
Abstract
Epigenetic change is part of the carcinogenic process and a deep reservoir for biomarker discovery. Reversible methylation of cytosines is noteworthy because it can be measured accurately and easily by various molecular methods and DNA methylation patterns are linked to important tumourigenic pathways. Clinically relevant methylation changes are known in common human cancers such as cervix, prostate, breast, colon, bladder, stomach and lung. Differential methylation may have a central role in the development and outcome of most if not all human malignancies. The advent of deep sequencing holds great promise for epigenomics, with bioinformatics tools ready to reveal large numbers of new targets for prognosis and therapeutic intervention. This review focuses on two selected cancers, namely cervix and prostate, which illustrate the more general themes of epigenetic diagnostics in cancer. Also discussed is differential methylation of specific human and viral DNA targets and laboratory methods for measuring methylation biomarkers.
Cancer biomarkers & the role of epigenetics
Biomarkers are fundamental tools in human healthcare with numerous supporting studies. A computer-based search in the Pubmed database for papers published prior to the end of December 2013 with the terms ‘biomarker or biomarkers’ produced more than 650,000 papers. There are thousands of newly proposed biomarkers but few are ready for use in clinical medicine. Large numbers of novel biomarkers are discovered each year; however, the job of credentialing, validating and qualifying biomarkers for use with patients is slow and has fallen behind the rate of discovery. Virtually all biomarkers have important limitations, and large gaps exist in our knowledge of disease progression at the level of individual patients. Vigorous discovery efforts need to continue in better synchrony with validation efforts. We need thousands of newly validated biomarkers to meaningfully realize the promise of personalized medicine. Escalating medical costs also need to be considered, and so there is a constant search for better, more highly multiplexed, less expensive and more automated tests. The focus of this review concerns the discovery and validation of clinically relevant DNA methylation biomarkers in two selected cancers: cervix and prostate, which will serve to illustrate the more general themes of epigenetic diagnostics in cancer.
DNA methylation is a very important information storage element of the cellular epigenetic machinery and is essential for normal development Citation[1–3]. Aberrant methylation is a central feature of carcinogenesis; it causes defective gene expression, faulty condensation and chromosomal instability, and is a hallmark of cellular defenses acting to silence foreign DNA. Methylation at position 5 of the cytosine ring in a CpG dyad is initiated and maintained by a set of DNA methyl transferases (DNMT), some of which are de-novo (DNMT3A, DNMT3B), while another (DNMT1) is a maintenance enzyme Citation[1]. Methylation is a noteworthy event because 5-methyl cytosine (5-mC) represents a 5th base, and levels of this molecule (and related molecules) in tissue can be measured quite accurately and easily Citation[2,4]. There are several other cytosine modifications that participate in DNA methylation and demethylation events; one of the more notable ones is 5-hydroxymethyl cytosine (5-HmC), which is generated by the Ten-eleven translocation complex of enzymes. 5-HmC appears to be important in certain organs such as the central nervous system and is also an intermediate on the active demethylation pathway for 5-mC Citation[5]. Bisulfite conversion of DNA followed by PCR does not distinguish between 5-mC and 5-HmC. In this review, methylated DNA refers to any region of DNA with one or more modified cytosines that behave like 5-mC after bisulfite conversion.
Figure 1. Examples of DNA methylation modifications on cytosines (C) of CpG dyads and the effects of these changes at the level of the gene and relationship to carcinogenesis. (A) Nucleotide C is shown on the left, this base can be methylated at position 5 of the aromatic ring by one of several DNMT to become 5-methyl cytosine shown on the right. The methylation is reversible and methyl modifications can be changed to hydroxymethyl or other groups or removed by various enzymes. (B) The gross effect of DNA hypermethylation in a gene promoter region, is to turn off transcription of the gene (shown by the curved arrow). Many tumor suppressor genes, such as APC and BCL2 are controlled by methylation, and if the genes are deactivated, then critical checks and balances in cells are removed, which can lead to apoptosis or to carcinogenesis. Similarly, oncogenes such as CCND2 can be activated by removal of methylation marks from important regulatory regions Citation[73,89]. (C) The methylation levels of a DNA region can be measured by several methods. One of the more comprehensive and convenient methods involves bisulfite conversion, which chemically changes nucleotide C into uracil (U). The U then pairs with adenine (A) and upon replication of DNA by PCR the nucleotide U is converted into thymine (T). This C to T change can be measured by sequencing and forms the basis of many DNA methylation quantitation tests. The affected C and T bases are shown in underlined boldface and a methylated C is shown as C*. The upper strand represents the native DNA sequence and the lower strand the same sequence replicated after PCR amplification.
![Figure 1. Examples of DNA methylation modifications on cytosines (C) of CpG dyads and the effects of these changes at the level of the gene and relationship to carcinogenesis. (A) Nucleotide C is shown on the left, this base can be methylated at position 5 of the aromatic ring by one of several DNMT to become 5-methyl cytosine shown on the right. The methylation is reversible and methyl modifications can be changed to hydroxymethyl or other groups or removed by various enzymes. (B) The gross effect of DNA hypermethylation in a gene promoter region, is to turn off transcription of the gene (shown by the curved arrow). Many tumor suppressor genes, such as APC and BCL2 are controlled by methylation, and if the genes are deactivated, then critical checks and balances in cells are removed, which can lead to apoptosis or to carcinogenesis. Similarly, oncogenes such as CCND2 can be activated by removal of methylation marks from important regulatory regions Citation[73,89]. (C) The methylation levels of a DNA region can be measured by several methods. One of the more comprehensive and convenient methods involves bisulfite conversion, which chemically changes nucleotide C into uracil (U). The U then pairs with adenine (A) and upon replication of DNA by PCR the nucleotide U is converted into thymine (T). This C to T change can be measured by sequencing and forms the basis of many DNA methylation quantitation tests. The affected C and T bases are shown in underlined boldface and a methylated C is shown as C*. The upper strand represents the native DNA sequence and the lower strand the same sequence replicated after PCR amplification.](/cms/asset/02d80529-eea5-4e0f-bf86-8a59eee0efee/iero_a_897610_f0001_b.jpg)
Changes in methylation at specific CpG positions in the human genome can turn genes on or off and have been linked to a wide variety of important normal and impaired molecular pathways ; thus DNA methylation is one of the most fertile and tractable reservoirs for new biomarker discovery. Cancer-related changes in methylation of cytosine often occur in areas with a relatively dense representation of CpGs called CpG islands Citation[2,3]. The islands are usually located at, or close to, promoters, and the expression of a very large proportion of genes may be controlled from CpG islands Citation[6]. There can be CpG islands upstream, within and downstream of a given gene and methylation of CpGs in any of these areas can have biomarker value Citation[7]. Methylation changes in CpG islands or at CpG island borders can turn off or turn on gene sets in a specific and co-ordinated way, with long-range effects spanning 1 Mb or more Citation[8]. The reversible silencing process and its effects are complex and extend well beyond a simple set of on/off switches. Although gene silencing by methylation is potentially reversible, it generally has a longer term effect than control of gene expression by transcription factors. Methylation and chromosomal condensation are intrinsically connected to embryogenesis and cell differentiation. Detailed knowledge of methylation patterns allows us to look further into the future of cell fates and glean more accurate information on individual outcomes Citation[7,9]. Changes in methylation of CpGs in human and viral DNA that are not in CpG islands have been observed and may have regulatory importance that needs to be taken into account Citation[6,10].
Epigenetic processes amplify the effects of mutations and can lead to disease in the absence of any detectable relevant genetic changes. Epigenetic pathways are affected by environmental stimuli and insults to a greater extent than classical genetic pathways. Some cancers have a CpG island methylator phenotype that can arise early and drive carcinogenesis. CpG island methylator phenotype varies in different malignancies and may confer poor prognosis Citation[11].
There are several aspects to epigenetics besides DNA methylation, for example, histone modifications (methylation or acetylation) and miRNA processes also have a role in carcinogenesis Citation[1,2,12,13]. These additional elements act more directly in a coordinated way with transcription factors to regulate shorter-term gene expression, while in concert with changes in DNA methylation they lead to more permanent effects on gene expression. Histone and miRNA epigenetics are not considered in this review because of technical complexities in their use as biomarkers that has resulted in slower development of validated clinical assays.
Quantitative DNA methylation assays for cervical cancer
Cervical cancer was one of the most important cancers in women before the advent of widespread cervical screening by cytology. In 2012, the GLOBOCAN Citation[14] cervical cancer estimates were an overall 528,000 cases and 266,000 deaths, of which 83,000 cases and 35,000 deaths were in more developed regions and 445,000 cases and 230,000 deaths were in less developed regions. However, the maintenance of low incidence levels of cervical cancer in developed regions requires frequent screening, most commonly at intervals of between 3 and 5 years. Persistent infection with high-risk human papillomavirus (hrHPV) is the main cause of cervical cancer. Testing for hrHPV DNA with new molecular instruments has excellent performance and reproducibility. Systematic reviews by a number of teams, including Cuzick et al., consistently show HPV testing with a sensitivity (this variable indicates the percentage of positive test results in a group of people who do have the disease or condition of interest) of 90–100% for pre-cancer, compared to a 50–80% sensitivity for screening cytology Citation[15,16]. HPV DNA testing is becoming important globally as the new reference standard for routine screening to detect women with high-grade cervical intraepthelial neoplasia (CIN 2/3). Appropriate triage, biopsy and treatment of these women can greatly lower their risk of cervical cancer for a decade or longer. Several large randomized controlled trials (RCT) have shown that the rate of CIN2/3 after a negative HPV test is considerably lower than the rate after negative cytology Citation[17–20]. In one European study, the cumulative risk of CIN3 or cancer (CIN3+) 6 years after a negative hrHPV test was only 0.27% as compared to a cumulative risk of 0.51% 3 years after a negative cytology Citation[20]. The rate of CIN3+ detected at the second round of screening (between 1 and 5 years after enrolment) was approximately 50% less in women originally screened by HPV testing compared to women screened by cytology Citation[17–20]. Thus by catching and treating more pre-cancer in the first round, the trials found that there was less disease detected in the second round. Recently, a combined analysis of several European RCTs by Ronco et al. which included a total of 176,464 screened women, looked at HPV versus cytology testing to prevent cervical cancer Citation[21]. After 5.5 years of follow-up, a negative HPV test at entry resulted in a cumulative incidence of invasive cancer per 100,000 women of 8.7 (95% CI: 3.3–18.6) compared to 36 (23.2–53.5) for negative cytology. In an RCT in Mexico on 25,061 women comparing HPV DNA testing on vaginal self-collected specimens versus routine cytology, Lazcano and colleagues reported that the HPV DNA test detected 2.4-times more CIN3 (the immediate precursor of cervical cancer), per protocol, at baseline than screening with cytology, which would likely translate into a corresponding decrease in incident invasive cancer in subsequent years Citation[22]. The HPV test detected 4.2-times more prevalent cancers, per protocol, which is also important because invasive cancers detected earlier by HPV testing gives women a better chance of cure.
Not surprisingly, HPV DNA testing has become one of the most common molecular assays in the routine clinical laboratory. With such very high volumes of testing, the impact of even small differences in assay performance are greatly magnified and at a population level, there can be a large cost to pursuing a testing strategy with an overall lower specificity (this variable indicates the percentage of negative test results in a group of people who do not have the condition or disease of interest). An important limitation of hrHPV testing is a comparatively lower specificity (90–93%) than for Papanicolaou cytology (95–98%) Citation[15,16,23]. There has been quite a lot of discussion about which HPV test is the most specific, but in fact a relatively more important point is the overall specificity of the triage algorithm. Consequently, there has been an active search for tests to triage HPV positive screening results so that women who need to be referred to colposcopy can be separated from those who do not. There are several existing and proposed triage tests including cytology, immunostaining for p16 and Ki-67, and genotyping for HPV16, HPV18, HPV33 and HPV45 or combinations Citation[16,24,25].
Tumor viruses are recognized by cells as foreign DNA and their genomes are subject to selective differential methylation and silencing by the defensive system, which more easily recognizes invading genomes at higher copy number. Methylation of viral DNA plays a role in the development of cancer. Entire viral genomes are subject to methylation, but certain regions of the genome, such as important regulatory and promoter regions, can escape silencing. Molecular evolution in a pool of genomes within the cell leads to the selection and survival of incompletely methylated molecules that allow expression of virus oncoproteins. Some viral genomes adapt to a low copy number balance in stem cells and avoid detection. Partially methylated genomes appear to be a determinant of successful persistence and allow the maintenance of tumorigenic stimuli Citation[10,26,27]. Carcinogenic viruses such as hepatitis B virus, Epstein–Barr virus, and Human T-lymphotropic virus exhibit increased methylation of certain genomic regions during the establishment of persistent infection. Maintenance of hypomethylation in the upstream regulatory region (URR) of HPV allows ongoing transcription and is important for the viral life cycle. In contrast, methylation of the viral late (L) regions is an indicator of the length of time that the genome spends in a given host tissue and appears to be a proxy for viral persistence, cell immortalization and progression, depending on the level of methylation on certain CpG sites Citation[10,27].
Differential methylation of the HPV genome was first shown in the 1980s and was studied with greater effort only within the last 10 years Citation[10,26–31]. More recently, the quantitative measurement of DNA methylation has become a leading candidate biomarker for triage of hrHPV positive women to colposcopy. Assays based on HPV DNA methylation have good sensitivity, specificity and positive predictive value (PPV), with the added benefits of a fully molecular reflex testing approach Citation[27,32]. Methylation assays are relatively easy to set up and perform and can be readily automated. Testing can be done directly from self-collected vaginal specimens, thus removing the need for an additional clinic visit to obtain a specimen for cytology or p16 testing, which requires well-preserved cells.
There are three basic approaches to DNA methylation-based triage of HPV positive women: testing of HPV genomes; testing of a set of human biomarker genes; and testing of a combination of HPV and human genes. Certain human genes and specific areas of the HPV genomes (mainly the late and some early regions but not the URR) show extensive methylation changes at multiple CpG sites during persistence.
Classifiers based on methylation of HPV DNA
Differential changes in HPV DNA methylation were first recognized by Burnet and Sleeman in 1984 Citation[26]. In their study of HPV1a, they reported increased methylation of the viral late regions and hypomethylation of the URR and early (E) region, and speculated that these patterns were consistent with the need for ongoing expression of viral early region tumourigenic genes. Recent research by a number of teams has extended these findings to HPV16, HPV18 and other hrHPV types and associated differential methylation changes with aspects of carcinogenesis Citation[28–33].
HPV16 has approximately 113 scattered CpG sites (depending on the variant) with some clustering of CpGs in the E1/ E2 regions but no classically defined CpG islands. Many of the CpGs in HPV16 are differentially methylated, and in particular, increased methylation of the LI, L2 and E2 genes is consistently observed and associated with cervical carcinogenesis Citation[10,27–35]. Our research team used multivariate logistic regression to develop a specific DNA methylation classifier, termed Score 1 (S1). S1 was initially developed as a classifier on a set of methylation data from Costa Rican women Citation[10], then applied under a strict predefined statistical analysis plan to triage HPV16 positive women with abnormal cytology undergoing follow-up in the Welsh routine screening program Citation[27]. SI includes information on the quantitative levels of methylation at CpG sites 6367 and 6389 in the LI gene and on CpG sites 4238, 4247, 4259, 4268 and 4275 in the L2 gene and has the form S1 = 36L1 + 64L2, where L1 and L2 refer to the mean methylation of the two sites in L1 and the proportion methylated of the five sites in L2, respectively. The classifier performs modestly in separating HPV16 infected women with CIN2/3 from infected women with CIN1 histology or normal cytology, with an area under the curve of 0.74, a sensitivity of 92% and a PPV of 44%. Application of the S1 classifier would allow 40% of HPV16 positive women to defer colposcopy to a later date and allow for possible elimination of the viral infection by the immune system. A similar study on a different group of women from Wales identified HPV16 CpG sites 5600 and 5609 as possible classifiers, giving an impressive area under the curve of 0.9 for separating normal women from those with severe dyskaryosis (the cytological equivalent of CIN2/3). The performance of CpG sites 5600/5609 needs to be validated in a large study of women that includes CIN1 combined with normals to more closely represent a triage setting Citation[34].
Methylation testing for HPV16 is an attractive triage option; however, it is only effective in the management of HPV16 positive women who represent about 50% of the risk for cervical cancer. HPV16 and HPV18 combined contribute to approximately 70% of cervical cancers, while HPV31 and HPV33 are among the next most prevalent types, causing approximately an additional 8% of the cancers Citation[33,36,37]. Expanding the methylation classifier to a panel of HPVs is worthwhile; ideally, a comprehensive assay would include the most important 13 or 14 hrHPV types if a suitable multiplex approach can be developed; however, at least HPV18, HPV31 and HPV33 should be included with HPV16 in a panel to give more complete coverage of the main types in cancer.
A number of other hrHPVs have been investigated for methylation, including HPV18, HPV31, HPV33, HPV45, HPV52 and HPV58. These types have been studied to a lesser extent than HPV16 and there are no methylation data available on many hrHPV types Citation[33,37,38]. The methylation patterns of investigated hrHPV genomes are quite similar. Methylation levels are low or absent in the URR and E6 regions in women with normal cytology but there is a gradual increase through E7, E1 and E2 to reach peak values in the L2 and L1 regions . This pattern is also mostly maintained in CIN lesions through to cancer, although the slopes become steeper and the peaks become higher with increasing disease severity. Average methylation values in the L1 region in normal tissues is in the range of 5–10%, while in cancer, the values are typically 40–80% Citation[10].
Figure 2. DNA methylation patterns across the genome of HPV16, showing the percentage median values for selected CpG sites in cancers (solid circles), cervical intraepthelial neoplasia 2/3 (triangles), and normal women who had transient HPV16 infections (open circles). Other hrHPVs have similar methylation patterns, with relative peaks in the L1 and L2 regions and little to no methylation in the URR. The circular genome is depicted as opened in the URR region. The specific peaks and valleys of the HPV16 genome methylation profile are quite reproducible in specimens from different geographic locations and may reflect the intrinsic relative positions of histones and other binding complexes.
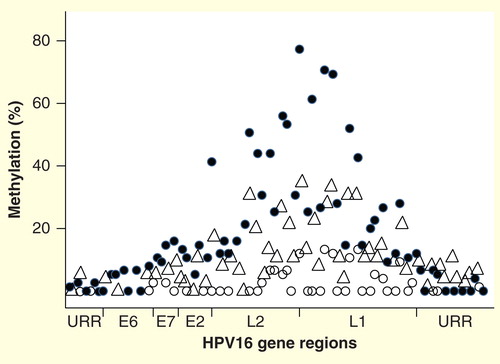
There is some evidence that methylation levels in the URR, particularly in important regulatory elements such as the E2 protein binding sites (E2BS), may be somewhat increased in the cancers, with levels of methylation perhaps related to the co-existence of different combinations of integrated and episomal genomes Citation[10,34,39–41]. There are four E2BS in the HPV16 URR enhancer, and binding of the E2 activator/repressor to certain E2BS elements can either activate or repress transcription of E6/E7 RNAs. Perhaps differential methylation of the repressive E2BS alters the ability of the E2 repressor to bind and allows continued production of the HPV early oncogenes Citation[39–41].
Classifiers based on methylation of human DNA
More than 80 human genes have been reported as possible biomarkers of cervical cancer (Box 1) Citation[42–48]. Most of the genes have only a single associated study. However, some groups have been more active and provided further support to earlier publications. Of particular interest are genes promoted by three well-known HPV research groups who study biomarker targets including CADM1, MAL, miR-124–2, EPB41L3, JAM3, TERT, C13ORF18, LMX1, SOX1, PAX1 and NKX6-1 Citation[42–46]. These genes have been highlighted as having possible utility for the routine clinical identification of high-grade cervical disease with good sensitivity and specificity. Three important limitations of currently published studies looking at human gene methylation in cervical cancer are: the sets of genes have been compared in only small studies; none of the genes in the different sets were compared to genes in other sets; and the clinical test performance of the methylated gene targets have not been validated by independent groups. Despite these caveats, the results of the studies are of interest. One of the more promising human gene pairs as DNA methylation classifiers are combinations of MAL and CADM1 from the team of Meijer et al. in the Netherlands, which for a sensitivity of 87% (75–94), gave a specificity of 43% (37–46), and for a sensitivity of 68% (50–81), gave a specificity of 75% (70–80) for CIN3+ in a small screening study Citation[44]. In the same set of women, the triage sensitivity and specificity values for cytology were 66% (50–79) and 79% (74–83), respectively, while cytology combined with genotyping for HPV16 and HPV18 had a sensitivity of 84% (72–93) and a specificity of 54% (47–59). Studies by the team of Yu et al. in Taiwan on the genes LMX1A, PAX1, NKX6-1 and SOX1 in various combinations gave a sensitivity of between 88 and 96% and specificity of between 77 and 87% for detection of CIN3+ in a retrospective case control study of patients in a hospital setting Citation[46]. Similarly, the team of Wisman showed that a combination of the genes EPB41L3, JAM3, TERT and C13ORF18, gave a sensitivity of 71% and specificity of 77% for detecting CIN2, CIN3 or cancer (CIN2+) in a convenience set of specimens from a Dutch tissue bank Citation[45].
Box 1. Genes proposed as candidate DNA methylation diagnostic biomarkers for cervical cancer.
APC, ASC, BRCA1, BRCA2, CADM1 , CAGE , CALCA, CAV1, CCNA1, CCND2, CDH1, CDH13, CDKN2A, CDKN2B, CHFR, COX2, CTNNB1, C13ORF18 , C15ORF48, DAPK1 , DcR1 , DcR2 , DLC1, DPYS, EDNRB, ESR1, EPB41L3 , FANCF, FHIT, GSTP1, HIC1, HLTF, miR-124-2 , HSPA2, HS3ST2, JAM3 , KLK10 , LHFPL4, LMX1 , LRRC3B , MAL , MDR1, MGMT, MLH1, MTIG, MYOD1, NKX6-1 , NOL4, PAX1 , PGR, POU2F3, PRDM2, PTEN, PTGS2, P14, RASSF1A , RARB, RB1, ROBO1, ROBO3, RRAD, RPRM, RUNX3 , SCGB3A1, SFN, SFRP1, SLIT1, SLIT2, SLIT3, SNCG , SOCS1, SOCS2, SOCS3, SOX1 , SPARC, SYK, TERT , TFP12, THBS1, TIMP2, TIMP3, TNFRSF10, TP73, TWIST1, VHL, ZMYND10
Classifiers based on a combination of human gene & HPV DNA methylation
Use of a combination of human genes and HPV DNA methylation diagnostic targets has the advantage of applicability to all CIN regardless of detectable HPV, with the added benefit of information provided by methylation levels if hrDNA is measurable. The combination may be a more effective triage tool to detect women at high risk of developing CIN2+ and also addresses the needs of women infected by less common hrHPV types. Kalantari et al. recently reported that a bisulfite cloning and sequencing method using a combination of methylation measurements of DAPK1, HPV16, HPV18, HPV31 and HPV45 provides for an interesting classifier; in one variation, they observed a sensitivity of 80% and a specificity of 89% for detecting CIN2+ Citation[32].
Brentnall et al. have developed a combination methylation test based on pyrosequencing (PSQ) of bisulfite converted DNA using HPV16, HPV18, HPV31 and the human gene EPB41L3 as targets to predict whether women had CIN2/3 at the time of sampling Citation[49]. The specimens were from two large HPV studies of colposcopy-referral populations in London UK, with biopsy-proven CIN2/3 status. The study included 1493 women and 556 CIN2/3. The new classifier was designed to predict CIN2/3 histology from specimens taken prior to colposcopy in women with prior abnormal cytology. A three-stage design of model development, validation and updating was used to mitigate issues associated with overfitting. The final model used methylation measurements of selected CpG sites in the human gene EPB41L3, the S1 classifier of HPV16 and CpG sites in HPV18-L2 and HPV31-L1. The S4 classifier was significantly associated with CIN2/3 harboring any of 14 hrHPV types (area under curve = 0.80, p < 0.0001). For a cutoff with 90% sensitivity, specificity was 36% (33–40) and PPV was 46% (43–48). Application of the classifier could allow more than one-third of the women to avoid an initial colposcopy and wait for possible clearance of the HPV infections. The caveats mentioned above for human gene methylation assays also apply to assays on HPV DNA methylation and to the combinations of HPV and human genes. Much additional research is required to determine the best assays for clinical use.
Quantitative DNA methylation assays for the diagnosis & prognosis of prostate cancer
Prostate cancer is one of the most common malignancies in men and the diagnosis has been increasing, especially in countries where more screening is taking place. In 2012, the GLOBOCAN Citation[14] prostate cancer estimates were an overall 1,112,000 cases and 307,000 deaths, of which 759,000 cases and 142,000 deaths were in more developed regions and 353,000 cases and 165,000 deaths were in less developed regions. Men with incident prostate cancer are typically either self-referred due to symptoms detected by digital rectal examination or screened by prostate specific antigen (PSA). Of these methods, PSA has the highest sensitivity and can detect a lot of occult disease. The main advantage of PSA is that it is a simple inexpensive serum test and as such is valuable for early detection of prostate cancer; however, the main disadvantage of PSA is its poor specificity. PSA screening decreases the absolute risk of death from prostate cancer at the price of many more invasive examinations and biopsy, overdiagnoses and overtreatment Citation[50,51]. A majority of the prostate cancer cases detected by modern screening methods are essentially harmless and will not result in morbidity or death if left untreated Citation[52]. It is estimated that to prevent one prostate cancer-specific death, 1400 men need to be screened, resulting in approximately 50 additional cases of unnecessarily diagnosed cancer Citation[51]. Thus, there has been an ongoing search for better diagnostic strategies. Major goals are to find biomarkers that would allow for improvements in diagnosis and prognosis. DNA methylation biomarkers have shown promise for the detection and prognosis of prostate cancer but are one among several approaches to improve clinical management Citation[2,4,6,7]. Other kinds of tests for improved diagnosis or prognosis include PCA3, Ki-67, CCP score or TMPRSS-ERG for prognosis Citation[53–56].
Many men with suspected prostate cancer, mainly related to elevated PSA, are subject to painful and risky needle biopsies, where up to a dozen or more cores may be taken in order to find the disease. The results of these biopsies are sometimes inconclusive and necessitate additional biopsies. Biomarkers to better direct the needles would be really useful; low-risk men could be identified from a simple serum or urine test and only men at a higher risk would receive biopsy. A related approach is a biomarker test on biopsy cores from men with a first set of inconclusive histopathological results, to contribute information for the decision as to which men need a re-biopsy. DNA methylation testing can be applied to both of these situations but an important question is which small set of methylated genes are the best biomarkers for a simple inexpensive assay to assist in disease diagnosis?
The GSTP1 gene has been one of the most studied DNA methylation biomarkers in prostate cancer and is hypermethylated in >90% of cancers Citation[57–67]. Many other genes are candidate methylation diagnostic biomarkers, a few of the more commonly mentioned are, RASSF1A Citation[58–61,65,66], RARB2 Citation[58,62,65,66], APC Citation[59,61,65,66], TIG1 Citation[65,66], BCL2 Citation[66] and SCGB3A1 (alias HIN1) Citation[68], but the list of interesting genes runs into the hundreds. From a genome-wide perspective, 2481 differentially methylated regions were identified in just one study of prostate tissue, any one of which may be a biomarker Citation[6]. Box 2 lists 91 genes, which includes some of the more common candidate genes proposed as diagnostic biomarkers in prostate cancer and investigated in at least some detail prior to the recent deluge of genome-wide data. To search for the best small set of genes, Vasiljević et al. Citation[69] compared DNA methylation in benign prostate hyperplasia (BPH) versus prostate cancer in a large set of candidate genes that covered a wide range of cellular pathways including genes shown as aberrantly methylated in prostate cancer and genes that have displayed interesting potential in cancers other than prostate. DNA methylation levels of these genes were measured by PSQ.
Box 2. Genes proposed as candidate DNA methylation diagnostic or prognostic biomarkers for prostate cancer.
ABHD9, APC , AR, BCL2 , CADM1, CAGE , CAR, CAV1, CCND2 , CDH1, CDH13 , CDKNIB, CDKN1C , CDKN2A, CD44, CRBP1, DAPK1, DcR1 , DcR2 , DKK3, DPYS , DRM, EDNRB , EGFR5 , ESR1 , ESR2, FHIT, FOXE3 , GSTP1 , GPR7, GPX3, HIC1, HLAa , HOXA1 , HOXA4 , HOXA5 , HOXA7 , HOXA9 , HOXA10 , HOXA11 , HOXA13 , HOXD3 , HOXD4 , HOXD9 , HPSE, HPP1, HSPB1 , HSPRY2, KBTBD6, LAM-A3, LAM-B3, LAM-C3, KLK10 , LRRC3B , MAL , MCAM, MDR1 , MGMT, MTIG, NEP, NKX2-5 , NOTCH1, NTRK2, PDLIM4 , PITX2 , PTGS2 , PXMP4, RASSF1A , RARB , RARRES1, RIZ1, RRAD, RTVP1/GLIPR, RUNX3 , SCGB3A1 , SERPINB5 , SFN, SFRP1, SLIT2, SNCG , SOCS1, S100A2 , S100A6, THBS1, THRB, TIG1 , TIMP3, TWIST1, VDR, WT1 , ZNF185
Twenty of the investigated genes, namely, RARB, SCGB3A1, BCL2, GSTP1, CCND2, EGFR5, APC, RASSF1A, MDR1, NKX2-5, CDH13, DPYS, PTGS2, EDNRB, MAL, PDLIM4, SERPINB5, HLAa, ESR1 and TIG1 were significantly more highly methylated in cancers than BPH at a false discovery risk of <1%, some genes (RARB2, GSTP1, SCGB3A1, APC, BCL2) had close to 100% accuracy for separating cases from controls, with quite large absolute differences in methylation levels. For several of these genes (RARB2, SCGB3A1, BCL2, CCND2, GSTP1) the mean DNA methylation levels in BPH were in the range of 0–10%, while in the cancers, the means were in the range of 40–60% Citation[69].
Prognosis in men with histopathologically diagnosed prostate cancer is another important area and a more difficult assignment for molecular biomarkers. Currently, the best prognostic tool for routine management, in terms of accuracy, ease-of-use and cost to the medical system is Gleason score. Histopathology is the established grading method and has many advantages, including a long history, broad base of experience, ease of application and relatively low costs; however, Gleason histopathology also has some limitations such as intra- and interobserver variability in reading Citation[70]. For needle biopsies, additional variability may arise due to difficulty in targeting cores precisely to the cancerous areas. These sources of variability can lead to inaccuracy in prognosis. A standardized quantifiable molecular biomarker assay could help improve disease management. Abnormal methylation contributes to the progression of prostate cancer Citation[2,4,6,7,57–69]. DNA methylated targets can be detected in body fluids such as urine and blood, opening up an attractive, non-invasive approach to diagnosis and prognosis. The development of methylation assays to diagnose and/or predict disease outcomes in cancer patients undergoing active follow-up with minimal intervention is a current hot topic. Numerous diagnostic genes shown to be hypermethylated in prostate cancer have also been proposed as prognostic biomarkers including GSTP1, APC1 and RARB Citation[2,7,71]. A majority of the studies focusing on the prognostic value of methylation have used time to biochemical recurrence (BCR) after surgical treatment as the primary end point. BCR has the advantage of providing an objective end point after follow-up of just a few years; however, an important disadvantage is that BCR is an indirect marker that represents prostate inflammation and is much less accurate at estimating the potential of death Citation[72]. Death from prostate cancer is the gold standard end point, which is available from few studies, with the Transatlantic Prostate Group (TAPG) study being the one with the longest follow-up. TAPG is a well-characterized cohort of men residing in the UK who did not receive any treatment at the time of diagnosis or within 6 months thereafter and experienced a high rate of prostate cancer-related deaths Citation[56,72].
We conducted a biomarker study on DNA extracted from TAPG biopsies to assess both univariately and multivariately the prognostic biomarker potential of DNA methylation in 13 promising genes selected from an initially much larger set Citation[2,7,69,73]. The association between gene methylation and death from prostate cancer was examined as the primary end point. A secondary aim was to investigate if methylation measurement improves the prognostic value of the current clinical reference variables, Gleason score and PSA. Univariate analysis showed that selected genes individually were modest predictors of death from prostate cancer. However, the multivariate analysis showed that methylation of DPYS, CCND2 and HSPB1 added substantial prognostic information not captured by any other measure Citation[7,73]. A model including PSA and methylation of DPYS, HSPB1, MAL and TIG1, but excluding Gleason score, was almost as good at predicting prostate cancer–related mortality as the full model including Gleason score. Although GSTP1 and APC were univariately associated with death, they were not selected multivariately in the final models due to exclusion by the more powerful classifiers.
A prostate cancer prognostic biomarker that has received a lot of recent attention is PITX2, which is classified as a tumor suppressor gene in the Wnt-pathway. Hypermethylation of PITX2 in prostate cancer has previously been shown as an independent prognostic biomarker of BCR and was also associated with reduced mRNA transcription Citation[74,75]. We studied methyation of PITX2 and risk of death from prostate cancer in the TAPG cohort and confirmed earlier studies. PITX2 was very strongly associated with death (p < 0.0001) and had a large effect size (odds ratio 2.7), which was independent of Gleason score, PSA or stage [Lorincz, Pers. Comm.]. PITX2 promises to be an important prognostic biomarker either alone or in combination with other genes.
Genes differentially methylated in cervix, prostate & other cancers
The a priori expectation with respect to comparisons between cervical and prostate cancer are that there would not be a big overlap in differentially methylated genes, certainly a lot less than between prostate and breast cancer. With respect to potential cancer diagnostic markers in cervix and prostate, we studied 19 candidate genes in both kinds of cancers and identified 4 genes (DPYS, EPB41L3, EDNRB and MAL) as biomarkers in common Citation[69]. Earlier studies also reported some differentially methylated genes in both cervical and prostate cancer including: RASSF1A, SCGB3A1, RUNX3, CAGE, DcR1, DcR2, SNCG, KLK10 and LRRC3B Citation[76–83]. Most of the genes are represented by a single investigation, and in our PSQ studies, we were unable to confirm differential methylation of SCGB3A1 or RASSF1A in cervical cancer. However, using the same tests, the genes were highly diagnostic between BPH and prostate cancer Citation[69], suggesting the possibility that genes commonly methylated in different cancers may show changes in disparate target regions.
A recent systematic review of genome-wide methylation studies in two hormonally regulated cancers, prostate and breast, found that of 95 genes reported as frequently methylated in a differential manner in prostate cancer, 54 (57%) are also often differentially methylated in breast cancer. Of 425 breast cancer genes, 13% were also differentially methylated in prostate cancer. The genes in common include many members of the HOX family (FOXE3, HOXA1, HOXA4, HOXA5, HOXA7, HOXA9, HOXA10, HOXA11, HOXA13, HOXD3, HOXD4, HOXD9 and WT1) and other frequently studied genes, such as APC, RASSF1A, GSTP1, CDH13, CDKN1C, CDKN2A, KLK10, RUNX3 and S100A2 (see Table 4 of reference Citation[84] for the full list). There are probably many more common variably methylated biomarkers to be identified from the thousands of prostate and breast differentially methylated regions. The greater number of apparent biomarkers in breast cancer likely reflects the differences in the number and size of included studies; there were 22 genome-wide studies regarding breast cancer versus 10 studies regarding prostate cancer Citation[84]. Of the genes showing varying methylation in both prostate and breast cancer, there were some also proposed as biomarkers in the cervix literature (Box 1). Examples of such genes are: RASSF1A, RUNX3 and KLK10. The kalikrein protein family appears to be epigenetically regulated in many cancers. KLK10 appears to be differentially methylated in breast, prostate and cervix cancer. Similarly, hypomethylation of KLK3, the gene coding for PSA, was shown as a potentially important control mechanism in prostate cancers Citation[8].
Expert commentary
DNA methylation shows variability related to mitotic age and an interesting approach is to focus on the evolution of methylation heterogeneity, which could provide additional information on disease outcomes Citation[85]. However, for the moment, the main approach for methylation assays is to focus on specific targets, similar to classical diagnostic approaches. New methylation assays can measure quite small changes (in the range of a few percent) in specific CpG targets with high precision Citation[2,4,7,10,27]. A concern raised about the overall value of looking for new methylation biomarkers is that methylation may simply mirror RNA transcription. However, gene-specific correlations between RNA and methylation signatures at the genomics level are variable and mostly only in the fair-to-good range Citation[84,86], showing that both RNA expression and DNA methylation change should be considered together to more fully understand the disease. Methylation at individual CpG sites can provide information on genes that are expressed only sporadically or at levels too low to be measured by a transcriptomic approach, for example, rare splice variants Citation[6]. Methylation can also silence genes that will be needed in future cell generations but that are not expressed at a given moment; in such a situation, the transcriptome will not reveal outcomes discernable from the methylome.
Diagnostically important DNA methylation changes have been reported in many common human cancers such as breast, colon, bladder, lung, prostate, cervix, stomach and so on. It is quite likely that epigenetics in general, and DNA methylation in particular, has a role in most, if not all, human cancers. The presence or absence of definitive methylation biomarkers for virtually every disease may be merely a function of how hard and how long researchers have looked to find the relevant epigenetic changes. Work on various cancers, including cervix and prostate, has revealed DNA methylation increases and decreases in specific genes or in viral genome regions that are quite promising as independent diagnostic and prognostic markers. We can already see how to construct new and improved classifiers from large pools of differentially methylated genes. Many of these markers are common to two or more cancers, while others appear unique, although they may be shared with cancers not yet investigated. This diversity provides some interesting opportunities for precise determination of the provenance of metastases of uncertain origin. The new information will enhance our understanding of relationships between diseases. For example, molecular similarities between morphologically disparate malignancies and differences among morphologically similar cancers may become clearer. Methylomic signatures are likely to contribute to signatures already established by transcriptomics, single nucleotide polymorphism analyses and other ‘omics’ classifiers.
With respect to laboratory practicalities, methylation assays are relatively simple, inexpensive and robust. Tests can be performed by average laboratory technicians, accompanied by robotic equipment, at rates of thousands of specimens per week. There are numerous opportunities for improvements in cancer detection, diagnosis, prognosis and prediction of response to therapy. The ramifications are not just for some aspects of certain diseases but for virtually every disease. Any sample that contains DNA – whether it be blood, urine, stool, aspirates, or others – is amenable to DNA methyl testing. Automated high-throughput technologies are available and the cost is decreasing.
However, many critical issues need to be addressed in order for things to go right for epigenetic diagnostics and we are not far along the road in completing these steps. First, there is a need for evaluation and selection of the most appropriate biomarker sets and the methods for assessment of each type of alteration, with a strict standardization of approach. This is then followed, in somewhat variable order and combination, by clinical validation, regulatory approvals, health technology assessments, professional medical group guidelines, and finally, qualification by clinical labs for routine use. It is important to not underestimate the complexity and time taken to complete these translational steps. There is a growing need for standardization and clinical validation of specific applications, a situation related in no small way to the novelty of the epigenetic approach, the constant creation of new technologies and the deluge of new biomarkers produced by genome-wide studies. These powerful forces may have a tendency to swamp the process and delay implementation of useful epigenetic biomarkers. From another perspective, the unsuspecting use of diverse assays with various technical limitations may give non-comparable results in different studies, also leading to the biomarkers being delayed or discarded. DNA methylation assays are still relatively complex and require great care in primer design and optimization. Rigorous quality control must be implemented to identify problems such as inadequate DNA, inhibitors and contamination. The assays need to be made more robust and simpler, and they need to be compared frequently in carefully designed inter-laboratory evaluations. Until the evaluation studies are done and the results generally available to relevant expert committees, the tests will not be ready for routine use in a clinical lab.
Epigenetic biomarkers need to be considered in the context of important differential diagnostic questions that clinicians face with their patients every day, for which they may already have trusted and reasonably effective approaches. It will be necessary for each epigenetic assay to prove itself superior in some way to what exists today. To take an example from the drug validation field, the relevant comparison of the new test is not to a placebo but to whatever is the best drug combination of the day. What is needed then is lots of very specific information, preferably in large well-designed and adequately powered studies to show the real-world performance of the new test. The information needs to come from many different independent sources, with all the strengths and limitations of the compared tests in full view.
Current genomics/epigenomics methods provide too much information of limited value at high cost. While these methods are excellent for research and epidemiological use, they should not be oversold and prematurely pushed into the clinic by advertising unsubstantiated benefits to the public. The clinical use of genomics will be restricted until the information can be integrated into a unified framework that allows elimination of other unnecessary tests and provides clear and meaningfully actionable information not previously available. A compromise is required that balances the importance, reliability and cost of new information provided by the genomics tests.
Five-year view
Research in DNA methylation as a source of cancer biomarkers has moved to the leading edge of translational research, especially for early detection and diagnosis. Harnessing the power of epigenetic analyses represents a paradigm shift in human healthcare that is happening now. The greatest need in epigenetic diagnostics today is to formally validate existing candidate biomarkers; the studies are ongoing and should start to yield fruit in the near future. Companies targeting personalized medicine are working closely with academic groups to validate epigenetic biomarkers. Several DNA methylation assays have already made it into the routine testing area; some examples are SEPT9 for colorectal cancer Citation[87], marketed under different names (Epi proColon, Epigenomics AG, Berlin Germany; ColoVantage, Quest Diagnostics, Teterborough, NJ, USA), SHOX2 for lung cancer (Epi proLung, Epigenomics AG), Vimentin assays for colorectal cancer (ColoSure™, LabCorp, Burlington, NC, USA) and various tests based on GSTP1, APC and other targets for prostate cancer (LabCorp). Recently, the team of Snijders et al. reported on the use of a quantitative methylation-specific PCR test for MAL-m1 and miR-124-2 that performed quite well in detecting CIN2+ in self-collected vaginal specimens and looks suitable for further validation and possible routine clinical use Citation[88].
Despite these modest successes, there are still relatively few clinical applications for DNA methylation markers and no major global test. This situation is related to various factors, including not finding the best markers, not targeting the best applications, inadequate studies, lack of sufficient interest from large diagnostic companies and weak company R&D, regulatory and marketing efforts. However, it may be expected that in the next 5 years, some important and highly performing epigenetic tests will make the transition to clinical use, either as local hospital lab developed tests, or in a more general way, into larger commercial labs in the USA and Europe. Most of the first batch of tests will be directed to detection, triage and diagnosis, such as tests to triage hrHPV positive women. It will take longer to prove the utility of prognostic markers.
A vast array of novel DNA methylation biomarkers is to be expected in 5 years and it will be a real challenge to sort through these to find the ones that deserve to be validated. Assay systems and costs are important issues; complex and expensive genomics tests will have limited appeal in the clinic until two main issues are resolved: the mass of information provided needs to be turned into something that the clinician can understand and is relevant to patient care; and their costs should be affordable to healthcare systems with already overstretched budgets. These are high barriers and major applications may not happen for 5 years or more; in the meantime, the door is wide open for directed and much less expensive epigenetic assays with small sets of highly informative genes.
Epigenetic information will feed into the development of innovative personalized therapeutic strategies. DNA methylation changes are dynamic and reversible and we can take advantage of these characteristics to design new anticancer agents. Some epigenetically targeted drugs have already been approved for hematological malignancies, with more and different drugs expected. Where possible, we need to move away from global methylation or demethylation inhibitors and find drugs that target pathways and one or a few known genes. Epigenetic drugs that are too broad spectrum will tend to have unacceptable side effects and risks of new cancers in the future. If all goes well, then within 5–15 years, personalized DNA methylation assays and companion drugs may be just another set of tools for use by the clinician in everyday practice.
Key issues
Aberrant methylation is a central feature of carcinogenesis and causes defective gene expression, faulty condensation and chromosomal instability and is a hallmark of cellular defenses acting to silence foreign DNA.
Epigenetic pathways are affected by environmental stimuli and insults to a greater extent than classical genetic pathways.
Diagnostically important DNA methylation changes have been reported in many common human cancers such as breast, colon, bladder, lung, prostate, cervix, stomach and others, and it is quite likely that epigenetics in general, and DNA methylation in particular, has a role in most, if not all, human cancers.
Methylation of the human papillomavirus (HPV) late regions is an indicator of the length of time that the genome spends in a given host tissue, and appears to be a proxy for viral persistence, cell immortalization and progression.
Tests based on HPV DNA methylation have good sensitivity, specificity and positive predictive value, with the added benefits of a fully molecular approach.
Triage of hrHPV positive women with a combined methylation classifier using both HPV and a human gene had an area under curve of 0.80 (p < 0.0001), 90% sensitivity, 36% specificity and 46% positive predictive value.
The methylation of some genes such as RARB2, GSTP1, SCGB3A1, APC and BCL2 have a high accuracy for separating prostate cancer from normal tissue.
Prostate cancer is a heterogeneous and mostly indolent disease for which epigenetic prognostic biomarkers offer major benefits.
Methylation of PITX2 was very strongly associated with prostate cancer death (p < 0.0001) and had quite a large effect size (odds ratio 2.7), which was independent of Gleason score, prostate specific antigen or stage.
The greatest need in epigenetic diagnostics today is to formally validate the hundreds of promising candidate biomarkers and create robust assays.
Financial & competing interests disclosure
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Notes
Genes specifically mentioned elsewhere in this review as of particular interest for cervical cancer are underlined.
Data taken from Citation[42–46,76–83].
Genes specifically mentioned elsewhere in this review as of particular interest for prostate cancer are underlined. The above list is only partial and focuses on the more frequently mentioned genes. There are hundreds of additional genes that have been reported as possible methylation markers for prostate cancer in genome-wide studies.
Data taken from Citation[7,57–69,71,73–84].
References
- Denis H, Ndlovu MN, Fuks F. Regulation of mammalian DNA methyltransferases: a route to new mechanisms. EMBO Rep 2011;12:647-56
- Lorincz AT. The promise and the problems of epigenetics biomarkers in cancer. Expert Opin Med Diagn 2011;5:375-9
- Illingworth RS, Bird AP. CpG islands – “a rough guide”. FEBS Lett 2009;583:1713-20
- Laird PW. Principles and challenges of genome-wide DNA methylation analysis. Nature Rev Genet 2010;11:191-203
- Liu S, Wang J, Su Y, et al. Quantitative assessment of Tet-induced oxidation products of 5-methylcytosine in cellular and tissue DNA. Nucleic Acids Res 2013;41:6421-9
- Kim JH, Dhanasekaran SM, Prensner JR, et al. Deep sequencing reveals distinct patterns of DNA methylation in prostate cancer. Genome Res 2011;21:1028-41
- Vasiljević N, Ahmad AS, Wu K, et al. Evaluation of diagnostic and prognostic potential of heat shock gene HSPB1 DNA methylation in prostate cancer. Prost Cance Prost Dis 2013;16:35-40
- Bert SA, Robinson MD, Strbenac D, et al. Regional activation of the cancer genome by long-range epigenetic remodelling. Cancer Cell 2013;23:9-22
- Heyn H, Carmona FJ, Gomez A, et al. DNA methylation profiling in breast cancer discordant identical twins identifies DOK7 as a novel epigenetic biomarker. Carcinogenesis 2013;34:102-8
- Mirabello L, Schiffman M, Ghosh A, et al. Elevated methylation of HPV16 DNA indicates increased risk for the development of high grade cervical intraepithelial neoplasia. Int J Cancer 2013;132:1412-22
- Hughes LAE, Melotte V, Schrijver J, et al. The CpG island methylator phenotype: what’s in a name. Cancer Res 2013;73:5858-68
- Cao R, Wang L, Wang H, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002;98:1039-43
- Jansson MD, Lund AH. MicroRNA and cancer. Mol Oncol 2012;6:590-610
- International Agency for Research on Cancer, World Health Organization. GLOBOCAN. 2012. Available from: http://globocan.iarc.fr/factsheets/cancers
- Cuzick J, Arbyn M, Sankaranarayanan R, et al. Overview of human papillomavirus-based and other novel options for cervical cancer screening in developed and developing countries. Vaccine 2008;26(Suppl 10):K29-41
- Cuzick J, Bergeron C, von Knebel Doeberitz M, et al. New technologies and procedures for cervical cancer screening. Vaccine 2012;30(Suppl 5):F107-16
- Naucler P, Ryd W, Tornberg S, et al. Human papillomavirus and Papanicolaou tests to screen for cervical cancer. N Engl J Med 2007;357(16):1589-97
- Rijkaart DC, Berkhof J, Rozendaal L, et al. Human papillomavirus testing for the detection of high-grade cervical intraepithelial neoplasia and cancer: final results of the POBASCAM randomised controlled trial. Lancet Oncol 2012;13(1):78-88
- Ronco G, Giorgi-Rossi P, Carozzi F, et al. Efficacy of human papillomavirus testing for the detection of invasive cervical cancers and cervical intraepithelial neoplasia: a randomised controlled trial. Lancet Oncol 2010;11(3):249-57
- Dillner J, Rebolj M, Birembaut P, et al. Long term predictive values of cytology and human papillomavirus testing in cervical cancer screening: joint European cohort study. BMJ 2008;337:a1754
- Ronco G, Dillner J, Elfstrom MK, et al. Efficacy of HPV-based screening for prevention of invasive cervical cancer: follow-up of four European randomised controlled trials. Lancet 2014;383(9916):524-32
- Lazcano-Ponce E, Lorincz AT, Cruz-Valdes A, et al. Self-collection of vaginal specimens for human papillomavirus testing in cervical cancer prevention (MARCH): a community-based randomised controlled trial. Lancet 2011;378:1868-73
- Szarewski A, Mesher D, Cadman L, et al. Comparison of seven tests for high-grade cervical intraepithelial neoplasia in women with abnormal smears: the Predictors 2 study. J Clin Microbiol 2012;50:1867-73
- Lorincz AT, Castanon A, Lim A, Sasieni P. New strategies for papillomavirus-based cervical screening. Womens Health 2013;9:443-52
- Carozzi F, Gillio-Tos A, Confortini M, et al. Risk of high-grade cervical intraepithelial neoplasia during follow-up in HPV-positive women according to baseline p16-INK4A results: a prospective analysis of a nested substudy of the NTCC randomised controlled trial. Lancet Oncol 2013;14(2):168-76
- Burnett ST, Sleeman J. Uneven distribution of methylation sites within the human papillomavirus 1a genome: possible relevance to viral gene expression. Nucleic Acids Res 1984;12:8847-60
- Lorincz AT, Brentnall AR, Vasiljević N, et al. HPV16 L1 and L2 DNA methylation predicts high grade cervical intraepithelial neoplasia in women with mildly abnormal cervical cytology. Int J Cancer 2013;133:637-44
- Kalantari M, Calleja-Macias IE, Tewari D, et al. Conserved methylation patterns of human papillomavirus type 16 DNA in asymptomatic infection and cervical neoplasia. J Virol 2004;78:12762-72
- Badal S, Badal V, Calleja-Macias IE, et al. The human papillomavirus-18 genome is efficiently targeted by cellular DNA methylation. Virology 2004;324:483-92
- Brandsma JL, Sun Y, Lizardi PM, et al. Distinct human papillomavirus type 16 methylomes in cervical cells at different stages of premalignancy. Virology 2009;389:100-7
- Clarke MA, Wentzensen N, Mirabello L, et al. Human papillomavirus DNA methylation as a potential biomarker for cervical cancer. Cancer Epidemiol Biomarkers Prev 2012;21:2125-37
- Kalantari M, Osann K, Galleja-Macias IE, et al. Methylation of human papillomavirus 16, 18, 31 and 45 L1 and L2 genes and the cellular DAPK gene: considerations for use as biomarkers of the progression of cervical neoplasia. Virology 2014;448:314-21
- Wentzensen N, Sun C, Ghosh A, et al. Methylation of HPV18, HPV31, and HPV45 genomes and cervical intraepithelial neoplasia grade 3. J Natl Cancer Inst 2012;104:1738-49
- Bryant D, Tristram A, Liloglou T, et al. Quantitative measurement of human papillomavirus type 16 L1/L2 DNA methylation correlates with cervical disease grade. J Clin Virol 2014;59(1):24-9
- Oka N, Kajita M, Nishimura R, et al. L1 gene methylation in high risk human papillomaviruses for the prognosis of cervical intraepithelial neoplasia. Int J Gynecol Cancer 2012;23:235-43
- Bosch FX, Lorincz A, Muñoz N, et al. The causal relation between human papillomavirus and cervical cancer. J Clin Pathol 2002;55:244-65
- Vasiljević N, Scibior-Bentkowska D, Brentnall A, et al. A comparison of methylation levels in HPV18, HPV31 and HPV33 genomes reveals similar associations with cervical precancers. J Clin Virol 2014;59(3):161-6
- Murakami I, Fujii T, Katsuaki D, et al. Methylation of human papillomavirus-52 and -58 is a candidate biomarker in cervical neoplasia. J Clin Virol 2013;58:149-54
- Chaiwongkot A, Vinokurova S, Pientong C, et al. Differential methylation of E2 binding sites in episomal and integrated HPV16 genomes in preinvasive and invasive cervical lesions. Int J Cancer 2013;132:2087-94
- Cheung JLK, Cheung TH, Yu MY, Chan PKS. Virological characteristics of cervical cancers carrying pure episomal form of HPV16 genome. Gynecol Oncol 2013;131:374-9
- Jacquin E, Baraquin A, Ramanah R, et al. Methylation of human papillomavirus type 16 CpG sites at E2-binding site 1 (E2BS1), E2BS2, and the Sp1-binding site in cervical cancer samples as determined by high-resolution melting analysis-PCR. J Clin Microbiol 2013;51:3207-15
- Wentzensen N, Sherman ME, Schiffman M, Wang SS. Utility of methylation markers in cervical cancer early detection: appraisal of the state-of-the science. Gynecol Oncol 2009;112:293-9
- Overmeer RM, Louwers JA, Meijer CJ, et al. Combined CADM1 and MAL promoter methylation analysis to detect premalignant cervical lesions in high-risk HPV-positive women. Int J Cancer 2010;129:2218-25
- Hesselink AT, Heideman DA, Steenbergen RD, et al. Combined promoter methylation analysis of CADM1 and MAL: an objective triage tool for high-risk human papillomavirus DNA-positive women. Clin Cancer Res 2011;17:2459-65
- Eijsink JJ, Lendvai A, Deregowski V, et al. A four-gene methylation marker panel as triage test in high-risk human papillomavirus positive patients. Int J Cancer 2012;130:1861-9
- Lai HC, Lin YW, Huang RL, et al. Quantitative DNA methylation analysis detects cervical intraepithelial neoplasms type 3 and worse. Cancer 2010;116:4266-74
- Brebi P, Maldonado L, Noordhuis MG, et al. Genome-wide methylation profiling reveals Zinc finger protein 516 (ZNF516) and FK-506-binding protein 6 (FKBP6) promoters frequently methylated in cervical neoplasia, associated with HPV status and ethnicity in a Chilean population. Epigenetics 2013;9(2):308-17
- Vidal AC, Henry NM, Murphy SK, et al. PEG1/MEST and IGF2 DNA methylation in CIN and cervical cancer. Clin Transl Oncol 2013;16(3):266-72
- Brentnall AR, Vasiljevic N, Scibior-Bentkowska D, et al. A human gene and HPV DNA methylation classifier for cervical pre-cancer. Int J Cancer 2014. [Epub ahead of print]
- Stamey TA, Yang N, Hay AR, et al. Prostate-specific antigen as a serum marker for adenocarcinoma of the prostate. N Engl J Med 1987;317(15):909-16
- Schroder FH, Hugosson J, Roobol MJ, et al. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med 2009;360(13):1320-8
- Wilt TJ, Brawer MK, Jones KM, et al. Radical prostatectomy versus observation for localized prostate cancer. N Engl J Med 2012;367(3):203-13
- Hessels D, Verhaegh GW, Schalken JA, Witjes JA. Applicability of biomarkers in the early diagnosis of prostate cancer. Expert Rev Mol Diagn 2004;4:513-26
- Mehra R, Tomlins SA, Yu J, et al. Characterization of TMPRSS2-ETS gene aberrations in androgen-independent metastatic prostate cancer. Cancer Res 2008;68:3584-90
- Berney DM, Gopalan A, Kudahetti S, et al. Ki-67 and outcome in clinically localised prostate cancer: analysis of conservatively treated prostate cancer patients from the Trans-Atlantic Prostate Group study. Br J Cancer 2009;100:888-93
- Cuzick J, Swanson GP, Fisher G, et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. Lancet Oncol 2011;12:245-55
- Lee WH, Morton RA, Epstein JI, et al. Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc Natl Acad Sci USA 1994;91:11733-7
- Kwabi-Addo B, Chung W, Shen L, et al. Age-related DNA methylation changes in normal human prostate tissues. Clin Cancer Res 2007;13:3796-802
- Bastian PJ, Ellinger J, Wellmann A, et al. Diagnostic and prognostic information in prostate cancer with the help of a small set of hypermethylated gene loci. Clin Cancer Res 2005;11:4097-106
- Maruyama R, Toyooka S, Toyooka KO, et al. Aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features. Clin Cancer Res 2002;8:514-19
- Yegnasubramanian S, Kowalski J, Gonzalgo ML, et al. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res 2004;64:1975-86
- Yamanaka M, Watanabe M, Yamada Y, et al. Altered methylation of multiple genes in carcinogenesis of the prostate. Int J Cancer 2003;106:382-7
- Jeronimo C, Henrique R, Hoque MO, et al. A quantitative promoter methylation profile of prostate cancer. Clin Cancer Res 2004;10:8472-8
- Vanaja DK, Ballman KV, Morlan BW, et al. PDLIM4 repression by hypermethylation as a potential biomarker for prostate cancer. Clin Cancer Res 2006;12:1128-36
- Rosenbaum E, Hoque MO, Cohen Y, et al. Promoter hypermethylation as an independent prognostic factor for relapse in patients with prostate cancer following radical prostatectomy. Clin Cancer Res 2005;11:8321-5
- Cho NY, Kim BH, Choi M, et al. Hypermethylation of CpG island loci and hypomethylation of LINE-1 and Alu repeats in prostate adenocarcinoma and their relationship to clinicopathological features. J Pathol 2007;211:269-77
- Nelson WG, De Marzo AM, Yegnasubramanian S. Epigenetic alterations in human prostate cancers. Endocrinology 2009;150:3991-4002
- Krop I, Player A, Tablante A, et al. Frequent HIN-1 promoter methylation and lack of expression in multiple human tumor types. Mol Cancer Res 2004;2:489-94
- Vasiljević N, Wu K, Brentnall A, et al. Absolute quantitation of DNA methylation of 28 candidate genes in prostate cancer using pyrosequencing. Dis Markers 2011;30:151-61
- Egevad L, Ahmad AS, Algaba F, et al. Standardization of Gleason grading among 337 European pathologists. Histopathology 2013;62(2):247-56
- Nelson WG, Yegnasubramanian S, Agoston AT, et al. Abnormal DNA methylation, epigenetics, and prostate cancer. Front Biosci 2007;12:4254-66
- Cuzick J, Fisher G, Kattan MW, et al. Long-term outcome among men with conservatively treated localised prostate cancer. Br J Cancer 2006;95:1186-94
- Vasiljević N, Ahmad AS, Thorat MA, et al. DNA methylation gene-based models indicating independent poor outcome in prostate cancer. BMC Cancer 2014; In press
- Vinarskaja A, Schulz WA, Ingenwerth M, et al. Association of PITX2 mRNA down-regulation in prostate cancer with promoter hypermethylation and poor prognosis. Urol Oncol 2013;31:622-7
- Dietrich D, Hasinger O, Banez LL, et al. Development and clinical validation of a real-time PCR assay for PITX2 DNA methylation to predict prostate-specific antigen recurrence in prostate cancer patients following radical prostatectomy. J Mol Diagn 2013;15(2):270-9
- Kuzmin I, Liu L, Dammann R, et al. Inactivation of RAS Association Domain Family 1A gene in cervical carcinomas and the role of human papillomavirus infection. Cancer Res 2003;63:1888-93
- Shigematsu H, Suzuki M, Takahashi T, et al. Aberrant methylation of HIN-1 (high in normal-1) is a frequent event in human malignancies. Int J Cancer 2005;113:600-4
- Kim TY, Lee HJ, Hwang KS, et al. Methylation of RUNX3 in various types of human cancers and premalignant stages of gastric carcinoma. Lab Invest 2004;84:479-84
- Cho B, Lee H, Jeong SW, et al. Promoter hypomethylaton of a novel cancer/testis antigen gene CAGE is correlated with its aberrant expression and is seen in premalignant stage of gastric carcinoma. Biochem Biophys Res Commun 2003;307:52-63
- Shivapurkar N, Toyooka S, Toyooka KO, et al. Aberrant methylation of TRAIL decoy receptor genes is frequent in multiple tumor types. Intl J Cancer 2004;109:786-92
- Liu H, Liu W, Wu Y, et al. Loss of epigenetic control of synuclein-γ gene as a molecular indicator of metastasis in a wide range of human cancers. Cancer Res 2005;65:7635-43
- Zhang Y, Bhat I, Zeng M, et al. Human kallikrein 10, a predictive marker for breast cancer. Biol Chem 2006;387(6):715-21
- Haraldson K, Kashuba VI, Dmitriev AA, et al. LRRC3B gene is frequently epigenetically inactivated in several epithelial malignancies and inhibits cell growth and replication. Biochimie 2012;94:1151-7
- Day TK, Bianco-Miotto T. Common gene pathways and families altered by DNA methylation in breast and prostate cancers. Endocr Relat Cancer 2013;20:R215-32
- Graham TA, Humphries A, Sanders T, et al. Use of methylation patterns to determine expansion of stem cell clones in human colon tissue. Gastroenterology 2011;140:1241-50
- Killian JK, Bilke S, Davis S, et al. Large-scale profiling of archival lymph nodes reveals pervasive remodeling of the follicular lymphoma methylome. Cancer Res 2009;69:758-64
- Church TR, Wandell M, Lofton-Day C, et al. Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut 2014;63(2):317-25
- Hesselink AT, Heideman DAM, Steenbergen RDM, et al. Methylation marker analysis of self-sampled cervico-vaginal lavage specimens to triage high-risk HPV-positive women for colposcopy. Int J Cancer 2014. [Epub ahead of print]
- Henrique R, Ribeiro FR, Fonseca D, et al. High promoter methylation levels of APC predict poor prognosis in sextant biopsies from prostate cancer patients. Clin Cancer Res 2007;13(20):6122-9