
Abstract
Introduction: Genetic skeletal diseases (GSDs) are a diverse and complex group of rare genetic conditions that affect the development and homeostasis of the skeleton. Although individually rare, as a group of related diseases, GSDs have an overall prevalence of at least 1 per 4,000 children. There are currently very few specific therapeutic interventions to prevent, halt or modify skeletal disease progression and therefore the generation of new and effective treatments requires novel and innovative research that can identify tractable therapeutic targets and biomarkers of these diseases.
Areas covered: Remarkable progress has been made in identifying the genetic basis of the majority of GSDs and in developing relevant model systems that have delivered new knowledge on disease mechanisms and are now starting to identify novel therapeutic targets. This review will provide an overview of disease mechanisms that are shared amongst groups of different GSDs and describe potential therapeutic approaches that are under investigation.
Expert opinion: The extensive clinical variability and genetic heterogeneity of GSDs renders this broad group of rare diseases a bench to bedside challenge. However, the evolving hypothesis that clinically different diseases might share common disease mechanisms is a powerful concept that will generate critical mass for the identification and validation of novel therapeutic targets and biomarkers.
1. Introduction
Genetic skeletal diseases (GSDs) are an extremely diverse and complex group of rare genetic conditions that primarily affect the development and homeostasis of the osseous skeleton Citation[1,2]. Although individually rare, as a group of related genetic diseases, GSDs have an overall prevalence of at least 1 per 4,000 children, which extrapolates to a minimum of 225,000 people in the European Union. This burden in pain and disability leads to poor quality of life and high healthcare costs.
There are more than 450 unique and well-characterized phenotypes that range in severity from relatively mild to severe and lethal forms and are described in detail in the 2011 Nosology and Classifications of the GSDs Citation[1]. Forty different diagnostic groups have been recognized to date, which are defined by a combination of molecular, biochemical and/or radiographic criteria. The 2011 Nosology includes 316 conditions associated with one or more of 226 different genes; however, the continued genetic and molecular characterization of GDSs has led to a better defined clinical-molecular classification and a greater understanding of their aetiology Citation[2]. The generation and in-depth analysis of relevant cell and animal models has also increased our understanding of disease mechanisms and has identified phenotype-specific disease signatures through ‘omics’-based analysis.
GSDs are difficult human diseases to treat, particularly when the pathological process begins before birth and can affect the entire skeletal system. Furthermore, since it is now known that the skeleton has close physiological relationships with many other tissue systems in the body, and mutant genes may have pleiotropic effects, patients affected by GSDs may also have serious complications with other organs, including the peripheral nervous system, brain, bone marrow, immune system, pancreas, kidney, heart, muscle and tendon.
The translation of state-of-the-art technology into quantifiable patient benefits such as the development of new treatments or effective biomarkers has been extremely limited for GSDs. The few notable exceptions include Biomarin’s drug candidate for achondroplasia (ACH), a C-type natriuretic peptide (CNP) analogue PG-CNP37 (BMN-111) Citation[3], bisphosphonate treatment for OI Citation[4] and fibrous dysplasia Citation[5], enzyme replacement in lysosomal storage diseases Citation[6] or hematopoietic stem cell transplantation for infantile osteopetrosis Citation[7]. This review will explore a select range of GSDs and propose shared disease mechanisms that hold the promise as potential therapeutic targets.
2. Genetic mouse models provide new insight into shared disease mechanisms
Over the last 20 years, the generation and in-depth analysis of transgenic, knock-out, knock-in and ENU-derived mice models of GSDs have generated valuable knowledge of disease mechanisms in vivo. Several recent reviews have highlighted both strengths and weaknesses of various modelling approaches Citation[8]; however, these methodologies are still the gold standard for generating relevant in vivo models to investigate skeletal pathobiology. These will also act as pre-clinical models when new therapeutic targets are identified and validated.
3. ER stress is a shared mechanism and therapeutic target in a range of GSDs resulting from dominant-negative mutations in cartilage structural proteins
The extracellular matrix (ECM) of cartilage is a highly organized composite material comprising numerous structural macromolecules such as collagens (Types II, IX, X and XI), proteoglycans (aggrecan) and glycoproteins (matrilin-3 and cartilage oligomeric matrix protein [COMP]). Mutations have now been identified in all the genes encoding the major structural components of the cartilage ECM and result in a diverse group of both dominant and recessive GSDs. These assorted mutations fall into two broad classes: qualitative mutations, such as those that have dominant-negative (antimorphic) effects, and quantitative mutations that result in haploinsufficiency and/or a complete loss of protein function. This section will focus specifically on dominant-negative (antimorphic) mutations, which affect conserved residues that are structurally and functionally important for normal protein folding and function ().
Table 1. Disease mechanisms and potential therapeutic targets in selected GSDs resulting from antimorphic mutations in cartilage structural proteins.
The endoplasmic reticulum (ER) is a distinct organelle of eukaryotic cells and plays the major role in the synthesis, folding and trafficking of proteins entering the secretory pathway. The ER has a highly sophisticated quality control mechanism for ensuring that misfolded mutant proteins do not accumulate in, or enter the secretory pathway. Eukaryotic cells have a homeostatic mechanism for maintaining the protein-folding equilibrium of the ER, which is known as the unfolded protein response (UPR) Citation[9]. However, the UPR has evolved to resolve short-term acute ER stress such as high protein load, heat shock or ischemia, but not prolonged ER stresses due to rare events such as the misfolding of mutant proteins in human genetic diseases Citation[10]. A prolonged UPR can eventually have detrimental effects on chondrocyte phenotype, differentiation and viability Citation[10].
An extensive allelic series of glycine and non-glycine substitutions in type II collagen have been introduced into mice to model a diverse range of type II collagenopathies Citation[11,12]. A common feature in many of these mouse models was the evidence of ER stress, translating in some cases into a reduction in chondrocyte proliferation and an increase in apoptosis. Moreover, the recent development of induced pluripotent stem (iPS) cells, and their differentiation into relevant cell types such as chondrocytes (iChon), has allowed the in-depth analysis of cells from patients with various type II collagenopathies and the testing of potential corrector molecules such as trimethylamine N-oxide (TMAO) Citation[13], which is a chemical chaperone that can alleviate mutant protein aggregation and ER stress Citation[14,15]. These studies have provided preliminary proof-of-principle evidence that molecular chaperones may serve as therapeutic drug candidates Citation[13].
The role of ER stress in GSDs might best be exemplified by mutations in COMP, matrilin-3 and type X collagen resulting in pseudoachondroplasia (PSACH), multiple epiphyseal dysplasia (MED) Citation[8] and metaphyseal chondrodysplasia type Schmid (MCDS), respectively ( & ) Citation[16]. Over the last 10 years, the extensive analysis of mouse models for the MATN3 (V194D) Citation[17], COMP (D469del, T585M) Citation[18,19] and COL10A1 (N617K) Citation[16] mutations has been performed, which has allowed a direct comparison of disease mechanisms Citation[8,20]. Furthermore, the application of ‘omics’-based investigations (mRNA and protein) has allowed genotype-specific disease signatures to be derived and either shared or discrete downstream genetic pathways to be identified Citation[8,18,21,22].
Figure 1. Schematic showing chondrocytes and pericellular cartilage matrix from the growth plate of a 1-week-old wild type mouse. Five fundamental disease mechanisms are highlighted along with a selection of associated genetic skeletal diseases.
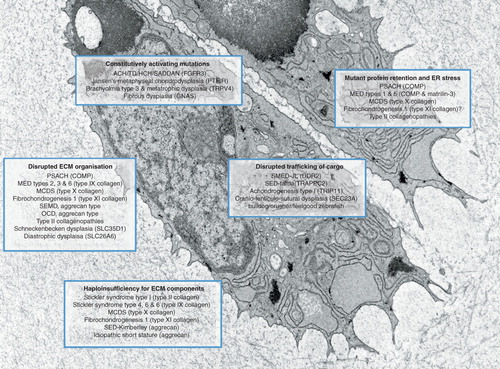
Interestingly, both Matn3 (V194D) and Col10a1 (N617K) mutations cause misfolding and retention of the relevant mutant protein, inducing ER stress and a classical UPR, primarily characterized by the up-regulation of ER chaperones BiP, Grp94 and a range of protein disulphide isomerases (PDIA) Citation[21,22]. Hartley and colleagues Citation[23] commented on a similar increase in specific PDIAs (PDIA1, 3, 4 and 6) in chondrocytes from Col10a1N617K and Matn3V194D mutant mice and also noted that two novel ER stress-related genes, Armet and Creld2, were also significantly upregulated in these two models Citation[23]. The cartilage-specific knock-out of both Armet and Creld2 has resulted in mice with growth plate dysplasia, thus confirming their important role in skeletal development (our unpublished observations). Moreover, the recent cartilage-specific knock-out of PDIA3 (also called ERP57/GRP58) caused ER stress resulting in reduced proliferation and accelerated apoptotic cell death of chondrocytes in the growth plate Citation[24]. Finally, the cartilage-specific ablation of an entire UPR branch (i.e. Xbp-1 signalling) also resulted in a chondrodysplasia that was characterized by reduced chondrocyte proliferation and leading to delayed cartilage maturation and mineralization Citation[25].
In contrast, the accumulation of mutant COMP has been demonstrated to result in the induction of novel stress pathways, which are characterized by changes in the expression of groups of genes implicated in oxidative stress (ER dependent), cell cycle regulation and apoptosis Citation[18,26,27]. In this context, Posey and colleagues have recently demonstrated that the postnatal administration of aspirin to a transgenic dox-induced COMP-overexpression model of PSACH abolished mutant COMP intracellular retention and had beneficial effects on chondrocyte proliferation, apoptosis and final bone length Citation[28]. However, this study failed to show increased secretion of wild type or mutant COMP upon treatment and also to identify a mechanism by which aspirin may reduce mutant COMP retention and modulate chondrocyte phenotype and bone growth in PSACH Citation[28]. Nevertheless, these are interesting findings that require further validation.
In summary, these recent studies using a complimentary group of genetically relevant mouse models and cartilage-specific knock outs have demonstrated the key role that ER stress plays in the initiation and progression of growth plate dysplasia and reduced bone growth (). Moreover, preliminary studies suggest that ER stress is a good therapeutic target that can be influenced through small molecule intervention, and to date, the use of trimethylamine N-oxide (TMAO) Citation[13], lithium, valproate Citation[29], sodium phenylbutyrate (SPB) Citation[22,29] and various antioxidant and anti-inflammatory agents Citation[28] have been tested in both cell and mouse models with varying degrees of success.
4. Disruption to protein trafficking in chondrocytes leads to various chondrodysplasia phenotypes through different mechanisms
Missense mutations in the discordin domain receptor 2 (DDR2) have been shown to cause the rare autosomal recessive spondyo-meta-epiphyseal dysplasia with short limbs and abnormal calcifications (SMED-SL) due to either protein trafficking defects or loss of ligand binding Citation[30]. DDR2 is a plasma membrane receptor tyrosine kinase that functions as a collagen receptor and missense mutations in DDR2 result in either its retention within the ER, or loss of collagen-binding activity and transmembrane signalling. Both mechanisms most likely lead to retention in the ER of the DDR2 cargo (e.g. fibrillar collagens), thereby further exacerbating ER stress due to mutant DDR2 alone Citation[30]. The deletion of DDR2 in mice has previously been reported to cause reduced chondrocyte proliferation and dwarfism Citation[31], suggesting a role for DDR2 in cell-matrix attachment and in pathological conditions where increased cell proliferation and ECM turnover are coupled () Citation[32].
Table 2. Disease mechanisms and potential therapeutic targets in selected GSDs resulting from a disruption to protein trafficking in chondrocytes.
Similarly, the ER export of procollagen is also controlled in part by the ubiquitously expressed Sedlin (TrappC2), and antimorphic or loss of function mutations in TRAPPC2 cause X-linked spondyloepiphyseal dysplasia Tarda (SEDT) Citation[33]. SEDT results from a chondrocyte-specific defect in the trafficking and secretion of cartilage structural proteins, specifically type II procollagen () Citation[34,35]. Moreover, mutations in essential components of COPII-coated vesicles, such as SEC23A, sec23a and sec24d, which transport secretory proteins from the ER to Golgi, result in cranio-lenticulo-sutural dysplasia (CLSD) Citation[36] and the Zebrafish mutants crusher Citation[37] and bulldog Citation[38], respectively. In all three conditions, there is accumulation of proteins, in particular fibrillar collagens, within the ER of relevant cell types (). Finally, loss-of-function mutations in Golgin GMAP-210 (TRIP11) causes a lethal skeletal dysplasia in both mice and humans (achondrogenesis type 1A) Citation[39], which is characterized by disrupted Golgi architecture and ER stress due to the intracellular accumulation of perlecan (but not aggrecan or type II collagen) eventually leading to abnormal chondrocyte differentiation and increased apoptosis () Citation[39].
In summary, these various genetic studies consistently demonstrate that ‘professionally secreting cells’ such as chondrocytes are highly susceptible to perturbations in ER homeostasis and defects in protein trafficking and secretion. This premise is supported by the observation that deletion or mutation of ubiquitously expressed components of the secretory pathway specifically causes chondrocyte disruption and cartilage defects (). Modulation of the secretory pathway in zebrafish mutants by brefeldin A treatment has recently been demonstrated in polycystic kidney disease by Le Corre and colleagues Citation[40], who have proposed that restoration of normal rates of secretory protein synthesis and secretion may be a new target in the treatment of autosomal dominant trafficking defects.
5. Incorporation of mutant proteins into the ECM leads to cartilage defects and GSDs
The retention of mutant protein in the ER of cells appears to be a major pathomolecular mechanism underpinning the disease aetiology in a range of GSDs (). Therefore, one of the potential targeting avenues suggested in the literature is the use of molecular chaperones, which are small molecules that could potentially aid in the degradation and/or secretion of the mutant proteins into the cartilage ECM Citation[41].
The potential effectiveness of such treatment has been demonstrated for type IV collagen mutations causing hemorrhagic stroke. In a study described by Murray and colleagues Citation[42], two related individuals carrying the same missense mutation presented with differing severity of cerebral haemorrhaging; whilst the parent appeared unaffected, his son showed symptoms of severe porencephaly. In fibroblast cell lines from both individuals, the mutant protein was secreted and incorporated into the ECM of the basement membrane causing morphological abnormalities. However, the cells from the severely affected son secreted less collagen IV and showed an increased retention of the mutant protein in the rER. Treatment with a molecular chaperone (sodium phenyl butyrate) appeared to alleviate the ER stress and restore protein secretion to some extent, thus presenting a promising thera eutic avenue Citation[42].
However, it is important to note that the incorporation of mutant proteins into the ECM may also have deleterious effects and lead to a skeletal condition, as highlighted by the PSACH and MED-causing mutations in the C-terminal domain (CTD) of COMP Citation[19]. In several-documented cases CTD mutant COMP protein is secreted, eliciting either a mild ER stress or no ER stress at all, yet still resulting in dysplasia. The incorporation of mutant COMP into the ECM results in abnormal fibrillar and proteoglycan composition, leading to abnormal chondrocyte clustering, a decrease in proliferation and eventually apoptosis Citation[19]. Moreover, changes in cartilage ECM composition has also been detected in some type II collagenopathies (SEDc and Kniest Dysplasia), diastrophic dysplasia (DTDST) where the cartilage shows signs of under-sulphation of proteoglycans and other ECM abnormalities Citation[43], and finally Schneckenbecken dysplasia where the ECM abnormalities result from a loss-of-function mutation to the SLC35D1 nucleotide-sugar transporter Citation[44]. In all these examples, the ECM defects lead to an altered tissue morphology and cellular organization, thereby affecting the chondrocyte columnar arrangement in the proliferative zone of the growth plate and ultimately impacting on long bone growth.
An in-depth analysis of the structural proteins in the cartilage ECM, in particular the proteins that have a bridging/adaptor function in the ECM, using transgenic mouse models has shown that some of these molecules are actually redundant in cartilage. For example, the genetic deletion of COMP Citation[45], matrilin-3 Citation[46], matrilin-1 Citation[47], type IX collagen Citation[48] and WARP Citation[49] had no effect on gross cartilage structure and long bone growth. Therefore, one can postulate that silencing the defective alleles may pose an exciting therapeutic avenue for skeletal dysplasias resulting from mutations in structural proteins Citation[50]; in fact, Posey and colleagues have tested this approach by using RNA interference Citation[51]. In this particular study, the authors used short hairpin RNA to knock down the expression of both wild-type and mutant COMP, thus reducing the presence of mutant COMP inside and outside the cell Citation[51]. However, this type of approach would not be applicable for the most important structural molecules of cartilage and bone, that is, the collagens and proteoglycans. For example, haplo-insufficiency for type II collagen and aggrecan leads to Stickler syndrome and SED-Kimberley, respectively (), whereas a lack of perlecan is associated with Schwartz-Jampel syndrome Citation[1]. Interestingly, allele-specific RNA silencing using siRNA has recently been described as potential therapy for Meesmann epithelial corneal dystrophy caused by a point mutation in the keratin gene. Therefore, pending the development of effective methods to deliver such therapies to the dense and largely avascular environment of cartilage, they nonetheless present an exciting future therapeutic avenue.
Table 3. Disease mechanisms in selected GSDs resulting from haploinsufficiency for cartilage structural proteins.
Mutations that result from aberrant splicing often lead to nonsense-mediated decay of mRNA and associated cell stress or the production of truncated molecules, which may have downstream dramatic effects. This is the case in some cases of Schwartz-Jampel syndrome (SJS) and in dyssegmental dysplasia, Silverman-Handmaker type (DDSH) where an exon skipping mutation leads to a truncated perlecan molecule Citation[2]. However, it is sometimes possible to predict a milder phenotype if the RNA splicing can be partially restored, resulting in a shorter molecule but with a majority of the domains in-frame and correctly folded. Such a rationale was recently applied to the treatment of Duchenne muscular dystrophy by attempting to restore partially functional dystrophin and thus reducing the severity of the disease to a milder Becker muscular dystrophy-like phenotype. Whilst this approach may not be suitable for all cases of GSD, it could be applicable in those diseases where haploinsufficiency and/or retention of the mutant protein leads to a severe phenotype, but a presence of the partially functional mutant molecule in the ECM may lead to a milder condition, and is therefore an interesting therapeutic avenue that could be explored in the future.
6. Mechanosensing is important in the pathobiology of GSDs
Primary cilium is an organelle existing on almost every cell type in the human body Citation[52]. In cartilage, cilia have been implicated in important signalling pathways (such as hedgehog and wnt signalling) and have been suggested as a mechanosensory organelle on the cells Citation[53]. Various ECM receptors, including several integrins, are located on or within a close vicinity of the primary cilia and may be important in regulating the cell–matrix interactions Citation[54]. Furthermore, the primary cilia length and prevalence are increased in osteoarthritic cartilage Citation[55] and changes to the cilia organization have been observed in skeletal dysplasias (our unpublished observations) and chondrosarcoma tissues Citation[56].
Disruptions to the columnar organization of proliferative chondrocytes have been described in several mouse models of rare skeletal conditions, in particular those where the ECM structure was affected Citation[57–61]. The change in cell alignment was not dissimilar from abnormalities seen in cartilage-specific integrin knock-out mice, which indicated that the disruption of the cell matrix interactions may in fact be the underlying pathology Citation[62,63]. Moreover, a truncating mutation in integrin α 10 leads to a canine chondrodysplasia Citation[64]. From a therapeutic perspective, it is interesting to speculate that the way a cell senses its environment could be modulated. Indeed, such therapies have in fact been tried in Crohn’s disease where the use of integrin antagonists has shown a dramatic improvement in disease severity Citation[65].
Mechanosensing is important for cartilage development and homeostasis and mutations involving primary cilia molecules have been discovered in several chondrodysplasias to date, including the Verma-Naumoff syndrome, Majewski syndrome, Jeune syndrome, Ellis-van Creveld syndrome, the Sensenbrenner syndrome and Weyers acrofacial dysostosis Citation[1]. In many cases of the skeletal ciliopathies, the underlying disease mechanism is the disruption to the hedgehog-signalling pathway, which is an important pathway regulating cartilage proliferation and differentiation. Interestingly, hedgehog signalling can be partially modulated and/or restored using small molecular treatment, and several reagents including inducible protein reagents based on the Gli1 and Gli3 transcription factors as well as purmorphamine (a small-molecule agonist of Smoothened) are able to activate the Hedgehog pathway and are the proposed reagents currently tested for future treatment of ciliopathies Citation[66].
7. Changes to extracellular signalling can also lead to skeletal dysplasia phenotypes
ECM molecules are important for the sequestration and diffusion of endocrine, paracrine and autocrine molecules in the dense cartilage tissue Citation[67]. Mutations inducing changes in the chemical and physical composition of the ECM may therefore have detrimental effects on the signalling pathways driving development and differentiation in cartilage. For example, changes in Indian hedgehog signalling have been detected in DTDST cartilage Citation[68] and in transgenic mice harbouring the deletion of exon 48 in the mouse alpha1(II) procollagen gene Citation[69].
Mutations in the genes encoding molecules important in cartilage signalling have also been implicated in GSDs. Eiken syndrome is a recessive skeletal condition resulting from a truncating mutation in the parathyroid hormone-related peptide type 1 receptor (PTHR1) and mutations in the PTHR1 have also been found in certain forms of Jansen dysplasia Citation[2]. SHOX haploinsufficiency leads to perturbed programmed cell death of hypertrophic chondrocytes and premature epiphyseal fusion of the distal radius in patients. In the case of truncating and loss of function mutations, silencing of the defective receptors, the use of soluble agonists and activators of the signalling pathways or the use of soluble receptors and enzyme replacement therapy (ERT) can all be proposed as potential therapeutic avenues. For example, in the case of hypophosphatasia due to loss of function mutations in the gene encoding tissue-nonspecific alkaline phosphatase, asfotase alfa (a first-in-class ERT) is undergoing evaluation Citation[70–72].
Conversely, numerous GSDs result from activating mutations affecting the signalling pathways in the cartilage Citation[2]. Mutations within the fibroblast growth factor receptors (FGFR) 1 – 3 cause a wide range of skeletal disorders, including diseases primarily characterized by craniosynostosis such as osteoglophonic dysplasia (FGFR1, Citation[73]), Apert (FGFR2, Citation[74]), Crouzon (FGFR2, Citation[75]), Pfeiffer (FGFR1 and FGFR2, Citation[76]), Beare-Stevenson cutis grata (FGFR2, Citation[77]) and Muenke (FGFR3, Citation[78]) syndromes, in addition to the dwarfing syndromes ACH, hypochondroplasia and thanatophoric dysplasia (all FGFR3, Citation[79]). FGFRs are plasma membrane receptor tyrosine kinases that mediate intracellular signalling upon ligand binding. While the phenotypes of these disorders vary, they all arise as a consequence of constitutive FGFR protein activation, and disruption to downstream signalling cascades.
Other GSDs share this common disease basis with activating mutations found in different genes causing diverse disorders such as Jansen’s metaphyseal chondrodysplasia (PTH1R) Citation[80], fibrous dysplasia (GNAS, Citation[81]) and brachyolmia type 3 and metatrophic dysplasia (TRPV4) () Citation[82]. Similar to FGFR1 – 3, PTH1R encodes a cell surface receptor (parathyroid hormone/parathyroid hormone-related peptide receptor); however, downstream signalling occurs through several different guanine-nucleotide binding proteins (G-proteins) Citation[83], while GNAS encodes the α subunit of one of these G-proteins, Gsα. Constitutive activation of both PTH/PTHrP and Gsα leads to the excessive synthesis of cyclic AMP, disrupted chondrocyte signalling and bone pathology Citation[80,84]. Activating mutations in transient receptor potential vallinoid family member 4 (TRPV4) leads to a delay in bone mineralization and a spectrum of diseases from autosomal dominant brachyolmia to lethal metatrophic dysplasia. TRPV4 (transient receptor potential vanilloid 4) encodes a calcium permeable cation channel and disease-causing mutations constitutively activate this channel resulting in an uncontrolled influx of calcium into chondrocytes, activation of follistatin Citation[85], increased inhibition of BMP activity Citation[86] and ultimately, improper bone formation (). Finally, one of the best examples of this class of GSD is fibrodysplasia ossificans progressive (FOP), which results from heterozygous activating mutations in Activin receptor A, type I/Activin-like kinase 2 (ACVR1/ALK2), a bone morphogenetic protein receptor. The genetic basis of FOP and relevant therapeutic targets and approaches has been extensively reviewed by Frederick Kaplan and colleagues Citation[87]. Briefly, targeting the bone morphogenetic protein signalling pathway has been proposed as a promising therapeutic target and palovarotene, a retinoic acid receptor γ agonist Citation[88], is currently in Phase II clinical trials for patients with FOP.
Existing therapies for GSDs caused by constitutively activating mutations remain limited, although insights into their molecular pathogeneses have yielded some exciting therapeutic targets (). A murine model introduced with a dominant-negative FGFR1 construct in utero prevented suture fusion within the skull Citation[89], providing proof-of-principle that the abnormal suture fusion of FGFR-related craniosynostosis may be modulated by engineered FGFR receptors. Perhaps more promising as potential therapies, both RNA interference of FGFR2 and pharmacological inhibition of MEK-ERK signalling have been shown to prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis and reduce cranial suture fusion Citation[90,91].
Table 4. Disease mechanisms and potential therapeutic targets in selected GSDs resulting from constitutively activating mutations.
A significant development in efforts to treat ACH was the discovery that CNP overexpression rescues the phenotype in an ACH mouse model by inhibiting FGFR3 downstream signalling through the MAPK pathway Citation[92]. Recent efforts have focused upon optimizing the pharmacological properties of CNP by designing and validating CNP analogues such as BMN 111 Citation[93,94], or improving production yields Citation[95]. An alternative to CNP-based therapy has involved the repositioned use of a FDA-approved drug (meclizine used for treatment of motion sickness) Citation[96]. In this study, human chondrosarcoma cells expressing constitutively active mutants of FGFR3 that cause thanatophoric dysplasia, SADDAN and achondrogenesis were treated in vitro with meclozine and this ameliorated abnormally suppressed cell proliferation. Comparison with the effect of CNP on the same cell models revealed that meclozine was as efficient as CNP in attenuating the abnormal FGFR3 signalling. Subsequent studies validated this result in a mouse model of ACH, showing that the bone lengths of both wild-type and mutant mice treated with meclozine were significantly longer than in untreated mice Citation[97]. Given that meclozine is already an FDA-approved drug, it is ideally positioned for assessment for clinical use in the treatment of short stature in achondrogenesis.
The use of statins to treat FGFR3-related disorders, ACH and tanatophoric dysplasia type I (TD1) has recently been proposed Citation[98]. Yamashita et al. have demonstrated that statins, previously used in the treatment of tumours and of disorders in the cardiovascular, nervous and immune systems, were able to upregulate the expression of sox9, type II collagen and aggrecan in cell and animal models of ACH and TD1 Citation[98]. They have also demonstrated a decrease in the protein levels of FGFR3 in the patient-derived induced pluripotent stem cells (iPSCs) differentiated into the chondrogenic lineage and in the Fgfr3Ach mouse model of ACH. Interestingly, the decrease in the protein level did not correlate with a decrease in mRNA level for FRFR3, which was actually slightly increased upon the statin treatment. Treatment of chondrocytes derived from the Fgfr3Ach mice with lovastatin in the presence of proteasomal inhibitors revealed that the statin partially upregulated the proteosomal degradation of FGFR3 in the treated cells and animals Citation[98].
Several approaches have been proposed as possible therapeutic interventions for TRPV4-related disease Citation[99]. Existing agents capable of blocking the calcium-permeable TRPV4 channel (GSK2193874 Citation[100] and HC-067047 Citation[101]) represent candidate drugs that would be highly appropriate for administration to mouse models of TRPV4-related disease Citation[102] and subsequent phenotypic analysis. Alternatively, inhibition of TRPV4 protein expression using shRNA might be another way to circumvent the harmful effect of uncontrolled calcium influx into chondrocytes.
Perhaps the most effective clinically approved treatment for a GSD caused by a constitutively activating mutation is the intravenous administration of bisphosphonates to treat fibrous dysplasia. This class of drugs possesses a common basic structure similar to pyrophosphate that inhibits bone resorption and has been used with success for decades. In addition to a decreased turnover of bone, the radiological aspect of existing bone lesions is improved, as is the experience of pain reported by patients Citation[103]; however, a subset of patients remain nonresponsive to bisphosphonate treatment. The relatively recent discovery of GNAS as the disease locus of fibrous dysplasia raises the possibility of targeting the constitutively active Gsα protein and downstream effectors in future therapeutic interventions.
8. Reduced chondrocyte proliferation and increased dysregulated apoptosis are the downstream effects and a shared disease mechanism of many GSD mutations
Several studies have recently demonstrated that reduced chondrocyte proliferation, increased and/or dysregulated apoptosis in the growth plates of mouse models is a major pathological component of various GSDs, including those resulting from mutations in genes encoding cartilage structural proteins (Comp, Matn3 and Col2a1) Citation[8,12,104], a sulphate transporter (Slc26a2) Citation[105] and components of the trans-golgi network (GMAP-210) Citation[32]. These pathomolecular mechanisms are particularly relevant to those GSDs that have a significant epiphyseal involvement, such as PSACH-MED, DTDST and the type II collagenopathies, but are perhaps not so relevant for metaphyseal chondrodysplasias such at MCDS where the pathology involves only non-proliferating hypertrophic chondrocytes Citation[16].
Defining the relative contribution of reduced chondrocyte proliferation, increased and/or dysregulated apoptosis to growth plate dysplasia and reduced bone growth is experimentally challenging; however, the study of novel ‘ER-stress phenocopies’ has recently provided new insight into the specific impact of these different disease mechanisms Citation[16,106,107]. The cartilage-specific expression of mutant forms of thyroglobulin has confirmed that reduced chondrocyte proliferation resulting from an intracellular stress caused by the accumulation of a misfolded protein and in the absence of perturbations to apoptosis was sufficient to cause a significant reduction in long bone growth Citation[107].
In summary, these innovative studies therefore defined reduced chondrocyte proliferation as a major determinant of reduced bone growth in epiphyseal dysplasias, which holds the promise of therapeutic intervention or as a robust readout of drug efficacy in these pre-clinical models of GSDs.
9. Soft tissue complications are a common factor in GSDs
Cartilage, bone, tendon, ligament and skeletal muscle are all tissues of the mesenchymal lineage. It is therefore not surprising that many of the structural molecules implicated in GSDs are also expressed in more than one of these tissues. Musculoskeletal complications are therefore an additional complication that is important to consider in terms of clinical management of GSDs. Patients with MED Citation[108,109], Camurati-Englemann disease Citation[110,111], Marfan syndrome Citation[112,113] and Schwartz-Jampel syndrome Citation[114,115] present with musculoskeletal complications (muscle weakness, muscle stiffness, joint laxity, easy fatigue) that are often difficult to diagnose upon biopsy alone. Understanding the disease mechanisms underlying these clinical complications is of utmost importance, especially considering that these symptoms can manifest prior to the onset of skeletal manifestations and can lead to an initial misdiagnosis. Moreover, mesenchymal tissues are highly mechanically responsive and able to regulate gene expression depending upon the mechanical stimulus Citation[116,117]. It is therefore interesting to speculate that the understanding of the musculoskeletal complications associated with GSDs may lead to a better physiotherapy regime and management of the patients in the future.
10. Biomarkers are important for monitoring disease progression and efficacy of treatment
A lack of relevant disease biomarkers has been a key factor to the delay in the development of new therapeutic targets, largely due the interdependency between biomarker and drug development pipelines Citation[118]. Worryingly there are currently no reliable and readily quantifiable biomarkers that allow pre-symptomatic diagnosis of most GSDs, or to monitor responses to therapeutic regimes. This lack of suitable specific biomarkers reflects the complexity of the biomarker pipeline, which involves the co-dependent processes of identification, verification, validation and reliable detection and quantification in easily obtained biological samples such as blood, urine and cell culture medium.
The complete ECM portrait of bone and cartilage comprises a limited number of ∼300 different components termed the ‘core matrisome’ Citation[118], which limits the available ‘pool’ of potential proteins and/or degradation products that may be found in serum. The determination of biomarkers in serum or urine is widely used to screen for specific pathologies of various organs. For example, individuals with skeletal disorders can be monitored for biomarkers of bone formation, bone resorption or cartilage degradation, which helps to discriminate between different types of skeletal pathologies. Moreover, the information obtained from this analysis can be relevant for individual treatment regimes, for example, not every patient displaying reduced bone mass will necessarily profit from the commonly used anti-resorptive medication. In addition to the well-established relevance of biomarkers in disease diagnosis and personalized treatment, their altered concentrations can also be causative for disease-associated pathologies, such as the case for pyridoxal-5′-phosphate and pyrophosphate in hypophosphatasia, TGFß in geleophysic dysplasia or for FGF23 in hypophosphatemic rickets. In particular, the latter example underscores the relevance of identifying disease-associated biomarkers since FGF23 is now considered to be one of the key regulators of phosphate homeostasis in humans Citation[118].
The use of Omics technologies and Systems analysis to identify phenotype and/or genotype-specific disease profiles promises to provide a plethora of novel putative biomarkers that can be fully investigated through relevant cell and animal models. Ultimately, personalized treatments and care strategies will require relevant biomarkers to monitor efficacy of treatment and disease progression. Therefore, novel biomarkers are urgently required for detecting pre-clinical disease, to monitor disease progression and as a prerequisite for clinical trials of new therapeutic targets. This lack of suitable biomarkers is recognized as a major hindrance in translating research into patient benefits.
11. European-wide networks generate critical mass for diagnostic and research excellence: the search for common therapeutic targets
In Europe, the skeletal genetics field has been fortunate over the last 15 years to secure significant funding from the European Commission, via its various Framework Programs, to establish three contiguous clinical and/or research networks focused on rare skeletal diseases. The origin of these successful large-scale collaborative projects lies with a Concerted Action (1995 – 2000), which provided the first opportunity for both clinical and research experts in GSDs to interact in a Pan-European environment and this ultimately fostered important collaborations and ground-breaking ideas that would lead to future GSD networks.
The FP5-funded European Skeletal Dysplasia Network (ESDN) for research and diagnosis (2002 – 2006) was one of the first networks of expertise in the field of rare diseases to use information and communications technology tools for the purposes of tele-expertise and medical diagnosis. Since September 2003, ESDN has received over 2000 referrals through an on-line Case Manager and 450 users have accessed ESDN from 45 different countries worldwide. During this initial funding period, research activities within ESDN developed several relevant mouse models of GSDs and identified the first disease mechanisms, which would later be defined as hallmark features of disease pathology and be tentatively proposed as potential therapeutic targets.
EuroGrow (2007 – 2010) was an FP6 project that focused on the phenotyping of mouse models of a select group of GSDs in order to develop and validate experimental approaches for deep-phenotyping including omics-based analysis. This project successfully identified a range of common disease mechanisms between phenotypically different GSDs and laid the foundation for future large-scale projects.
The FP7-funded SYBIL project (Systems Biology for the functional validation of genetic determinants of skeletal diseases; http://www.sybil-fp7.eu) is a large-scale collaborative project that aims to functionally validate genetic determinants of common and rare skeletal diseases to gain a mechanistic understanding of the disease processes and age-related changes and to deliver new and validated therapeutic targets. The outcomes of this project will include the generation and deep phenotyping of a diverse range of cell and animal models of GSDs that will generate new knowledge on disease mechanisms. The major strength of this coordinated and multidisciplinary approach is the use of System Biology to underpin the extensive ‘omics’-based analysis that will identify common disease mechanisms and potential therapeutic targets in an unbiased and iterative process.
Finally, National GSD networks such as the Skeletal Dysplasia Group (UK), SKELNET (Germany) and Skeldys.org (Switzerland) have provided a combination of both formal and informal discussion and diagnostic forums, whilst the establishment within several EU countries of National Centers of Excellence for specific GSDs has generated critical mass for delivering clinical best practice and translational research.
In summary, trans-European networks of clinical and research excellence in GSDs have a proven track record in delivering an efficient world class diagnostic service and generating new knowledge on disease mechanisms that will translate into novel therapeutic targets that might even be shared amongst groups of different disease (i.e. Common amongst the Rare). Moreover, interactions with patient self-support groups, the International Rare Diseases Research Consortium (IRDiRC) and the establishment of a European Reference Network in the GSD domain will all help to accelerate and deliver translational research through broad international collaborations and the sharing of best practice.
12. Clinical utility and patient expectations
The development of potential therapeutic targets to the point at which clinical trials can be performed and pharmacological agents ultimately brought to market is a notoriously challenging process with high attrition rates Citation[119]. The low prevalence of individual GSDs is a further challenge to the viable commercial development of products in this area. In some cases, it has been possible to apply drugs already used for the treatment of related conditions to rare GSDs. For example, the use of bisphosophonates in the treatment of adults and particularly children with Osteogenesis Imperfecta (OI) has been credited with reducing fracture frequency and improving quality of life Citation[120,121]; however, recent studies have called this into question Citation[122]. The example of the bisphosphonates illustrates some of the challenges of demonstrating clinical utility in the use of drugs in this group of diseases. This is even more difficult in conditions where, unlike OI, the relatively hard end points of fracture frequency and changes in vertebral morphology are not applicable. It has been suggested that changes in final adult height might be a relevant measure. It is unlikely that changes of sufficient magnitude to truly alter functional outcome will be achieved in many of the GSDs, particularly when the condition is associated with very significant reductions not only in stature but also in reach. Alternative measures of improved symptom control, particularly reduction in pain and neurological symptoms with improvements in mobility, will need to be assessed. A particular challenge in this area will be the lack of availability of baseline data from which assessments of change can be made, even for the most common of the GSDs.
Recent discussions with support groups around the clinical trials currently being carried out for the BioMarin product BMN-111 have emphasized the importance of these issues. Concerns have been expressed that undue focus on stature is inappropriate in the context of the symptoms people with ACH have on a daily basis, whilst a reduction of the neurological impairment associated with neural axis compression would be extremely welcome.
The rarity of the GSDs will pose significant challenges not only to those developing therapeutic agents but also to regulators and healthcare commissioners and providers. The relatively small market available for drugs targeted at specific molecular targets or even specific genetic pathways limits the financial viability of such agents and is part of the wider debate around the development of treatments for rare diseases Citation[123].
13. Expert opinion
The extensive clinical variability and genetic heterogeneity of GSDs, coupled with complex disease mechanisms, renders this extensive group of rare diseases a bench to bedside challenge. Indeed, this large number of different and highly complex phenotypes makes the identification, validation and development of potential therapies almost impossible for anything other than the most common GSDs. As an alternative approach, we might consider identifying genotype- and/or phenotype-independent ‘core disease mechanisms’ that are shared amongst families of clinically unrelated GSDs. This approach would allow the focusing of resources into several areas of concerted investigation that have the potential to identify and validate therapeutic targets with a broad application to GSDs, inherited connective tissues as a whole and rare genetic disease in general. Indeed, Jürgen Spranger first suggested the idea of ‘bone dysplasia families’ in 1985 Citation[124] and proposed that phenotypes with a similar clinical and radiographic phenotype would likely have a similar disease mechanism. Thirty years later, we can now expand upon this pioneering concept and propose that common disease mechanisms can also be shared amongst clinically different phenotypes (‘common amongst the rare’).
In this context, ER stress has been associated with a diverse range of genetic diseases and chronic conditions such as skeletal dysplasia (as discussed in this review), myopathy Citation[125], cerebro-vascular Citation[42], kidney Citation[126], ischaemia and cardiovascular diseases Citation[127]. Moreover, ER stress is emerging as a very attractive target that is being successfully exploited in a broad range of diseases including neuropathy, juvenile-onset open-angle glaucoma, obesity, diabetes, asthma and epidermolysis bullosa, to name but a few. Historically many GSDs were considered diseases of the ECM and proposed therapeutic interventions involved the removal and/or correction of the relevant mutated gene or abnormal gene product. This was particularly the case with dominant-negative mutations in the large structural proteins of the cartilage ECM such as type II collagen Citation[50]. However, emerging knowledge suggests that the primary genetic defect may be less important than the cells’ response to the expression of the mutant gene product Citation[107]. Moreover, the largely overlooked response of a cell (i.e. chondrocyte) to the abnormal extracellular environment is also important for disease progression as illustrated by several GSDs discussed in this review.
It is important that ‘omics’-based approaches and technologies are systematically applied to the study of rare GSDs so that definitive reference profiles and disease signatures are generated for each phenotype. These can then be used in a Systems Biology approach to identify both common and dissimilar pathological signatures and disease mechanisms. This approach is entirely dependent upon relevant in vitro and in vivo models (and also novel ‘disease-mechanism phenocopies’ Citation[107]) for testing new diagnostic and prognostic tools and for determining the molecular mechanisms that underpin the pathophysiology so that effective therapeutic treatments can be developed and validated. This approach will eventually lead to personalized treatments and care strategies centred on shared disease mechanisms with the use of relevant biomarkers to monitor the efficacy of treatment and disease progression.
It is vital that all relevant stakeholders are involved from the outset in defining the appropriate outcomes of any potential therapeutic regime. The perceptions of a successful therapy can differ widely between the clinical academic community and the relevant patient-support groups and it is vital that there is engagement on all these issues.
In summary, the identification of causative genes and mutations for GSDs over the last 20 years, coupled with the generation and in-depth analysis of a plethora of relevant cell and mouse models, has derived new knowledge on disease mechanisms and suggested potential therapeutic targets. The fast-evolving hypothesis that clinically disparate diseases can share common disease mechanisms is a powerful concept that will generate critical mass for the identification and validation of novel therapeutic targets and biomarkers.
Endoplasmic reticulum stress is a shared disease mechanism and potential therapeutic target in a diverse range of Genetic skeletal diseases (GSDs) resulting from dominant-negative mutations in cartilage structural proteins.
Disruption to protein trafficking in chondrocytes leads to a variety of chondrodysplasia phenotypes, which highlights that ‘professionally secreting cells’ such as chondrocytes are highly susceptible to perturbations in ER homeostasis and defects in protein trafficking and secretion.
The incorporation of mutant proteins into the extracellular matrix leads to changes in the composition and properties of cartilage.
Reduced chondrocyte proliferation, increased and/or dysregulated apoptosis are common downstream effects for a range of different GDS and are robust readouts for pre-clinical studies.
Numerous GSDs result from mutations affecting signalling pathways in cartilage and these are the targets of new therapeutic interventions.
Declaration of interest
The author’s work presented in this review was supported by the Wellcome Trust (no. 084353/Z/07/Z) and the European Community’s Seventh Framework Programme under grant agreement no. 602300 (SYBIL). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Notes
This box summarizes key points contained in the article.
Related Research Data
Bibliography
- Warman ML, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A 2011;155A(5):943-68
- Krakow D, Rimoin DL. The skeletal dysplasias. Genet Med 2010;12(6):327-41
- Yasoda A, Nakao K. Translational research of C-type natriuretic peptide (CNP) into skeletal dysplasias. Endocr J 2011;57(8):659-66
- Glorieux FH. Bisphosphonate therapy for severe osteogenesis imperfecta. J Pediatr Endocrinol Metab 2000;13 Suppl 2:989-92
- Makitie AA, Tornwall J, Makitie O. Bisphosphonate treatment in craniofacial fibrous dysplasia--a case report and review of the literature. Clin Rheumatol 2008;27(6):809-12
- Valayannopoulos V, Wijburg FA. Therapy for the mucopolysaccharidoses. Rheumatology (Oxford) 2012;50(Suppl 5):v49-59
- Corbacioglu S, Honig M, Lahr G, et al. Stem cell transplantation in children with infantile osteopetrosis is associated with a high incidence of VOD, which could be prevented with defibrotide. Bone Marrow Transplant 2006;38(8):547-53
- Briggs MD, Bell PA, Pirog KA. The utility of mouse models to provide information regarding the pathomolecular mechanisms in human genetic skeletal diseases: The emerging role of endoplasmic reticulum stress (Review). Int J Mol Med 2015;35(6):1483-92
- Boot-Handford RP, Briggs MD. The unfolded protein response and its relevance to connective tissue diseases. Cell Tissue Res 2009;339(1):197-211
- Tsang KY, Chan D, Bateman JF, Cheah KS. In vivo cellular adaptation to ER stress: survival strategies with double-edged consequences. J Cell Sci 2010;123(Pt 13):2145-54
- Yang RC, Chen MH, Chen PY, et al. A mutation of the Col2a1 gene (G1170S) alters the transgenic murine phenotype and cartilage matrix homeostasis. J Formos Med Assoc 2014;113(11):803-12
- Liang G, Lian C, Huang D, et al. Endoplasmic reticulum stress-unfolding protein response-apoptosis cascade causes chondrodysplasia in a col2a1 p.Gly1170Ser mutated mouse model. PLoS One 2014;9(1):e86894
- Okada M, Ikegawa S, Morioka M, et al. Modeling type II collagenopathy skeletal dysplasia by directed conversion and induced pluripotent stem cells. Hum Mol Genet 2015;24(2):299-313
- Cameron NE. Role of endoplasmic reticulum stress in diabetic neuropathy. Diabetes 2013;62(3):696-7
- Gong B, Zhang LY, Pang CP, et al. Trimethylamine N-oxide alleviates the severe aggregation and ER stress caused by G98R alphaA-crystallin. Mol Vis 2009;15:2829-40
- Rajpar MH, McDermott B, Kung L, et al. Targeted induction of endoplasmic reticulum stress induces cartilage pathology. PLoS Genet 2009;5(10):e1000691
- Leighton MP, Nundlall S, Starborg T, et al. Decreased chondrocyte proliferation and dysregulated apoptosis in the cartilage growth plate are key features of a murine model of epiphyseal dysplasia caused by a matn3 mutation. Hum Mol Genet 2007;16(14):1728-41
- Suleman F, Gualeni B, Gregson HJ, et al. A novel form of chondrocyte stress is triggered by a COMP mutation causing pseudoachondroplasia. Hum Mutat 2012;33(1):218-31
- Pirog-Garcia KA, Meadows RS, Knowles L, et al. Reduced cell proliferation and increased apoptosis are significant pathological mechanisms in a murine model of mild pseudoachondroplasia resulting from a mutation in the C-terminal domain of COMP. Hum Mol Genet 2007;16(17):2072-88
- Posey KL, Alcorn JL, Hecht JT. Pseudoachondroplasia/COMP - translating from the bench to the bedside. Matrix Biol 2014;37:167-73
- Cameron TL, Bell KM, Tatarczuch L, et al. Transcriptional profiling of chondrodysplasia growth plate cartilage reveals adaptive ER-stress networks that allow survival but disrupt hypertrophy. PLoS ONE 2011;6(9):e24600
- Nundlall S, Rajpar MH, Bell PA, et al. An unfolded protein response is the initial cellular response to the expression of mutant matrilin-3 in a mouse model of multiple epiphyseal dysplasia. Cell Stress Chaperones 2010;15(6):835-49
- Hartley CL, Edwards S, Mullan L, et al. Armet/Manf and Creld2 are components of a specialized ER stress response provoked by inappropriate formation of disulphide bonds: implications for genetic skeletal diseases. Hum Mol Genet 2013;22(25):5262-75
- Linz A, Knieper Y, Gronau T, et al.R stress during the pubertal growth spurt results in impaired long bone growth in chondrocyte-specific ERp57 knockout mice. J Bone Miner Res 2015;30(8):1481-93
- Cameron TL, Gresshoff IL, Bell KM, et al. Cartilage-specific ablation of XBP1 signaling in mouse results in a chondrodysplasia characterized by reduced chondrocyte proliferation and delayed cartilage maturation and mineralization. Osteoarthritis Cartilage 2015;23(4):661-70
- Posey KL, Coustry F, Veerisetty AC, et al. Chop (Ddit3) is essential for D469del-COMP retention and cell death in chondrocytes in an inducible transgenic mouse model of pseudoachondroplasia. Am J Pathol 2012;180(2):727-37
- Coustry F, Posey KL, Liu P, et al. D469del-COMP retention in chondrocytes stimulates caspase-independent necroptosis. Am J Pathol 2012;180(2):738-48
- Posey KL, Coustry F, Veerisetty AC, et al. Antioxidant and anti-inflammatory agents mitigate pathology in a mouse model of pseudoachondroplasia. Hum Mol Genet 2015;24(14):3918-28
- Posey KL, Coustry F, Veerisetty AC, et al. Chondrocyte-specific pathology during skeletal growth and therapeutics in a murine model of pseudoachondroplasia. J Bone Miner Res 2014;29(5):1258-68
- Ali BR, Xu H, Akawi NA, et al. Trafficking defects and loss of ligand binding are the underlying causes of all reported DDR2 missense mutations found in SMED-SL patients. Hum Mol Genet 2010;19(11):2239-50
- Labrador JP, Azcoitia V, Tuckermann J, et al. The collagen receptor DDR2 regulates proliferation and its elimination leads to dwarfism. EMBO Rep 2001;2(5):446-52
- Kawai I, Hisaki T, Sugiura K, et al. Discoidin domain receptor 2 (DDR2) regulates proliferation of endochondral cells in mice. Biochem Biophys Res Commun 2012;427(3):611-17
- Tiller GE, Hannig VL, Dozier D, et al. A recurrent RNA-splicing mutation in the SEDL gene causes X-linked spondyloepiphyseal dysplasia tarda. Am J Hum Genet 2001;68(6):1398-407
- Venditti R, Scanu T, Santoro M, et al. Sedlin controls the ER export of procollagen by regulating the Sar1 cycle. Science 2012;337(6102):1668-72
- Choi MY, Chan CC, Chan D, et al. Biochemical consequences of sedlin mutations that cause spondyloepiphyseal dysplasia tarda. Biochem J 2009;423(2):233-42
- Boyadjiev SA, Fromme JC, Ben J, et al. Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat Genet 2006;38(10):1192-7
- Lang MR, Lapierre LA, Frotscher M, et al. Secretory COPII coat component Sec23a is essential for craniofacial chondrocyte maturation. Nat Genet 2006;38(10):1198-203
- Sarmah S, Barrallo-Gimeno A, Melville DB, et al. Sec24D-dependent transport of extracellular matrix proteins is required for zebrafish skeletal morphogenesis. PLoS ONE 2010;5(4):e10367
- Smits P, Bolton AD, Funari V, et al. Lethal skeletal dysplasia in mice and humans lacking the golgin GMAP-210. N Engl J Med 2010;362(3):206-16
- Le Corre S, Eyre D, Drummond IA. Modulation of the secretory pathway rescues zebrafish polycystic kidney disease pathology. J Am Soc Nephrol 2014;25(8):1749-59
- Bateman JF, Boot-Handford RP, Lamande SR. Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nat Rev Genet 2009;10(3):173-83
- Murray LS, Lu Y, Taggart A, et al. Chemical chaperone treatment reduces intracellular accumulation of mutant collagen IV and ameliorates the cellular phenotype of a COL4A2 mutation that causes haemorrhagic stroke. Hum Mol Genet 2014;23(2):283-92
- Gualeni B, de Vernejoul MC, Marty-Morieux C, et al. Alteration of proteoglycan sulfation affects bone growth and remodeling. Bone 2013;54(1):83-91
- Furuichi T, Kayserili H, Hiraoka S, et al. Identification of loss-of-function mutations of SLC35D1 in patients with Schneckenbecken dysplasia, but not with other severe spondylodysplastic dysplasias group diseases. J Med Genet 2009;46(8):562-8
- Svensson L, Aszodi A, Heinegard D, et al. Cartilage oligomeric matrix protein-deficient mice have normal skeletal development. Mol Cell Biol 2002;22(12):4366-71
- Ko Y, Kobbe B, Nicolae C, et al. Matrilin-3 is dispensable for mouse skeletal growth and development. Mol Cell Biol 2004;24(4):1691-9
- Aszodi A, Bateman JF, Gustafsson E, et al. Mammalian skeletogenesis and extracellular matrix: what can we learn from knockout mice? Cell Struct Funct 2000;25(2):73-84
- Hagg R, Hedbom E, Mollers U, et al. Absence of the alpha1(IX) chain leads to a functional knock-out of the entire collagen IX protein in mice. J Biol Chem 1997;272(33):20650-4
- Allen JM, Zamurs L, Brachvogel B, et al. Mice lacking the extracellular matrix protein WARP develop normally but have compromised peripheral nerve structure and function. J Biol Chem 2009;284(18):12020-30
- Arnold WV, Fertala A. Skeletal diseases caused by mutations that affect collagen structure and function. Int J Biochem Cell Biol 2013;45(8):1556-67
- Posey KL, Liu P, Wang HR, et al.Ai reduces expression and intracellular retention of mutant cartilage oligomeric matrix protein. PLoS ONE 2010;5(4):e10302
- Berbari NF, O’Connor AK, Haycraft CJ, Yoder BK. The primary cilium as a complex signaling center. Curr Biol 2009;19:R526–35.
- Whitfield JF. The solitary (primary) cilium–A mechanosensory toggle switch in bone and cartilage cells. Cell Signal 2008;20:1019–24.
- McGlashan SR, Jensen CG, Poole AC. Localization of extracellular matrix receptors on the chondrocyte primary cilium. J Histochem Cytochem 2006;54:1005–14.
- McGlashan S, Cluett E, Jensen C, Poole C. Primary cilia in osteoarthritic chondrocytes: from chondrons to clusters. Dev Dyn 2008;237:2013–20.
- de Andrea C, Wiweger M, Prins F, Bovee J, Romeo S, Hogendoorn P. Primary cilia organization reflects polarity in the growth plate and implies loss of polarity and mosaicism in osteochondroma. Labor Invest 2010;90:1091–101.
- Pirog-Garcia KA, Meadows RS, Knowles L, et al. Reduced cell proliferation and increased apoptosis are significant pathological mechanisms in a murine model of mild pseudoachondroplasia resulting from a mutation in the C-terminal domain of COMP. Human Mol Genet 2007;16:2072–88.
- Hiraoka S, Furuichi T, Nishimura G, et al. Nucleotide-sugar transporter SLC35D1 is critical to chondroitin sulfate synthesis in cartilage and skeletal development in mouse and human. Nat Med 2007;13:1363–7.
- Lowe DA, Lepori-Bui N, Fomin PV, et al. Deficiency in perlecan/HSPG2 during bone development enhances osteogenesis and decreases quality of adult bone in mice. Calcified Tissue Int 2014;95:29–38.
- Arita M, Fertala J, Hou C, et al. Mechanisms of aberrant organization of growth plates in conditional transgenic mouse model of spondyloepiphyseal dysplasia associated with the R992C substitution in collagen II. Am J Pathol 2015;185:214–29.
- Forlino A, Piazza R, Tiveron C, et al. A diastrophic dysplasia sulfate transporter (SLC26A2) mutant mouse: morphological and biochemical characterization of the resulting chondrodysplasia phenotype. Human Mol Genet 2005;14:859–71.
- Bengtsson T, Aszódi A, Nicolae C, et al. Loss of alpha10beta1 integrin expression leads to moderate dysfunction of growth plate chondrocytes. J Cell Sci 2005;118:929–36.
- Aszódi A, Hunziker E, Brakebusch C, Faessler R. Beta1 integrins regulate chondrocyte rotation, G1 progression, and cytokinesis. Genes Dev 2003;17:2465–79.
- Kyostila K, Lappalainen AK, Lohi H. Canine chondrodysplasia caused by a truncating mutation in collagen-binding integrin alpha subunit 10. PLoS One 2013;8:e75621.
- Smith MA, Mohammad RA. Vedolizumab: an alpha4beta7 integrin inhibitor for inflammatory bowel diseases. Ann Pharmacother 2014;48(12):1629–35.
- Lewis C, Krieg PA. Reagents for developmental regulation of Hedgehog signaling. Methods 2014;66(3):390–7.
- Slater M. Dynamic interactions of the extracellular matrix. Histol Histopathol 1996;11(1):175–80.
- Gualeni B, Facchini M, De Leonardis F, et al. Defective proteoglycan sulfation of the growth plate zones causes reduced chondrocyte proliferation via an altered Indian hedgehog signalling. Mat Biol 2010;29(6):453–60.
- Barbieri O, Astigiano S, Morini M, et al. Depletion of cartilage collagen fibrils in mice carrying a dominant negative Col2a1 transgene affects chondrocyte differentiation. Am J Physiol Cell Physiol 2003;285(6):C1504–12.
- Kosnik-Infinger L, Gendron C, Gordon CB, et al. Enzyme replacement therapy for congenital hypophosphatasia allows for surgical treatment of related complex craniosynostosis: a case series. Neurosurg Focus 2015;38(5):E10
- Ozono K. [Enzyme replacement therapy for hypophosphatasia]. Clin Calcium 2014;24(2):257-63
- de la Croix Ndong J, Makowski AJ, Uppuganti S, et al. Asfotase-alpha improves bone growth, mineralization and strength in mouse models of neurofibromatosis type-1. Nat Med 2014;20(8):904-10
- White KE, Cabral JM, Davis SI, et al. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet 2005;76(2):361-7
- Wilkie AO, Slaney SF, Oldridge M, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet 1995;9(2):165-72
- Reardon W, Winter RM, Rutland P, et al. Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nat Genet 1994;8(1):98-103
- Robin NH, Feldman GJ, Mitchell HF, et al. Linkage of Pfeiffer syndrome to chromosome 8 centromere and evidence for genetic heterogeneity. Hum Mol Genet 1994;3(12):2153-8
- Przylepa KA, Paznekas W, Zhang M, et al. Fibroblast growth factor receptor 2 mutations in Beare-Stevenson cutis gyrata syndrome. Nat Genet 1996;13(4):492-4
- Muenke M, Gripp KW, McDonald-McGinn DM, et al. A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. Am J Hum Genet 1997;60(3):555-64
- Naski MC, Wang Q, Xu J, Ornitz DM. Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nat Genet 1996;13(2):233-7
- Bastepe M, Raas-Rothschild A, Silver J, et al. A form of Jansen’s metaphyseal chondrodysplasia with limited metabolic and skeletal abnormalities is caused by a novel activating parathyroid hormone (PTH)/PTH-related peptide receptor mutation. J Clin Endocrinol Metab 2004;89(7):3595-600
- Weinstein LS, Shenker A, Gejman PV, et al. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med 1991;325(24):1688-95
- Rock MJ, Prenen J, Funari VA, et al. Gain-of-function mutations in TRPV4 cause autosomal dominant brachyolmia. Nat Genet 2008;40(8):999-1003
- Schwindinger WF, Fredericks J, Watkins L, et al. Coupling of the PTH/PTHrP receptor to multiple G-proteins. Direct demonstration of receptor activation of Gs, Gq/11, and Gi(1) by [alpha-32P]GTP-gamma-azidoanilide photoaffinity labeling. Endocrine 1998;8(2):201-9
- Regard JB, Cherman N, Palmer D, et al. Wnt/beta-catenin signaling is differentially regulated by Galpha proteins and contributes to fibrous dysplasia. Proc Natl Acad Sci U S A 2011;108(50):20101-6
- Leddy HA, McNulty AL, Lee SH, et al. Follistatin in chondrocytes: the link between TRPV4 channelopathies and skeletal malformations. FASEB J 2014;28(6):2525-37
- Saitta B, Passarini J, Sareen D, et al. Patient-derived skeletal dysplasia induced pluripotent stem cells display abnormal chondrogenic marker expression and regulation by BMP2 and TGFbeta1. Stem Cells Dev 2014;23(13):1464-78
- Kaplan FS, Pignolo RJ, Shore EM. From mysteries to medicines: drug development for fibrodysplasia ossificans progressive. Expert Opin Orphan Drugs 2013;1(8):637-49
- Hind M, Stinchcombe S. Palovarotene, a novel retinoic acid receptor gamma agonist for the treatment of emphysema. Curr Opin Investig Drugs 2009;10(11):1243-50
- Greenwald JA, Mehrara BJ, Spector JA, et al. In vivo modulation of FGF biological activity alters cranial suture fate. Am J Pathol 2001;158(2):441-52
- Shukla V, Coumoul X, Wang RH, et al. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat Genet 2007;39(9):1145-50
- Kim HJ, Lee MH, Park HS, et al. Erk pathway and activator protein 1 play crucial roles in FGF2-stimulated premature cranial suture closure. Dev Dyn 2003;227(3):335-46
- Yasoda A, Komatsu Y, Chusho H, et al. Overexpression of CNP in chondrocytes rescues achondroplasia through a MAPK-dependent pathway. Nat Med 2004;10(1):80-6
- Lorget F, Kaci N, Peng J, et al. Evaluation of the therapeutic potential of a CNP analog in a Fgfr3 mouse model recapitulating achondroplasia. Am J Hum Genet 2012;91(6):1108-14
- Wendt DJ, Dvorak-Ewell M, Bullens S, et al. Neutral endopeptidase-resistant C-type natriuretic peptide variant represents a new therapeutic approach for treatment of fibroblast growth factor receptor 3-related dwarfism. J Pharmacol Exp Ther 2015;353(1):132-49
- Long S, Wendt DJ, Bell SM, et al. A novel method for the large-scale production of PG-CNP37, a C-type natriuretic peptide analogue. J Biotechnol 2012;164(2):196-201
- Matsushita M, Kitoh H, Ohkawara B, et al. Meclozine facilitates proliferation and differentiation of chondrocytes by attenuating abnormally activated FGFR3 signaling in achondroplasia. PLoS One 2013;8(12):e81569
- Matsushita M, Hasegawa S, Kitoh H, et al. Meclozine Promotes Longitudinal Skeletal Growth in Transgenic Mice with Achondroplasia Carrying a Gain-of-Function Mutation in the FGFR3 Gene. Endocrinology 2015;156(2):548-54
- Yamashita A, Morioka M, Kishi H, et al. Statin treatment rescues FGFR3 skeletal dysplasia phenotypes. Nature 2014;513(7519):507-11
- McNulty AL, Leddy HA, Liedtke W, Guilak F. TRPV4 as a therapeutic target for joint diseases. Naunyn Schmiedebergs Arch Pharmacol 2015;388(4):437-50
- Thorneloe KS, Cheung M, Bao W, et al. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci Transl Med 2012;4(159):159ra148
- Chen Y, Williams SH, McNulty AL, et al. Temporomandibular joint pain: a critical role for Trpv4 in the trigeminal ganglion. Pain 2013;154(8):1295-304
- Weinstein MM, Tompson SW, Chen Y, et al. Mice expressing mutant Trpv4 recapitulate the human TRPV4 disorders. J Bone Miner Res 2014;29(8):1815-22
- Chapurlat RD, Hugueny P, Delmas PD, Meunier PJ. Treatment of fibrous dysplasia of bone with intravenous pamidronate: long-term effectiveness and evaluation of predictors of response to treatment. Bone 2004;35(1):235-42
- Arita M, Fertala J, Hou C, et al. Mechanisms of aberrant organization of growth plates in conditional transgenic mouse model of spondyloepiphyseal dysplasia associated with the R992C substitution in collagen II. Am J Pathol 2015;185(1):214-29
- De Leonardis F, Monti L, Gualeni B, et al. Altered signaling in the G1 phase deregulates chondrocyte growth in a mouse model with proteoglycan undersulfation. J Cell Biochem 2014;115(10):1779-86
- Kung LH, Rajpar MH, Preziosi R, et al. Increased classical endoplasmic reticulum stress is sufficient to reduce chondrocyte proliferation rate in the growth plate and decrease bone growth. PLoS One 2015;10(2):e0117016
- Gualeni B, Rajpar MH, Kellogg A, et al. A novel transgenic mouse model of growth plate dysplasia reveals that decreased chondrocyte proliferation due to chronic ER stress is a key factor in reduced bone growth. Dis Model Mech 2013;6(6):1414-25
- Jakkula E, Lohiniva J, Capone A, et al. A recurrent R718W mutation in COMP results in multiple epiphyseal dysplasia with mild myopathy: clinical and pathogenetic overlap with collagen IX mutations. J Med Genet 2003;40:942–8.
- Jackson GC, Marcus-Soekarman D, Stolte-Dijkstra I, et al. Type IX collagen gene mutations can result in multiple epiphyseal dysplasia that is associated with osteochondritis dissecans and a mild myopathy. Am J Med Gen 2010;152A(4):863–9.
- Behan WM, Longman C, Petty RK, et al. Muscle fibrillin deficiency in Marfan’s syndrome myopathy. J Neurol Neurosur Psychiat 2003;74(5):633–8.
- Voermans NC, Bonnemann CG, Huijing PA, et al. Clinical and molecular overlap between myopathies and inherited connective tissue diseases. Neuromuscul Disord 2008;18(11):843–56.
- Bondestam J, Pihko H, Vanhanen SL, et al. Skeletal dysplasia presenting as a neuromuscular disorder - report of three children. Neuromuscul Disord 2007;17(3):231–4.
- Yoshioka H, Mino M, Kiyosawa N, et al. Muscular changes in Engelmann’s disease. Arch Dis Child 1980;55(9):716–19.
- van Huffelen AC, Gabreels FJ, Luypen-vd Horst JS, et al. Chondrodystrophic myotonia. A report of two unrelated Dutch patients. Neuropadiatrie 1974;5(1):71–90.
- Schwartz O, Jampel RS. Congenital blepharophimosis associated with a unique generalized myopathy. Arch Ophthalmol 1962;68:52–7.
- Ho FC, Zhang W, Li YY, Chan BP. Mechanoresponsive, omni-directional and local matrix-degrading actin protrusions in human mesenchymal stem cells microencapsulated in a 3D collagen matrix. Biomaterials 2015;53:392–405.
- Schoenau E. From mechanostat theory to development of the Functional Muscle-Bone-Unit. J Musculoskelet Neuronal Interact 2005;5(3):232–8.
- Mobasheri A. Osteoarthritis year 2012 in review: biomarkers. Osteoarthritis Cartilage 2012;20(12):1451-64
- Kakkis ED, O’Donovan M, Cox G, et al. Recommendations for the development of rare disease drugs using the accelerated approval pathway and for qualifying biomarkers as primary endpoints. Orphanet J Rare Dis 2015;10(1):16
- Sousa T, Bompadre V, White KK. Musculoskeletal functional outcomes in children with osteogenesis imperfecta: associations with disease severity and pamidronate therapy. J Pediatr Orthop 2014;34(1):118-22
- Alcausin MB, Briody J, Pacey V, et al. Intravenous pamidronate treatment in children with moderate-to-severe osteogenesis imperfecta started under three years of age. Horm Res Paediatr 2013;79(6):333-40
- Hald JD, Evangelou E, Langdahl BL, Ralston SH. Bisphosphonates for the prevention of fractures in osteogenesis imperfecta: meta-analysis of placebo-controlled trials. J Bone Miner Res 2015;30(5):929-33
- Drummond M, Towse A. Orphan drugs policies: a suitable case for treatment. Eur J Health Econ 2014;15(4):335-40
- Spranger J. Pattern recognition in bone dysplasias. Prog Clin Biol Res 1985;200:315-42
- De Palma S, Capitanio D, Vasso M, et al. Muscle proteomics reveals novel insights into the pathophysiological mechanisms of collagen VI myopathies. J Proteome Res 2014;13(11):5022-30
- Cybulsky AV. The intersecting roles of endoplasmic reticulum stress, ubiquitin- proteasome system, and autophagy in the pathogenesis of proteinuric kidney disease. Kidney Int 2013;84(1):25-33
- Sozen E, Karademir B, Ozer NK. Basic mechanisms in endoplasmic reticulum stress and relation to cardiovascular diseases. Free Radic Biol Med 2015;78:30-41