
Abstract
The Polo-like kinase 1 (Plk1) plays a key role in regulating a broad spectrum of critical cell cycle events. Plk1 is a marker of cellular proliferation and has prognostic potential in different types of human tumors. In a series of preclinical studies, Plk1 has been validated as a cancer target. This prompted many pharmaceutical companies to develop small-molecule inhibitors targeting the classical ATP-binding site of Plk1 for anticancer drug development. Recently, FDA has granted a Breakthrough Therapy designation to the Plk inhibitor BI 6727 (volasertib), which provided a survival benefit for patients suffering from acute myeloid leukemia. Remarkably, a new generation of Plk1 inhibitors that target the second druggable domain of Plk1, the Polo-box domain, is currently being tested preclinically. Since various ATP-competitive compounds of Plk1 inhibit also the activities of Plk2 and Plk3, which act as tumor suppressors, the roles of closely related Plk-family members in cancer cells need to be considered carefully. In this article, the authors highlight recent insights into the biology of Plks in cancer cells and discuss the progress in the development of small-molecule Plk1 inhibitors. The authors believe that the greatest therapeutic benefit might come through leukemic cells that are in direct contact with the inhibitor in the blood stream. The identification of biomarkers and studies that document Plk activities in treated patients would also be beneficial to better understand the role of Plk inhibition in tumor development and anticancer therapy.
1. Introduction
Microtubule-targeting agents (MTAs) that change microtubule dynamics such as the taxanes and vinca alkaloids are considered as mainstays in the clinical treatment of cancer. Despite the proven success of many MTAs, issues of toxicity need to be considered when treating cancer patients. A major limitation in the use of MTAs is the high rate of neuropathy which manifests itself as a painful and debilitating peripheral neuropathy for which there is currently no effective symptomatic treatment. This type of toxicity has been a limiting factor in the development of different MTAs. In addition, myeloid toxicity is frequently observed with neutropenia as the most frequent and/or severe side-effect in regimens that include MTAs. Although neutropenia is one of the dose-limiting toxicities, it is usually manageable. Further intriguing issues concern the resistance to MTAs and the possible mutagenic properties that include the liability that these agents may increase the risk of secondary tumors. Since many clinical trials include patients whose life expectancy is short, it is difficult to establish whether these drugs are potentially carcinogenic per se.
Considering the limitations associated with the use of MTAs in cancer trials, the identification of centrosomal and mitotic regulators that function during S, G2, and M phase of the cell cycle provided many novel pharmaceutical targets, such as mitotic kinases or the mitotic spindle apparatus, required for the division of duplicated sets of chromosomes. The robust rate of discovery and the development of many mitosis-selective inhibitors has opened many attractive avenues for cancer therapy. Novel targets can be categorized according to their specific function during cell cycle progression: cell cycle entry, mitotic entry, spindle assembly, mitotic checkpoint, and mitotic exit. In this article, we will focus predominately on Polo-like kinase 1 (Plk1) which is a major regulator of spindle formation ().
Figure 1. Polo-like kinase 1 localizes to centrosomes during mitosis.
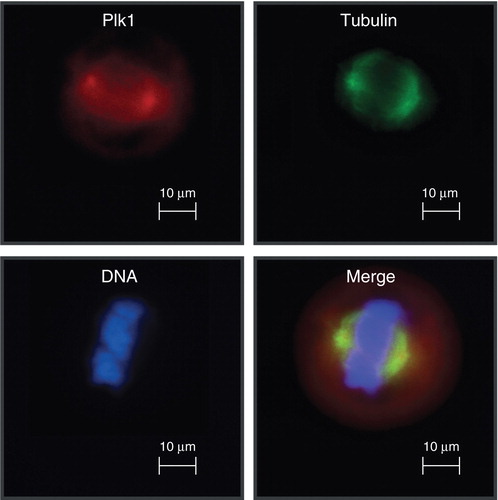
2. Biological functions of Plk1
The human serine/threonine kinase Plk1 is the best characterized cancer target of the Plk family Citation[1]. In eukaryotic cells from yeast to men, orthologs of Plk1 play cardinal roles for the regulation of mitosis and meiosis Citation[2,3]. In vertebrate cells, Plk1 has been implicated in multiple steps of mitotic progression including the activation of Cdk1/cyclin B1, centrosome maturation, release of cohesion from chromosomes, activation of the anaphase-promoting complex/cyclosome (APC/C), formation of bipolar spindles, and the regulation of the spindle assembly checkpoint (SAC) Citation[2,3]. Correlating with its multiple mitotic functions, Plk1 as marker of cellular proliferation is only detectable in a few adult tissues containing proliferating cells Citation[4,5]. Remarkably, the levels of Plk1 in different types of tumors correlate inversely with patient survival providing evidence for the role of Plk1 as prognostic marker Citation[6] and/or marker of metastatic disease Citation[7-9]. Recent genome-wide RNA interference (RNAi) screens confirmed that cancer cells are addicted to a high level of nononcogenic Plk1 for their viability Citation[10]. Inhibition of Plk1 function results in severe spindle defects, most frequently monopolar spindles associated with mitotic arrest due to activation of SAC. This arrest correlates with increased apoptosis in a spectrum of cancer cell lines. Remarkably, the recent analysis of an inducible RNAi model in transgenic mice demonstrated that cancer cells and primary cells differ clearly in their dependency to Plk1 Citation[11-14].
3. Targeting Plk1 for cancer therapy
The deep cavity in the ATP-binding domain of protein kinases is an obvious target in the search for ATP-competitive inhibitors. High-throughput screening and structure-based modeling have been used successfully to identify inhibitors of Plks. However, the high degree of structural conservation among ATP-binding pockets makes the identification of specific inhibitors a challenging endeavor because of the high probability of off-target effects. Still, the clinical success associated with the application of MTAs prompted many pharmaceutical companies worldwide to develop Plk1-specifc small molecule inhibitors targeting its ATP binding site. The development of various compounds that block Plk1 activity efficiently enabled many labs to unravel novel cellular functions of Plk1. BI 2536 developed by Boehringer Ingelheim can be considered as a first-in-class clinical inhibitor of Plk1 () Citation[15]. Cancer cells treated with BI 2536 formed monopolar spindles that do not stably attach to kinetochores. Mad2 accumulates at kinetochores, and cells arrest with an activated SAC. The observations suggested that accumulation of Plk1 at centrosomes and kinetochores depends on its own activity and that this activity is required for maintaining centrosome and kinetochore function. The results also demonstrated that an inhibition of Plk1 delays transition to prometaphase, and that Emi1 destruction in prometaphase is not essential for APC/C-mediated cyclin A degradation. In subsequent pharmaceutical efforts, novel Plk1 inhibitors like BI 6727, GSK461364A, GW843682X, TAK-960, MLN0905, NMS-P937, and ZK-Thiazolidinone have all been developed to inhibit Plk1 with an IC50 in the low nanomolar range and exert a strong antiproliferative effect on a variety of cancer cells Citation[1].
Table 1. Polo-like kinase 1 inhibitors in clinical trials. TKM-080301 represents Plk1 specific siRNA using nanoparticles as cargo.
4. Off-targets of ATP-competitive Plk1 inhibitors
Nevertheless, in vitro kinase assays revealed that most ATP-competitive Plk1 inhibitors show considerable activity against Plk2 and Plk3. The analysis of knock-out mice has demonstrated that Plk2 is not required for postnatal growth, but Plk2−/− embryos show retarded growth and skeletal development late in gestation Citation[16]. Cultured Plk2−/− embryonic fibroblasts grow more slowly than normal cells and show delayed entry into S phase suggesting a role for Plk2 in the cell cycle. Depletion of the Plk2 mRNA in cancer cells treated with taxol or nocodazole significantly increases apoptosis, suggesting that Plk2 may prevent mitotic catastrophe following spindle damage indicating that Plk2 may have important functions for spindle damage Citation[17]. In addition, Plk2 may also play a role in the DNA damage checkpoint, since Plk2 mRNA expression is induced by DNA-damaging agents Citation[17].
Plk3 mRNA, which is expressed ubiquitously, shows protein levels that remain relatively constant during the cell cycle Citation[18,19]. Opposing biological roles can be attributed to Plk3 and Plk1, because overexpression of Plk1 in murine cells results in oncogenic transformation, whereas ectopic expression of human Plk3 induces apoptosis Citation[20]. Plk3 kinase activity is rapidly increased upon DNA damage and cellular stress, with expression levels remaining unchanged. ATM is involved in upregulation of Plk3 kinase activity specifically after DNA damage leading to the phosphorylation of p53 and the promotion of its tumor suppressor function Citation[21]. The phosphorylation of p53 at Ser-20, a residue which is also phosphorylated by Chk1 and Chk2, results in p53 stabilization by interaction with HDM2 and cell cycle arrest. Various lines of evidence suggest that Plk3 functionally links DNA damage to cell cycle arrest and apoptosis via the p53 pathway.
Plk3(-/-) mice displayed an increase in weight and developed tumors in various organs at an advanced age Citation[22]. Many tumors in Plk3(-/-) mice were large in size, exhibiting enhanced angiogenesis. In addition, Plk3 is frequently downregulated in human tumors (lung, head, and neck) which could be due to a loss of heterozygosity of the Plk3 gene locus or a homologous deletion. After all, it is known that the absence of heterozygosity is an important characteristic of tumor suppressor genes. The fact that phosphorylation of several tumor suppressor proteins (p53, Chk2, PTEN) by Plk3 leads to their activation or stabilization, and that the absence of the Plk3 protein, results in tumor development at least in mice, demonstrate that it has tumor suppressor functions Citation[1]. Increasing evidence suggests that Plk2 and Plk3 are activated upon different types of stress thereby preventing further cell cycle progression to warrant repair of any damage or normalization of the stress level. Since both genes can be considered as tumor suppressor genes, which have the function to protect a cell from one step on the path to cancer, the pharmacological inhibition of the corresponding tumor suppressor protein might cause a loss or reduction in its function, a process, which allows the cell to progress to cancer, usually in combination with other genetic changes. Therefore, highly selective Plk1 inhibitors are desirable to exclude unwanted tumor-promoting effects. Furthermore, a recent quantitative chemical proteomics study revealed a Plk1 inhibitor–compromised cell death pathway in human cells Citation[23]. This investigation revealed novel targets of the clinical inhibitor BI 2536 including the death-associated protein kinase. Careful dose-finding studies in cancer trials targeting Plk1 seem to be necessary to prevent the inhibition of DAP kinases, which counteract the proapoptotic effect of Plk1 inhibition. Thus, a prerequisite of the design of an optimized cancer therapy is our understanding of the kinase selectivity of novel kinase inhibitors.
5. Targeting the Polo-box domain of Plk1
The targeted inactivation of disease-relevant protein kinases is often accomplished with ATP-competitive small-molecule inhibitors. However, a significant proportion of patients who are under long-term treatment with ATP-competitive inhibitors develop resistance as they acquire mutations in the kinase domain. In addition, due to the structural homology among the catalytic domains of protein kinases, it is a challenge to develop Plk1-specific inhibitors. Therefore, efforts are now focused on identifying novel inhibitors that do not compete with ATP to inhibit particular kinases, but instead compete with their substrates. Several studies have demonstrated that Polo-box domain (PBD) is crucial for substrate interaction; for targeting Plk1 to specific subcellular multiprotein complexes, such as the centrosomes, kinetochore, spindle apparatus, and Golgi; and for regulating the kinase activity of Plk1. First evidence for the PBD as a domain that could be targeted for the inhibition of Plk1’s function came from a study that utilized peptides representing a partial PBD of Plk1 linked to an Antennapedia peptide Citation[24]. The treatment of cancer cells with this peptide caused mitotic arrest, misaligned chromosomes, and multiple centrosomes indicating that membrane-permeable Polo-box peptides can inhibit cancer cell proliferation efficiently. The first reported small molecule inhibitor targeting the PBD of Plk1 is Poloxin (PBD inhibitor), which has antiproliferative activity in an array of cancer cells in cell culture and in xenograft models () Citation[25,26]. In a fluorescence polarization assay based on binding of the Plk1 PBD to a fluorophore-labeled peptide encompassing its optimal recognition motif Citation[27], Poloxin interfered with the function of the Plk1 PBD (apparent IC50 of 4.8 ± 1.3 μM) Citation[28]. In the same assay, the IC50 values against the PBDs of Plk2 and Plk3 were found to be approximately fourfold and 11-fold higher, respectively (apparent IC50: Plk2 PBD: 18.7 ± 1.8 μM; Plk3 PBD: 53.9 ± 8.5 μM). Moreover, the related compound Thymoquinone showed a stronger inhibitory effect on the function of the Plk1 PBD compared to Poloxin (apparent IC50: 1.14 ± 0.04 μM). A crystallographic investigation demonstrated that Thymoquinone and its analogue Poloxime occupy the binding pocket at the phosphoserine/phosphothreonine recognition site Citation[29]. However, in an ELISA-based assay that analyzed the interaction between the Plk1 PBD and a ligand from the p-T78 motif of PBIB1 (also known as centromeric protein Q, CENP-50) Poloxin did not prevent binding significantly Citation[30]. Supporting evidence for the binding of Poloxin to Plk1, which could arise from experimental approaches involving co-incubation of both proteins, subsequent tryptic/chymotryptic digest, and identification of peptides by mass spectrometry, is currently underway.
Table 2. Inhibitors of the polo-box domain of Plk1.
Alternative approaches for the development of PBD inhibitors based on peptides or peptidic inhibitors converted into more pharmaceutically relevant compounds are underway: The PBD represents a phosphoepitope binding domain that modulates serine- or threonine-phosphorylated peptides with the invariable serine residue at the -1 position (S-p-S/T motif) Citation[27]. The analysis of the interaction of Plk1 with PBIP1, which regulates the centromeric localization of Plk1, demonstrated that the PBD of Plk1 binds to the Thr78 region in PBIP1 in a phosphorylation-dependent manner Citation[31,32]. In subsequent investigations, minimal phospho-peptides derived from the Thr78 region of PBIP1 were identified that show high levels of affinity and specificity for the PBD of Plk1. This analysis supported an improved understanding of critical determinants that are required for binding of the Plk1 PBD including the core SpT motif as high-affinity anchor, the N-terminal hydrophobic residue, glycerol, and a network of contacting water molecules. Remarkably, the N-terminal residue of the optimal PBIP1-peptide is crucial for the affinity by forming polar contact with Arg516 in the Plk1 PBD. Plk2 and Plk3 posses a lysine residue at the position analogues to Plk1 Arg516; as a consequence, they may be unable to interact with Plk1-specific peptides. Recent work on a Plk1-PBD binding peptide derived from Cdc25C supported the critical role of residues that are located N-terminal to the core SpT motif for high affinity binding Citation[33]. With the knowledge of critical determinants of peptide binding within the PBD of Plk1 the ‘replacement with partial ligand alternative through computational enrichment’ (REPLACE) strategy was applied to develop inhibitors of Plk1 protein–protein interactions Citation[33]. By using this strategy fragment alternatives to the N-terminal tripeptide derived from Cdc25C were identified. The resulting fragment-ligated inhibitory peptides reduced the centrosomal localization of Plk1 and induced an antiproliferative effect in cancer cells. The resulting cellular phenotype was characterized by the existence of monopolar spindles, which could indicate that the Plk1-inhibitory effect is more pronounced compared to small-molecule inhibitors of the PBD Citation[25,34]. Both studies provide a better understanding of the PBD interaction, which is a prerequisite for the development of PBD binding agents and for the discovery of a new class of Plk1-specific anticancer therapeutics.
Plk1 represents an ideal protein-kinase target for cancer drug development because in addition to its kinase domain, which is related to many members of the protein-kinase superfamily, it also encompasses the unique PBD. It will be exciting to study whether more promiscuous kinase- or more specific PBD-inhibitors of Plk1 are advantageous in cancer cell killing, in preclinical and clinical trials.
6. Expert opinion
Can we improve on microtuble-targeting drugs? This is the major question that we will have to answer in the coming years. Compounds that alter microtuble function have proved to be highly active in patients with cancer. Vinca alkaloids and taxanes are currently administered in a large range of malignant diseases. However, an important issue that is discussed is that of toxicity, as many microtuble-targeting drugs cause considerable neurological toxicity. This and other aspects prompted, in the field of targeted drugs, the development of mitosis-specific agents including those that target mitotic kinases. Key finding in this field include the identification of various highly specific ATP-competitive Plk1 inhibitors that kill cancer cells in culture and in xenograft-experiments efficiently. Moreover, the high affinity of drugs synthesized and developed clinically, many of which have Ki values in the nM range, demonstrated the high quality of novel Plk1 inhibitors. The emergence of neutropenia as side-effect of novel Plk1 inhibitors is strong evidence that the pharmacodynamics were excellent and that their pharmacokinetics are likely favorable.
To date, clinical success of Plk1-specific agents was predominately seen in trials treating acute myeloid leukemia (AML) patients. FDA has granted a Breakthrough Therapy designation to the Plk inhibitor BI 6727 (volasertib) after significant benefit was observed in treating patients with AML in combination with the established therapy of low-dose cytarabine (LDAC). AML is an aggressive disease of the bone marrow and blood. It accounts for approximately one-third of all adult leukemias in the Western world and has one of the lowest survival rates of all leukemias. AML is primarily a disease of later adulthood; the average age of an AML patient is 65 – 70 years. The recommended standard of care is currently intensive chemotherapy, but many patients due to age and comorbidities cannot tolerate this therapeutic approach. For them, options are limited and their prognosis is typically poor. Volasertib is currently being investigated in this specific patient population Citation[35]. The Boehringer Ingelheim compound volasertib, a highly potent and selective inhibitor of Plks, was recently used in a randomized Phase II trial for the treatment of older patients with AML (median age 75 years), who were not considered suitable for intensive induction therapy. In this trial low-dose cytarabine (LDAC) was compared with or without volasertib. Response rate (complete remission [CR] and CR with incomplete blood count recovery [CRi]) was higher for LDAC + volasertib versus LDAC (31.0 vs 13.3%; odds ratio, 2.91; p = 0.052). Responses in the LDAC + volasertib arm were observed across all genetic groups. Median event-free survival was significantly prolonged by LDAC + volasertib compared with LDAC (5.6 vs 2.3 months). These encouraging data has prompted the initiation of a Phase III trial of volasertib in AML. The improved pharmakokinetic profile of volasertib might be one reason for the successful development of this Plk inhibitor. Further clinical trials investigating volasertib treatment as single agent or in combination with other compounds, will benefit from an improved understanding of the mode of action of Plk inhibition and the use of appropriate biomarkers.
In addition, a new generation of Plk1 inhibitors is at the horizon: Compounds that target the PBD of Plk1. This domain can be found only within the Plk family and it is very divergent comparing individual members of the Plk family. Therefore, drugs targeting the PBD of Plk1 will be highly specific. Preclinical experiments have already demonstrated their efficacy Citation[26]. Future studies are required to investigate their pharmacokinetic profile and additional clinically relevant parameters.
Numerous target validation trials using a spectrum of cancer cells in culture and in xenograft models came to the result that Plk1 is an excellent target in many different cancer cell types. However, in both experimental systems, cancer cells proliferate at high speed that is, have a higher percentage of cells in mitosis compared to some slow growing human solid cancers.
While the long doubling times of many primary cancer cells ranges from 100 to 200 days, their time spent in mitosis is only between 0.5 and 2.5 h. A targeted mitotic therapy can only be effective if the target is present. Mitotic kinases are predominantly expressed in G2 and M-phase of the cell cycle. Hence, only a small fraction of cells is vulnerable to a drug aimed at Plk1 that is expressed only transiently. The biggest challenge will be the use of Plk1 inhibitors for the induction of apoptosis in slow growing human tumors. However, recent data document that Plk1 plays also an important role during S-phase indicating that the window for targeting Plk1 is not restricted to S phase, but might be much wider as originally thought.
The most therapeutic benefit might be reached in leukemic cells that are in direct contact with the inhibitor in the blood stream and that often proliferate faster compared to some slow growing solid tumors. In addition, treatment schedules need to consider this aspect by prolonging the application of the drug. The identification of biomarkers and studies that documents Plk activities in treated patients would be beneficial to better understand the role of Plk inhibition in tumor development and anticancer therapy. Furthermore, analyzing the effect of Plk1-specific drugs on the neovascularization should also be an issue for future research activities.
Declaration of interest
This work was supported by grants from the German Cancer Consortium (DKTK, Heidelberg), Carls-Stiftung, Deutsche Krebshilfe, and BANSS Stiftung. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Bibliography
- Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov 2010;9:643-60
- Barr FA, Sillje HH, Nigg EA. Polo-like kinases and the orchestration of cell division. Nat Rev Mol cell biol 2004;5:429-40
- Archambault V, Glover DM. Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol cell biol 2009;10:265-75
- Holtrich U, Wolf G, Brauninger A, et al. Induction and down-regulation of PLK, a human serine/threonine kinase expressed in proliferating cells and tumors. Proc Natl Acad Sci USA 1994;91:1736-40
- Yuan J, Horlin A, Hock B, et al. Polo-like kinase, a novel marker for cellular proliferation. Am J Pathol 1997;150(4):1165-72
- Wolf G, Hildenbrand R, Schwar C, et al. Polo-like kinase: a novel marker of proliferation: correlation with estrogen-receptor expression in human breast cancer. Pathol Res Pract 2000;196(11):753-9
- Kneisel L, Strebhardt K, Bernd A, et al. Expression of polo-like kinase (PLK1) in thin melanomas: a novel marker of metastatic disease. J Cutan Pathol 2002;29(6):354-8
- Strebhardt K, Kneisel L, Linhart C, et al. Prognostic value of pololike kinase expression in melanomas. JAMA 2000;283:479-80
- Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer 2006;6:321-30
- Luo J, Emanuele MJ, Li D, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009;137:835-48
- Raab M, Kappel S, Kramer A, et al. Toxicity modelling of Plk1-targeted therapies in genetically engineered mice and cultured primary mammalian cells. Nat Commun 2011;2:395
- Kappel S, Matthess Y, Kaufmann M, Strebhardt K. Silencing of mammalian genes by tetracycline-inducible shRNA expression. Nature Protoc 2007;2:3257-69
- Matthess Y, Kappel S, Spankuch B, et al. Conditional inhibition of cancer cell proliferation by tetracycline-responsive, H1 promoter-driven silencing of PLK1. Oncogene 2005;24:2973-80
- Kappel S, Matthess Y, Zimmer B, et al. Tumor inhibition by genomically integrated inducible RNAi-cassettes. Nucleic Acids Res 2006;34:4527-36
- Lenart P, Petronczki M, Steegmaier M, et al. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr biol 2007;17:304-15
- Ma S, Charron J, Erikson RL. Role of Plk2 (Snk) in mouse development and cell proliferation. Mol Cell Biol 2003;23:6936-43
- Burns TF, Fei P, Scata KA, et al. Silencing of the novel p53 target gene Snk/Plk2 leads to mitotic catastrophe in paclitaxel (taxol)-exposed cells. Mol Cell Biol 2003;23:5556-71
- Holtrich U, Wolf G, Yuan J, et al. Adhesion induced expression of the serine/threonine kinase Fnk in human macrophages. Oncogene 2000;19:4832-9
- Ouyang B, Pan H, Lu L, et al. Human Prk is a conserved protein serine/threonine kinase involved in regulating M phase functions. J Biol Chem 1997;272:28646-51
- Wang Q, Xie S, Chen J, et al. Cell cycle arrest and apoptosis induced by human Polo-like kinase 3 is mediated through perturbation of microtubule integrity. Mol Cell Biol 2002;22:3450-9
- Bahassi el M, Conn CW, Myer DL, et al. Mammalian Polo-like kinase 3 (Plk3) is a multifunctional protein involved in stress response pathways. Oncogene 2002;21:6633-40
- Yang Y, Bai J, Shen R, et al. Polo-like kinase 3 functions as a tumor suppressor and is a negative regulator of hypoxia-inducible factor-1 alpha under hypoxic conditions. Cancer Res 2008;68:4077-85
- Raab M, Pachl F, Kramer A, et al. Quantitative chemical proteomics reveals a Plk1 inhibitor-compromised cell death pathway in human cells. Cell Res 2014;24(9):1141-5
- Yuan J, Kramer A, Eckerdt F, et al. Efficient internalization of the polo-box of polo-like kinase 1 fused to an Antennapedia peptide results in inhibition of cancer cell proliferation. Cancer Res 2002;62:4186-90
- Reindl W, Yuan J, Kramer A, et al. Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem Biol 2008;15:459-66
- Yuan J, Sanhaji M, Kramer A, et al. Polo-box domain inhibitor poloxin activates the spindle assembly checkpoint and inhibits tumor growth in vivo. Am J Pathol 2011;179(4):2091-9
- Elia AE, Cantley LC, Yaffe MB. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 2003;299:1228-31
- Reindl W, Strebhardt K, Berg T. A high-throughput assay based on fluorescence polarization for inhibitors of the polo-box domain of polo-like kinase 1. Anal Biochem 2008;383:205-9
- Yin Z, Song Y, Rehse PH. Thymoquinone blocks pSer/pThr recognition by Plk1 Polo-box domain as a phosphate mimic. ACS Chem Biol 2013;8:303-8
- Liao C, Park JE, Bang JK, et al. Exploring potential binding modes of small drug-like molecules to the polo-box domain of human polo-like kinase 1. ACS med Chem Lett 2010;1:110-14
- Kang YH, Park JE, Yu LR, et al. Self-regulated Plk1 recruitment to kinetochores by the Plk1-PBIP1 interaction is critical for proper chromosome segregation. Mol Cell 2006;24:409-22
- Yun SM, Moulaei T, Lim D, et al. Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1. Nat Struct Mol Biol 2009;16:876-82
- McInnes C, Estes K, Baxter M, et al. Targeting subcellular localization through the polo-box domain: non-ATP competitive inhibitors recapitulate a PLK1 phenotype. Mol Cancer Ther 2012;11:1683-92
- Reindl W, Yuan J, Kramer A, et al. A pan-specific inhibitor of the polo-box domains of polo-like kinases arrests cancer cells in mitosis. ChemBioChem 2009;10:1145-8
- Dohner H, Lubbert M, Fiedler W, et al. Randomized, phase 2 trial comparing low-dose cytarabine with or without volasertib in AML patients not suitable for intensive induction therapy. Blood 2014;124(9):1426-33