MS, but not from crystals of the liganded enzyme, informed medicinal chemistry efforts by suggesting specific chemical modifications that improved the inhibitor’s potency and pharmacokinetics to realize a potential therapeutic Citation[24]. Given the mounting interest in functionally selective or ‘biased’ drugs whose molecular pharmacology is capable of routing information output away from signaling pathways that elicit adverse events and toward those that are most therapeutically relevant Citation[32], HDX-MS has been used to identify and distinguish ligand-induced receptor conformational responses associated with distinct molecular pharmacologies (e.g., receptor agonist vs antagonist vs inverse agonist) Citation[28,29,33] and modes of ligand engagement (e.g., covalent vs noncovalent enzyme inhibitors) Citation[22] that are therapeutically beneficial. In these studies Citation[22,28,29,33], HDX-MS analyses enabled definition of the molecular determinants of the interactions between ligands as candidate drugs and therapeutic targets, data that helped tune the resulting information output to salutary ends. This information positively impacted the design of novel ligands that modulate in a therapeutically beneficial manner the function of key receptors and enzymes associated with diseases representing significant unmet medical needs and major unsolved health problems ().
Table 1. Exemplary discovery-related hydrogen–deuterium exchange mass spectrometry studies focused on disease-related proteins or protein therapeutics.
3. Functional annotation of mammalian proteomes: activity-based enzyme chemoproteomics
Enzymes catalyze discrete biological reactions that allow molecular transformations to take place within time frames compatible with the physiological activities and homeostasis essential to living systems. Modulation of enzyme function is a principal mode of (especially small-molecule) drug action, making the identification of pathologically significant enzymes and validation of their therapeutic relevance prominent, long-standing concerns of drug discovery and development Citation[34]. These considerations lend great importance to the annotation of enzyme function in health and disease and definition of the interaction specificity or promiscuity of designer small-molecule modulators with enzymes, the significance of which is likely to intensify, given our deepening appreciation of the complexities that underlie enzymatic regulation of metabolic pathways and information networks key to disease etiology and treatment Citation[35]. Specifically, detailed knowledge of the interaction spectrum of a potential drug candidate within a cellular/tissue proteome would be of great value in uncovering unwanted off-target interactions and as indicator of target/signaling pathway selectivity, information that can help guide medicinal chemistry efforts for optimizing these properties in a lead compound to therapeutic ends.
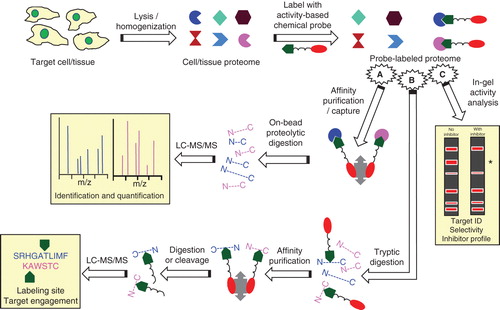
For these purposes, activity-based protein profiling (ABPP) is being embraced as a proteomic platform for chemically interrogating and quantifying the functional state of enzyme families in native biological samples. As detailed in reviews Citation[36-38] and illustrated in , prototypic ABPP technology utilizes active site-directed covalent probes that target a specific enzyme subset in a cell/tissue proteome characterized by shared mechanistic features underlying catalytic activity. Probe design incorporates a reactive ‘warhead’ (e.g., an electrophilic or photoreactive group) to label covalently active enzymes linked to an analytical (e.g., fluorophore or affinity) tag for detection and/or enrichment of the probe-labeled enzymes. The warhead binding group is designed to direct the probe toward select enzyme classes. Probe design allows for wide variations in the stringency of probe specificity among enzymes that share a similar catalytic mechanism and/or substrate selectivity. The resultant, probe-labeled proteome may then be analyzed in various ways. After affinity purification by exploiting a suitable analytical tag on the probe (e.g., with avidin beads for a biotin-functionalized probe and on-bead digestion), the targets of ABPP probe engagement can be quantified and identified by multidimensional liquid chromatography–tandem mass spectrometry (LC–MS/MS) with MS protein-sequence algorithm software. Alternatively, the probe-labeled proteome can be digested, and the labeled peptides affinity purified, cleaved from the affinity matrix and subjected to LC–MS/MS to identify directly the sites of probe labeling and target engagement in the tagged enzyme(s). Another ABPP variant, ‘competitive ABPP,’ can be used to screen for enzyme inhibitors by incubating the proteome under study with a putative inhibitor prior to introducing a fluorescent ABPP probe. Comparison by PAGE with a parallel sample reacted with the ABPP probe allows for quantitative in-gel activity analysis by virtue of the reduction in fluorescence band intensity at the inhibited enzyme(s), affording rapid identification of the inhibitor’s target(s) [and off-targets(s)] ().
Figure 2. Schematic outline of prototypical ABPP enzyme analyses. A cell/tissue proteome is incubated with a chemical probe () bearing both a warhead designed to react covalently with catalytically active enzymes in a specific enzyme/enzyme class (
) and a fluorophore or affinity moiety (
). The probe-labeled proteome can then be subjected to various analyses, including: (A) isolation of the probe-labeled proteins on affinity beads followed by on-bead enzymatic digestion and LC–MS/MS analysis to identify the modified proteins; (B) tryptic digestion of the labeled proteome followed by affinity purification, enzymatic digestion or cleavage, and LC–MS/MS to identify the sites of probe modification/target engagement at the amino acid level; (C) ‘competitive ABPP’ involving electrophoretic resolution of the proteome on polyacrylamide gels and in-gel fluorophore detection of the labeled proteins (red bands). Prior incubation of the proteome with a putative enzyme inhibitor (‘with inhibitor’) will block the inhibited enzyme’s reaction with the ABPP probe and obviate fluorescence in that band as compared to the proteome sample not previously treated with putative enzyme inhibitor (‘no inhibitor’), as indicated by the *symbol to the right of the electrophoretograms, affording profiling of the inhibitor’s target and selectivity within the proteome studied.
ABPP technology has allowed initial functional and mechanistic characterization of enzymes, elucidation of the roles of enzymes in metabolic control and cell-signaling information pathways, and definition of the influence of natural products/metabolites on enzyme activity Citation[39-44]. With regard to drug discovery, ABPP aided identification of several druggable enzymes of pathological relevance and development of novel, therapeutically promising enzyme inhibitors: Competitive ABPP played a critical role in demonstrating the involvement of monoacylglycerol lipase (MGL) in aggressive cancer and designing novel, selective MGL inhibitors that show preclinical efficacy against oncological end points in vitro and in vivo Citation[45,46]. Although excessive myeloperoxidase (MPO) activity has been implicated in the etiology of several diseases (e.g., rheumatoid arthritis, atherosclerosis, Parkinson’s disease), druggable MPO inhibitors have proven elusive. ABPP recently enabled the first identification of a mechanism-based MPO inhibitor with sufficient selectivity to serve as a tool compound for studies of the role of MPO in disease and as a template for rational drug design Citation[47]. Competitive ABPP proved instrumental in identifying novel carboxylesterase 3 (Ces3) inhibitors effective preclinically against metabolic syndrome in vivo Citation[48], thereby substantiating murine gene-modification data implicating Ces3 as a driver of pathogenic lipid accumulation associated with obesity and diabetes Citation[49] and pointing the way to pharmacological inhibition of the mouse enzyme’s human ortholog, Ces1, as a therapeutic modality for metabolic disease. Perhaps of greater overall significance for enabling drug discovery, ABPP applied to a preliminary cell-based (adipocyte) screen of a compound library allowed unambiguous identification of Ces3 as the enzyme whose inhibition was responsible for the salutary metabolic effects observed Citation[48], demonstrating a clear niche for ABPP in bridging target-based and phenotypic discovery approaches for annotating and deconvoluting disease-causing enzymes, finding directed therapeutic modulators with high confidence, and illuminating disease mechanisms.
4. Modeling complex human diseases: precision genome editor technologies
Living systems that display phenotypic features of a human disease have made uncontestable contributions to new therapeutics development. Yet cellular and rodent disease models conventionally used for drug discovery are intrinsically limited in scope and relevance. The range of standard immortalized cell lines and human cells able to be sampled noninvasively and studied in primary culture hardly begins to approach the variety of cell types involved in human disease. Likewise, decades of pharmacological data from murine disease models (commonly, single-gene-disruption ‘knock-out’ animals) have underscored the myriad metabolic, biochemical and physiological differences between rodents and humans that render such systems far from translationally optimal Citation[50]. The prognostic value of the current stock of cell- and animal-based systems has been considered sufficiently tenuous to engender increasingly noisome concern regarding the reliability with which results from extant disease models may be translated to humans Citation[51]. This situation invites improved experimental disease models for preclinical drug profiling that allow extrapolation to the clinic with heightened confidence for predicting the clinical outcome and human impact of potential diagnostic stratagems and therapies Citation[52]. In particular, living systems that display multiple contributors to disease etiology rather than one single characteristic of a human-disease phenotype should be most useful for deepening our understanding of mechanisms underlying therapeutic activity, drug–drug interactions and adverse-event profiles.
Beyond attempts at deriving curative, gene- and cell-based therapies and augmenting production-agriculture yields Citation[53,54], precision genome editing (PGE) has greatly enhanced the prospect of improved, tailor-made models of (especially multifactorial) human diseases. Nuclease-mediated PGE technology enables targeted introduction of discrete, single or multiple disease-related gene modifications into the chromosomal DNA of human cells and laboratory animals (including nonhuman primates), endowing these living systems with disease-specific signatures Citation[54,55]. The historical and technological details of genome modification through nuclease-mediated PGE are available in print overviews Citation[56-59] and on video (http://www.youtube.com/watch?v=zDkUFzZoQAs). Essentially, the most popular nuclease-mediated PGE technologies utilize one of three gene-editing enzymes – zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) or clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated (Cas) nuclease-based RNA-guided DNA endonucleases (also known as the type-II CRISPR system) – as ‘molecular scissors.’ The resulting double-strand DNA breaks recruit endogenous DNA damage-repair mechanisms that either induce local mutations or stimulate homologous recombination with exogenously provided donor DNA. Site-specificity in the ZFN and TALEN systems relies upon custom-made, programmable DNA-binding protein domains, whereas the CRISPR-Cas9 system attains specificity by virtue of an engineered RNA that guides the endonuclease. These PGE platforms enable custom, predetermined DNA sequence changes within natural chromosomal contexts at defined genomic sites as well as transgene addition at specific genomic loci virtually anywhere in complex genomes with a level of control allowing introduction of both single and multiple disease-relevant point mutations.
As demonstrated in several proof-of-principle studies discussed elsewhere Citation[59-62], PGE nuclease tools have been used to introduce targeted gene modifications into diverse cell types (including primary cells, [transformed human] cell lines, and adult or embryonic human stem cells) and animal species routinely (e.g., rat, mouse, hamster, zebra fish) and less frequently (e.g., pigs, monkeys, marmosets) employed in laboratory research. With respect to pathologically relevant cell systems, nuclease-based PGE has opened revolutionary possibilities in hereditable cellular reprogramming for introducing discrete disease mutations into (human) cells that can then be differentiated in culture to display aberrant phenotypic manifestations indicative of a specific disease Citation[63-72]. Particularly valuable in (patient-specific) disease situations where relevant human tissue sampling/in vivo experimentation is impractical or ethically questionable (e.g., many neurodegenerative syndromes Citation[63]), the resultant cell-based ‘disease-/patient-in-a-dish’ systems have already helped inform drug discovery in several ways: screening for and profiling (potential) therapies and mechanisms of drug resistance Citation[65,67,70,71]; selecting and monitoring pathologically relevant therapeutic targets Citation[63-65,67]; answering mechanistic questions of disease pathophysiology Citation[63-65,69]; and determining the effects of genetic contributions and drug sensitivities on disease phenotypes/human genetic disorders Citation[63,66,68] (). Likewise, mutations introduced into various laboratory animals by nuclease-mediated PGE have enabled construction of in vivo models of human diseases involving single or multiple mutations that have been used for key discovery-related activities, including profiling of potential therapeutics, identification of the genetic basis of disease and definition of the causal relationships between diseases and human gene variants identified in genome-wide association studies () Citation[73-80]. This last application of nuclease-mediated PGE is particularly intriguing for therapeutics invention, since it represents a potentially more relevant and informative ‘bedside-to-bench’ approach distinct from the typical, marketing-driven ‘bench-to-bedside’ discovery route.
Table 2. Exemplary discovery-related cell and animal systems generated through nuclease-mediated precision genome editing.
5. Conclusion
Research is characterized by change and unpredictability, both generally and with regard to the technology marketplace. HDX-MS, ABPP and targetable, nuclease-mediated PGE exemplify progressive technologies whose current reach and information yield position them as increasingly important and useful architects of the evolving discovery landscape and portend even more significant contributions to lead identification/characterization in drug discovery.
6. Expert opinion
Insights already afforded by the relatively new technologies of HDX-MS, ABPP and targetable nuclease-mediated PGE do not allow their dismissal by the discovery community as mere academic ‘solutions looking for problems’ Citation[81]. Rather, the author opines that these evolving technologies will continue to strengthen and diversify drug discovery links with the disciplines of structural biology, chemical proteomics and functional genomics to enhance our understanding of what potential therapeutics do and how they act in more disease-relevant experimental contexts prior to the huge investment ramp-up mandated by human studies and so often lost along the clinical development pathway. HDX-MS, ABPP and targetable nuclease-mediated PGE can be applied to interrogate biological pathways and information systems (e.g., proteomes, disease and drug-sensitive networks, functional genome-wide screening) in multiplex fashion, since they span across levels of biological organization (from molecules to mammals). This feature opens avenues beyond the reductionist strictures of target-focused pharmacophoric/chemotype searching, an ingrained practice often driven more by feasibility than by evidence of its being a route to competitive advantage in drug discovery. This proposition is enticing, given that integrative, systems-based phenotypic screening was the source of most first-in-class small-molecule medications approved between 1999 and 2008 Citation[1]. The fact that insufficient efficacy was the most frequent cause of Phase II clinical failures occurring during 2008 – 2011 and regulatory rejection of new molecular entities by the United States FDA during 2000 – 2012 Citation[82,83] strongly suggests that limitations of established disease models within canonical, target-based discovery paradigms have jeopardized clinical success rates by providing candidate identification/characterization insufficient for clinical development Citation[84]. By extension, enhanced understanding of therapeutically relevant biology and pharmacology of lead compounds as afforded by HDX-MS and ABPP in the preclinical context of pathologically relevant, PGE-derived disease models should help inform critical-path ‘go/no-go’ decision-making around compound attributes (e.g., efficacy, selectivity, mode of action, off-target interactions) important to populating discovery pipelines, assessing the potential for lead advancement into development, and reducing the inherently high risk associated with drug innovation.
The author posits that continued advances along these and other lines will intensify ongoing efforts to broaden the range of applications for HDX-MS, ABPP and nuclease-mediated PGE and will enhance the benefits these technologies bring to drug discovery. New applications should be fostered by relationships to both biological value and therapeutic impact – not by hype or technophilia – and would likely encompass the following:
6.1 Hydrogen–deuterium exchange mass spectrometry
Biopharmaceutical analysis and quality control; identification of new, functionally important protein ligand-binding (e.g., allosteric) sites to guide structure-based drug design; definition of protein structural pathology (e.g., aggregation domains) suggesting new therapeutic modalities.
6.2 Activity-based protein profiling
Biomarker discovery for in vivo diagnostics/imaging; annotation of the (therapeutic) activity of natural products and deconvolution of their biological targets; screening drugs for promiscuity/off-target effects; disease-related proteasome mapping in living systems.
6.3 Nuclease-mediated PGE
Defining the genetic etiology of complex disease phenotypes in vivo; generating tissue-specific, knock-in ‘humanized’ disease models; translating emerging data suggestive of gene-based correction therapies (e.g., mutation-focused drugs) Citation[85,86]; producing mammalian cells, tissues and organs for regenerative-medicine and synthetic-biology applications; high-resolution mapping of disease interactomes.
Even with their rich information yields and future potential, HDX-MS, ABPP and/or nuclease-mediated PGE per se cannot be expected to solve current pharma-industry woes Citation[2]. Nor are these technologies necessarily relevant or applicable to every discovery program, for inappropriate utilization of even the most time-honored techniques can hinder research progress. Every technology has limits and can be improved/refined. In this latter regard, recent HDX-MS procedural and automation refinements have reduced analysis time, facilitated data acquisition/handling, enhanced spatial and temporal resolution, and allowed examination of heterogeneous protein samples, with ongoing initiatives to reduce the dependence on available protein structural information for interpretation of peptide-level data Citation[15-19]. While definition of critical amino acid residues and binding poises essential to ligand engagement and therapeutic functional output by protein drug targets has proven useful to guide lead evolution and therapeutics design Citation[87], state-of-the-art HDX-MS is at the amino acid level and does not attain the atomic resolution of X-ray crystallography. Advancements in probe design and diversity and detection instrumentation have improved ABPP sensitivity for low-abundance proteins and made it possible to envision in vitro and in vivo ABPP applications to protein classes (ion channels, receptors, etc.) beyond those enzymes now amenable to analysis Citation[37,38,41]. In its current elaboration, ABPP perhaps best serves as a proteomic-level extension of – rather than a substitute for – direct enzyme assay to determine inhibitor constants for detailed structure–activity profiling. Improvements in monitoring genome-editing quality control (i.e., PGE nuclease specificity and target-site recognition) and the capacity and accuracy of high-throughput sequencing have enhanced the robustness, uniformity and reproducibility of nuclease-mediated PGE, as have emerging details about the structural basis of CRISPR technology Citation[88-90]. Although CRISPR-Cas9 is now considered the leading PGE platform because of its accessibility, applications versatility and simplicity as compared to the older ZFN and TALEN PGE technologies that require complicated protein engineering to ensure correct zinc-finger targeting, CRISPR-Cas9 faces potential hurdles (especially for direct therapeutic application) from off-target guide-RNA binding to DNA and the nature of Cas9 as a bacterial protein that could provoke an immune response in mammals Citation[91]. Complementarities among HDX-MS, ABPP, nuclease-mediated PGE and other technologies in structural biology, chemoproteomics, and functional genomics and limitations associated with each technology have invited concerted application of experimental approaches to amplify information yield relevant to drug discovery. For example, integration of HDX-MS with several other structural biology and protein-analysis techniques (mass, infrared and nuclear magnetic resonance spectrometries; circular dichroism; calorimetry; X-ray crystallography; peptide mapping) was essential for demonstrating that a candidate biosimilar was compatible with an originator monoclonal-antibody drug, a key requirement for regulatory approval Citation[92].
As John Donne (1572 – 1631) reminds us: ‘No man is an island.’ Now more than ever, drug discovery is a group endeavor requiring both diverse constituencies that share professional and technical knowledge and appreciable front-loaded financial and intellectual resources. In this regard, it is noteworthy that HDX-MS, ABPP, nuclease-mediated PGE originated from the public (academic/research institute) sector Citation[93-95], which would suggest opportunities for precompetitive public–private (i.e., industrial) collaboration to optimize these technologies and expand their burgeoning drug-discovery applications. Despite current opportunities such as the United States National Institute of Health’s Shared Instrumentation Grant Citation[96] and Instrument Development for Biomedical Applications Programs Citation[97] and non-governmental initiatives such as the Biomedical Resource and Technology Development grants offered in the United Kingdom by the Wellcome Trust Citation[98], support for fundamental research on discovery-relevant technology has been historically limited. Increased levels of funding and greater numbers of scientists dedicated to actualizing and refining these (and other developing) discovery technologies are urgently needed and could encourage research collaborations and facilitate wider technology access by researchers Citation[2]. Precompetitive collaborations with (big) pharma and other companies having vested interest in methods application and/or commercialization could serve as another spur to technology actualization for drug discovery. Startup contract organizations for nuclease-mediated PGE are but one current embodiment of this thinking Citation[99].
The only real success in drug discovery is delivery of a therapeutic into the clinic as an approved medication that addresses an unmet medical need safely and efficaciously. Integration of any technology into a discovery effort is no guarantee that this litmus test will be met. Nonetheless, as illustrated herein, application of technological advancements early in the discovery process can provide novel information that enriches candidate profiling in more disease-relevant models and deepens our understanding of disease processes. The beleaguered state of the pharmaceutical industry clearly requires discovery approaches beyond legacy groupthink that are open to developing, supporting and embracing innovative technologies. To quote the Canadian philosopher Marshall McLuhan (1911 – 1980): ‘It is the framework which changes with each new technology and not just the picture within the frame.’
Declaration of interest
D Janero has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Acknowledgment
The expert bibliographic assistance of Mr. Mitchel Ayer is gratefully acknowledged.
Notes
Bibliography
- Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov 2011;10:507-19
- Janero DR. Productive university, industry, and government relationships in preclinical drug discovery and development: considerations toward a synergistic lingua franca. Expert Opin Drug Discov 2012;7:449-56
- Jarvis LM. The year in new drugs. Chem Eng News 2014;92:10-13
- Jacunski A, Tatonetti NP. Connecting the dots: applications of network medicine in pharmacology and disease. Clin Pharmacol Ther 2013;94:659-69
- Kenakin T, Bylund DB, Toews ML, et al. Replicated, replicable and relevant-target engagement and pharmacological experimentation in the 21st century. Biochem Pharmacol 2014;87:64-77
- Hay M, Thomas DW, Craighead JL, et al. Clinical development success rates for investigational drugs. Nat Biotechnol 2014;32:40-51
- Bhogal N, Balls M. Translation of new technologies: from basic research to drug discovery and development. Curr Drug Discov Technol 2008;5:250-62
- Benovic JL. G-protein-coupled receptors signal victory. Cell 2012;151:1148-50
- Zheng H, Hou J, Zimmerman MD, et al. The future of crystallography in drug discovery. Expert Opin Drug Discov 2014;9:125-37
- Kang HJ, Lee C, Drew D. Breaking the barriers in membrane protein crystallography. Int J Biochem Cell Biol 2013;45:636-44
- Topiol S. X-Ray structural information of GPCRs in drug design: what are the limitations and where do we go? Expert Opin Drug Discov 2013;8:607-20
- Preininger AM, Meiler J, Hamm HE. Conformational flexibility and structural dynamics in GPCR-mediated G protein activation: a perspective. J Mol Biol 2013;425:2288-98
- Schramm VL. Transition states, analogues, and drug development. ACS Chem Biol 2013;8:71-81
- Marcsisin SR, Engen JR. Hydrogen exchange mass spectrometry: what is it and what can it tell us? Anal Bioanal Chem 2010;397:967-72
- Wei H, Mo J, Russell RJ, et al. Hydrogen/deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: methodology and applications. Drug Discov Today 2014;19:95-102
- Jaswal SS. Biological insights from hydrogen exchange mass spectrometry. Biochim Biophys Acta 2013;1834:1188-201
- Landgraf RR, Chalmers MJ, Griffin PR. Automated hydrogen/deuterium exchange electron transfer dissociation high resolution mass spectrometry measured at single-amide resolution. J Am Soc Mass Spectrom 2012;23:301-9
- Resetca D, Wilson DJ. Characterizing rapid, activity-linked conformational transitions in proteins via sub-second hydrogen deuterium exchange mass spectrometry. FEBS J 2013;280:5616-25
- Iacob RE, Engen JR. Hydrogen exchange mass spectrometry: are we out of quicksand? J Am Soc Mass Spectrom 2012;23:1003-10
- Balasubramaniam D, Komives EA. Hydrogen-exchange mass spectrometry for the study of intrinsic disorder in proteins. Biochim Biophys Acta 2013;1834:1202-9
- Shan Y, Arkhipov A, Kim ET, et al. Transitions to catalytically inactive conformations in EGFR kinase. Proc Natl Acad Sci USA 2013;110:7270-5
- Karageorgos I, Wales TE, Janero DR, et al. Active-site inhibitors modulate the dynamic properties of human monoacylglycerol lipase: a hydrogen exchange mass spectrometry study. Biochemistry 2013;52:5016-26
- Nasr ML, Shi X, Bowman AL, et al. Membrane phospholipid bilayer as a determinant of monoacylglycerol lipase kinetic profile and conformational repertoire. Protein Sci 2013;22:774-87
- Yu W, Chory EJ, Wernimont AK, et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun 2012;3:1-11
- Lim SM, Westover KD, Ficarro SB, et al. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew Chem Int Ed 2014;53:199-204
- Sowole MA, Alexopoulos JA, Cheng YQ, et al. Activation of ClpP protease by ADEP antibiotics: insights from hydrogen exchange mass spectrometry. J Mol Biol 2013;425:4508-19
- Boerma LJ, Xia G, Qui C, et al. Defining the communication between agnostic and coactivator binding in the retinoid x receptor alpha ligand binding domain. J Biol Chem 2014;289:814-26
- Ahmed AH, Ptak CP, Fenwick MK, et al. Dynamics of cleft closure of the GluA2 ligand-binding domain in the presence of full and partial agonists revealed by hydrogen-deuterium exchange. J Biol Chem 2013;288:27658-66
- West GM, Chien EY, Katritch V, et al. Ligand-dependent perturbation of the conformational ensemble for the GPCR β2 adrenergic receptor revealed by HDX. Structure 2011;19:1424-32
- Tiyanont K, Wales TE, Siebel CW, et al. Insights into Notch3 activation and inhibition mediated by antibodies directed against its negative regulatory region. J Mol Biol 2013;425:3192-204
- Iacob RE, Bou-Assaf GM, Makowski L, et al. Investigating monoclonal antibody aggregation using a combination of H/DX-MS and other biophysical measurements. J Pharm Sci 2013;102:4315-29
- Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov 2013;12:205-16
- Zhang J, Chalmers MJ, Stayrook KR, et al. Hydrogen/deuterium exchange reveals distinct agonist/partial agonist receptor dynamics within vitamin D receptor/retinoid X receptor heterodimer. Structure 2010;18:1332-41
- Wang K, Yang T, Wu Q, et al. Chemistry-based functional proteomics for drug target deconvolution. Expert Rev Proteomics 2012;9:293-310
- Copley SD. Toward a systems biology perspective on enzyme evolution. J Biol Chem 2012;287:3-10
- Simon GM, Cravatt BF. Activity-based proteomics of enzyme superfamilies: serine hydrolases as a case study. J Biol Chem 2010;285:11051-5
- Nodwell MB, Sieber SA. ABPP methodology: introduction and overview. Top Curr Chem 2012;324:1-42
- Li N, Overkleeft HS, Florea BI. Activity-based protein profiling: an enabling technology in chemical biology research. Curr Opin Chem Biol 2012;16:227-33
- Wang R, Borazjani A, Matthews AT, et al. Identification of palmitoyl protein thioesterase 1 in human THP1 monocytes and macrophages and characterization of unique biochemical activities for this enzyme. Biochemistry 2013;52:7559-74
- Wang C, Weerapana E, Blewett MM, et al. A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat Methods 2014;11:79-85
- Nan L, Kuo CL, Paniagua G, et al. Relative quantification of proteasome activity by activity-based protein profiling and LC-MS/MS. Nat Protoc 2013;8:1155-68
- Moellering RE, Cravatt BF. Functional lysine modification by an intrinsically reactive primary glycolytic metabolite. Science 2013;341:549-53
- Krysiak J, Breinbauer R. Activity-based protein profiling for natural product target discovery. Top Curr Chem 2012;324:43-84
- Bottcher T, Pitscheider M, Sieber SA. Natural products and their biological targets: proteomic and metabolomic labeling strategies. Angew Chem Int Ed 2010;49:2680-98
- Nomure DK, Dix MM, Cravatt BF. Activity-based protein profiling for biochemical pathway discovery in cancer. Nat Rev Cancer 2010;10:630-8
- Chang JW, Cognetta AB, Niphakis MJ, et al. Proteome-wide reactivity profiling identifies diverse carbamate chemotypes tuned for serine hydrolase inhibition. ACS Chem Biol 2013;8:1590-9
- Ward J, Spath SN, Pabst B, et al. Mechanistic characterization of a 2-thioxanthine myeloperoxidase inhibitor and selectivity assessment utilizing click chemistry-activity-based protein profiling. Biochemistry 2013;52:9187-201
- Dominguez E, Galmozzi A, Chang JW, et al. Integrated phenotypic and activity-based profiling links Ces3 to obesity and diabetes. Nat Chem Biol 2014;10:113-21
- Lian J, Quiroga AD, Li L, et al. Ces3/TGH deficiency improves dyslipidemia and reduces atherosclerosis in Ldlr-/- mice. Circ Res 2012;111:982-90
- Lin JH. Applications and limitations of genetically modified mouse models in drug discovery and development. Curr Drug Metab 2008;9:419-38
- van der Worp HB, Howells DW, Sena ES, et al. Can animal models of disease reliably inform human studies? PLoS Med 2010;7:e10000245
- Breyer MD. Improving productivity of modern-day drug discovery. Expert Opin Drug Discov 2014;9:115-18
- Hongmei LL, Nakano T, Hotta A. Genetic correction using engineered nucleases for gene therapy applications. Dev Growth Differ 2014;56:63-77
- Tan W, Carlson DF, Walton MW, et al. Precision editing of large animal genomes. Adv Genet 2012;80:37-97
- Sakuma T, Woltjen K. Nuclease-mediated genome editing: at the front-line of functional genomics technology. Dev Growth Differ 2014;56:2-13
- Perez-Pinera P, Ousterout DG, Gersbach CA. Advances in targeted genome editing. Curr Opin Chem Biol 2012;16:268-77
- Sun N, Zhao H. Transcription activator-like effector nucleases (TALENs): a highly efficient and versatile tool for genome editing. Biotechnol Bioeng 2013;110:1811-21
- Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods 2013;10:957-63
- Mashimo T. Gene targeting technologies in rats: zinc finger nucleases, transcription activator-like effector nucleases, and clustered regularly interspaced short palindromic repeats. Dev Growth Differ 2014;56:46-52
- Wirt SE, Porteus MH. Development of nuclease-mediated site-specific genome modification. Curr Opin Immunol 2012;24:609-16
- Wei C, Liu J, Yu Z, et al. TALEN or Cas9 – Rapid, efficient and specific choices for genome modifications. J Genet Genomics 2013;40:281-9
- Gaj T, Gersbach CA, Barbas CF. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 2013;31:397-405
- Musunuru K. Genome editing of human pluripotent stem cells to generate human cellular disease models. Dis Model Mech 2013;6:896-904
- Reinhardt P, Schmid B, Burbulla LF, et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013;12:354-67
- Toscano MG, Anderson P, Muñoz P, et al. Use of zinc-finger nucleases to knock out the WAS gene in K562 cells: a human cellular model for Wiskott-Aldrich syndrome. Dis Model Mech 2013;6:544-54
- Zhang X, Li H, Mao Y, et al. An over expression APP model for anti-Alzheimer disease drug acreening created by zinc finger nuclease technology. PLoS One 2013;8:e75493
- Frank S, Skryabin BV, Greber B. A modified TALEN-based system for robust generation of knock-out human pluripotent stem cell lines and disease models. BMC Genomics 2013;14:773
- Voit RA, Hendel A, Pruett-Miller SM, et al. Nuclease-mediated gene editing by homologous recombination of the human globin locus. Nucleic Acids Res 2014;42:1365-78
- Ochiai H, Miyamoto T, Kanai A, et al. TALEN-mediated single-base-pair editing identification of an intergenic mutation upstream of BUB1B as causative of PCS (MVA) syndrome. Proc Natl Acad Sci USA 2014;111:1461-6
- Horii T, Tamura D, Morita S, et al. Generation of an ICF syndrome model by efficient genome editing of human induced pluripotent stem cells using the CRISPR system. Int J Mol Sci 2013;14:19774-81
- Shalem O, Sanjana NE, Hartenian E, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014;343:84-7
- Wang T, Wei JJ, Sabatini DM, et al. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014;343:80-4
- Li H, Haurigot V, Doyon Y, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature 2011;475:217-21
- Watanabe M, Nakano K, Matsunari H, et al. Generation of interleukin-2 receptor gamma gene knockout pigs from somatic cells genetically modified by zinc finger nuclease-encoding mRNA. PLoS One 2013;8:e76478
- Ferguson C, McKay M, Harris RA, et al. Toll-like receptor 4 (Tlr4) knockout rats produced by transcriptional activator-like effector nuclease (TALEN)-mediated gene inactivation. Alcohol 2013;47:595-9
- Carlson DF, Tan W, Lillico SG, et al. Efficient TALEN-mediated gene knockout in livestock. Proc Natl Acad Sci USA 2012;109:17382-7
- Wefers B, Meyer M, Ortiz O, et al. Direct production of mouse disease models by embryo microinjection of TALENs and oligodeoxynucleotides. Proc Natl Acad Sci USA 2013;110:3782-7
- Tan W, Carlson DF, Lancto CA, et al. Efficient nonmeiotic allele introgression in livestock using custom endonucleases. Proc Natl Acad Sci USA 2013;110:16526-31
- Zhou J, Shen B, Zhang W, et al. One-step generation of different immunodeficient mice with multiple gene modifications by CRISPR/Cas9 mediated genome engineering. Int J Biochem Cell Biol 2014;46:49-55
- Niu Y, Shen B, Cui Y, et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 2014;156:1-8
- Townes CN. The first laser. In: Garwin L, Lincoln T, editors, A century of Nature: twenty-one discoveries that changed science and the world. University of Chicago Press, Chicago, USA; 2003. p. 107-12
- Prinz F, Schlange T, Asadullah K. Believe it or not: how much can we rely on published data on potential drug targets? Nat Rev Drug Discov 2011;10:712-13
- Sacks LV, Shamsuddin HH, Yasinkaya YI, et al. Scientific and regulatory reasons for delay and denial of FDA approval of initial applications for new drugs, 2000-2012. JAMA 2014;311:378-84
- Patel AC. Clinical relevance of target identity and biology: implications for drug discovery and development. J Biomol Screen 2013;18:1164-85
- Yin H, Xue W, Chen S, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol 2014. [ Epub ahead of print]
- Tebas P, Stein D, Tang WW, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med 2014;370:901-10
- Szymanski DW, Papanastasiou M, Melchior K, et al. Mass spectrometry-based proteomics of human cannabinoid receptor 2: covalent cysteine 6.47(257)-ligand interaction affording megagonist receptor activation. J Proteome Res 2011;10:4789-98
- Zhang F, Wen Y, Guo X. CRISPR/Cas9 for genome editing: progress, implications, and challenges. Hum Mol Genet 2014. [ Epub ahead of print]
- Sternberg SH, Redding S, Jinek M, et al. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014;507:62-7
- Jinek M, Jiang F, Taylor DW, et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 2014;343(6176):1247997
- Owens B. Zinc-finger nucleases make the cut in HIV. Nat Rev Drug Discov 2014;13:321-2
- Visser J, Feuerstein I, Stangler T, et al. Physicochemical and functional comparability between the proposed biosimilar rituximab GP2013 and originator rituximab. BioDrugs 2013;27:495-507
- Englander SW. Hydrogen exchange and mass spectrometry: a historical perspective. J Am Soc Mass Spectrom 2006;17:1481-9
- Jessani M, Cravatt BF. The development and application of methods for activity-based protein profiling. Curr Opin Chem Biol 2004;8:54-9
- Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013;339:819-23
- Department of Health and Human Services. Shared instrumentation grant program. 2014. Available from: www.grants.nih.gov/grants/guide/pa-files/PAR-13_008.html [Last accessed 14 May 2014]
- Department of Health and Human Services. Instrument development for biomedical applications. 2012. Available from: www.grants.nih.gov/grants/guide/rfa-files/PAR-RR-11-005.html [Last accessed 14 May 2014]
- Wellcome Trust. Biomedical resource and technology development grants. 2014. Available from: www.wellcome.ac.uk/Funding/Biomedical-science/Funding-schemes/Strategic-awards-and-initiatives/WTDV031727.htm [Last accessed 14 May 2014]
- Shen H. CRISPR technology leaps from lab to industry. Nature. 2013. Available from: www.natutre.com/news/cripsr-technoilogy-leaps-from-lab-to-industry-1.14299 [Last accessed 14 May 2014]