Abstract
A significant number of adult patients with congenital heart disease suffer from pulmonary arterial hypertension leading to a markedly increased morbidity and mortality. Some defects may be eligible for operative or interventional repair in adulthood but careful selection of candidates is crucial. With the emergence of disease-targeting therapies, symptomatic improvement and stabilization have become possible while the impact on survival currently remains unclear.
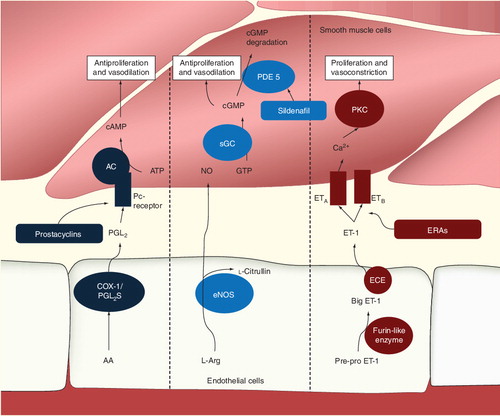
Prostacyclin (PGI2) is synthesized from arachidonic acid by COX-1 and PGI2 synthase. PGI2 activates the receptor of smooth muscle cells and in turn activates adenylate cyclase. This leads to increased production of cAMP from ATP with the effect of vasodilation and antiproliferation. Endothelial nitric oxide (NO) synthase produces NO from l-arginine. NO stimulates soluble guanylate cyclase resulting in increased production of cyclic guanylate monophosphate and thus antiproliferation and vasodilation. Preproendothelin is converted to big-endothelin (ET)-1 and subsequently ET-1 by furin-like enzyme and ET-converting enzymes, respectively. ET-1 activates ET-A and ET-B receptors, leading to calcium release and activation of protein kinase C, which leads to vasoconstriction and proliferation of smooth muscle cells.
Pulmonary hypertension (PH) was first described over one and a half centuries ago based on pathological studies. However, until recently, the underlying pathophysiology was not well understood and treatment options were very limited. Only in the last two decades, therapies have become available that have been proven to improve hemodynamics, augment exercise capacity and reduce mortality in patients with PH. The model disease for PH and the prime condition included in clinical trials remains idiopathic pulmonary arterial hypertension (iPAH). However, PH associated with other conditions is also a major source of morbidity and mortality. One of those is PAH associated with congenital heart disease (CHD) and will be the focus of the current review. The authors aim to provide an overview over the definition and prevalence of PAH in CHD, as well as its current management and briefly highlight possible future developments in the field.
Definition & classification
The current definition and classification of PH was updated at the 4th World Symposium on Pulmonary Hypertension in Dana Point (CA, USA) in 2008. PH – as a general term for a wide spectrum of disease entities with increased pulmonary pressures – has been defined as a mean pulmonary arterial pressure of greater than 25 mm Hg at rest. Pulmonary capillary wedge pressure should be below or equal to 15 mm Hg in all groups except for PH due to left heart disease or pulmonary venous obstruction (‘postcapillary PH’) Citation[1]. Even slightly elevated mean pulmonary pressures in the range of 21–25 mm Hg have been shown to have clinical significance. For this group of patients, the term ‘borderline PH’ has been proposed. A previously used cut-off value for pulmonary pressures during exercise has been removed from the definition as pulmonary pressures in healthy individuals have been shown to be highly variable on exertion Citation[2]. The purpose of all classifications of PH is to group disease entities according to their pathophysiological mechanisms, clinical appearance and response to medical therapies. While this facilitates clinical practice and research to a certain degree, it is of course an oversimplification as significant differences exist within groups and subgroups (Box 1). While the first group in the current classification is termed PAH the other four are described as PH owing to left heart disease, lung disease, chronic thromboembolic and multifactorial mechanisms, respectively. PAH associated with CHD (CHD-PAH) is part of group 1 together with idiopathic and heritable PAH, drug-induced PAH and PAH associated with connective tissue diseases.
In addition, the subclassification of CHD-PAH has been updated, now considering PAH severity, defect size, shunt direction and whether the patient has already undergone repair (Box 2) Citation[1]. The most extreme form of PAH in this setting is the Eisenmenger syndrome (ES) with arterial pressures reaching systemic levels, leading to flow reversal usually through large unrestrictive defects at ventricular or arterial level and accompanied by cyanosis at rest and consecutive multiorgan involvement Citation[3].
Prevalence
Adult CHD patients with PAH form a heterogenous population. There are hemodynamic differences due to the size and location of the underlying cardiac defects. In the Dutch CONCOR registry, the overall prevalence of PH defined as systolic pressure >40 mm Hg was 4.2%. In the population at risk, patients with systemic to pulmonary shunt and patients with surgical shunts, the prevalence was 10%. The occurrence of PAH varied with the underlying defect with a prevalence of 100% for aortopulmonary window, 41% for atrioventricular septal defects, 11% for ventricular septal defects and 8% for secundum atrial septal defects (ASD). In the Euro Heart Survey, a prevalence of 28% was found for PH in patients with septal defects Citation[4] again defining PAH by a systolic pulmonary artery pressure greater 40 mm Hg. Differences may be caused by incomplete data, inconsistent definitions and unrepresentative samples. In a recent retrospective longitudinal cohort study using Canadian healthcare data of 38,430 adults with CHD, 5.8% had the additional diagnosis of PH coded Citation[5].
ES has been shown to develop typically in large unrestrictive posttricuspid defects – that is, in patients with a communication at ventricular or aortopulmonary level but very rarely in ASD or anomalous pulmonary venous return (pretricuspid) Citation[3,6,7]. In the CONCOR registry, the overall prevalence of ES in all CHD patients was 1.1%. Of the patients with PAH due to septal defects, 58% had an ES Citation[8].
The goal of early corrective surgery is to avoid the development or progression of pulmonary vascular disease by eliminating pressure and volume overload of the pulmonary circulation. The development of PAH has been reported to be rare when ventricular shunts are repaired before the age of 2 years Citation[9]. Nevertheless, contemporary patients with operated septal defects still had a prevalence of PH of 3% in the CONCOR registry Citation[8].
Although pulmonary artery pressure increases with age in patients with unoperated ASD and the majority has PH when reaching age 60 and beyond Citation[10], they rarely develop severe pulmonary vascular disease. In a recent study, Huang et al. Citation[11] reported patients with pulmonary vascular resistance (PVR) of 5 Wood units or greater in 6.6% of their ASD population. The rare patients with this defect who present with severe pulmonary vascular disease are typically young adults, predominantly female and share similarities with iPAH Citation[12].
In the last few decades, surgical progress has allowed for earlier and more complex corrective operations leading to a decrease in mortality with more patients reaching adulthood Citation[13]. There has been a notable demographic change with an increasing number of patients with simple lesions not developing PAH while the relative number of complex CHD-PAH patients is increasing Citation[14]. While CHD-PAH usually has a homogeneous pressure distribution, a segmental PAH may sometimes be seen, especially in complex postsurgical CHD patients Citation[15].
Pathophysiology of PH: CHD
As highlighted in the introduction, the current understanding of the pathophysiology of PH is founded on both (histo)pathological and physiologic findings. PH is a chronic disease involving different mechanisms that, in concert, may lead to PH. It would be beyond the scope of this review to comprehensively discuss all known or suspected factors involved in the etiology of PH; however, it should be highlighted that while vasodilators were widely used to treat the condition and vasoconstriction is certainly one part of the underlying pathophysiology of PH, the condition is associated with pronounced histological changes at the level of the lung vessels. These include hypertrophy of vascular smooth muscle cells, plexiform lesions and displacement of smooth muscle cells distally into normally nonmuscular arterioles Citation[16,17]. In addition, these changes may be associated with in situ thrombosis. In the setting of CHD, the pathophysiology of PAH involves pulmonary volume and pressure overload, associated with shear stress at the level of pulmonary vascular endothelium. This induces the expression of local mediators, leading to the aforementioned remodeling of the vascular wall Citation[18]. It has been long appreciated, however, that not every patient with left-to-right shunt lesions and pulmonary overcirculation will develop PH. This has lead to the insight that the development of PH depends both on the magnitude of the exogenous trigger as well as intrinsic factor – that is, the susceptibility of the individual, chiefly determined by genetic factors Citation[19]. The central role of the endothelium in the development of CHD-PAH is due to its exposure to shear stress caused by increased pulmonary blood flow with or without increased pulmonary pressures. The specific pathophysiologic factors involved in translating shear stress into sustained chronic changes of the pulmonary microvasculature have been comprehensively discussed in detail by Rabinovich among others Citation[20]. The histologic alteration involves smooth muscle cell proliferation and hypertrophy, a reduction in apoptosis pathways, release of growth factors and degradation of extracellular matrix leading to the known histopathological changes in PAH. Endothelial abnormalities also favor platelet adherence and activation, immune inflammation and activation of coagulation pathways. Endothelial abnormalities are also central to the imbalance of mediators with vasoconstrictive and vasodilator action. Among the plethora of vasoactive mediators are three groups of substances that are of particular clinical interest as they form the basis of current medical therapy of the condition. These mediators include nitric oxide (NO), endothelin (ET) and prostacyclins.
NO is a vasodilator produced in tissue by one of the various isoforms of NO synthase. NO synthase III is the main form of the enzyme expressed in endothelial cells. The enzyme is activated by elevated intracellular calcium concentrations through a calcium–calmodulin interaction. NO diffuses to adjacent smooth muscle cells where it leads to increased intracellular levels of cyclic guanylate monophosphate (cGMP), inducing vasodilation and inhibiting cell proliferation Citation[21]. Intracellular cGMP is subsequently cleaved by phosphodiasterases (PDEs). Therefore, the pharmacological inhibition of PDEs, increases intracellular cGMP levels leading to vasodilation. Sildenafil represents the prime example of a PDE-5 inhibitor employed in the routine therapy of PH Citation[22].
Another main target of current PH pharmacotherapy is ET. ET is among the most potent vasoconstrictors. In addition, the substance is promitotic and induces fibrosis as well as inflammation. The metabolism of ET is complex and involves initial synthesis by endothelial cells as prepro-ET and is subsequently processed by furin-like enzymes to big-ET-1. The latter requires cleavage by ET-converting enzymes to active ET-1 Citation[23]. ET-1 activates ET receptors (A and B), mainly located on vascular smooth muscle cells inducing to calcium release and activation of protein kinase C. ET receptor antagonists (ERAs) have been developed and have demonstrated beneficial effects on hemodynamics in patients with PAH Citation[24].
Prostacyclin (PGI2) represents another mediator involved in the pathophysiology of the condition that is amenable to pharmacological modification. Prostacyclin is produced by cyclooxygenase-1 and prostacyclin synthase from arachidonic acid cleaved from membrane-bound lipids by phospholipase A2. Activation of prostacyclin receptors on smooth muscle cells leads to the stimulation of adenylate cyclase, increasing cellular cAMP levels Citation[25]. This in turn induces vasodilation and inhibits cellular proliferation. Various PGI2-analogs (epoprostenol, iloprostat and treprostinil) are currently routinely used in the treatment of PH patients. demonstrates the three major pathophysiological pathways.
Outcome & outcome predictors
While the short-term prognosis of CHD-PAH patients is often good, a markedly increased morbidity and mortality has been shown with increasing patient age. The degree of PAH is associated with an increase in New York Heart Association class in patients with septal defects. In a study based on data from the Euro Heart Survey, the presence of PAH was associated with an eightfold-increased probability of functional limitations in patients with open defects. In the presence of ES, there was a further sixfold increase. Similar limitations could be shown in patients with PAH despite a closed defect. During a 6-year follow-up, the presence of PAH was associated with an increased mortality. The worst survival, however, was shown for patients with ES, for who the median estimated mortality during the 5-year follow-up period was 20.6% Citation[4].
In an observational study with 171 ES patients, it was estimated that the average ES patient dies about 20 years earlier than a healthy age-matched peer. In the case of a complex lesion, life expectancy is further reduced by approximately 20 years Citation[26].
In an effort to identify prognostic markers that may guide treatment decisions for patients with PAH in general, a great number of parameters have been studied in recent years. WHO functional class, 6-min walking distance (6MWD), peak VO2, right ventricular function, hemodynamic parameters and plasma levels of brain natriuretic peptide (BNP) have all been found to influence prognosis Citation[27]. Recently, it has been proposed that multivariable risk models might better address a patient’s overall clinical status and outcome. Based on data from 2716 PAH patients from the REVEAL Registry, Benza et al. integrated multiple prognostic factors into a prognostic equation that estimates 1-year survival for a given PAH patient Citation[28]. Variables associated with an increased hazard ratio were similar to those reported in other studies including, but not limited to, type of PAH subgroup, functional class, 6MWD, renal insufficiency, BNP, presence of pericardial effusion and hemodynamic parameters. With only 319 CHD patients included in the study population (11.8% of all subjects included in the study), the significance of this model for CHD-PAH remains unclear. Interestingly, in this study, the authors could not find a better survival for CHD patients compared with other PAH groups. It is a matter of debate whether this is due to a more frequent use of disease-targeting therapies (DTT) in other PAH groups or just a matter of lacking statistical power in a subgroup with relatively few events Citation[29].
In ES patients, higher functional class, a history of arrhythmias, patient age, right ventricular dysfunction, syncope, elevated uric acid levels and the presence of Down’s syndrome have been found to be of value in predicting a poor outcome Citation[26,30–34]. In a very recent study based on data of 181 ES patients, a composite echocardiographic score was also highly predictive of clinical outcome Citation[35]. Another very recent study demonstrated the predictive value of BNP in ES Citation[36].
Acute vasolidator testing in the setting of CHD-PAH is not required since calcium antagonists have no role in the treatment of these patients. However, two previous studies have shown that 29% of patients with ES show at least partial responsiveness to acute vasodilator testing with inhaled NO which may be related to mid-term outcomes in this setting Citation[37,38]. The clinical utility remains unclear at present.
Management
Surgical therapy
Surgical repair is contraindicated in patients who have developed ES Citation[39]. Patients with shunt lesions and PAH that still show a left-to-right shunt have to be carefully evaluated for surgery. It is beyond the scope of this review to discuss this in detail. In brief, shunt lesions can safely be closed and patients expected to benefit as long as PVR does not exceed 5 Wood units. ASD patients who exceed this value should only be considered for defect closure when significant left-to-right shunt can still be proven with or without vasodilator challenge/therapy Citation[39]. Post-tricuspid shunts can be closed with higher PVR; however, as soon as systolic pressure or resistance exceed two-thirds of systemic values, evaluation requires particular care and experience including vasoreagibility testing and sophisticated shunt quantification Citation[39]. The decision of whether or not to perform surgery should be restricted to specialist centers.
Medical therapy: general aspects
The advent of DTT around the turn of the century has opened a new era of PAH therapy in CHD patients. Until then, treatment options were restricted to supportive therapies. This included addressing infections, symptomatic relief of volume overload using diuretics, treatment of arrhythmias, digoxin and oxygen therapy. This was largely based on clinical experience but did not address the underlying cause of the problem – that is, the pulmonary vascular disease, directly. Secondary erythrocytosis, commonly found in cyanotic CHD-PAH patients, represents a physiologic adaptation to improve oxygen delivery to the tissue Citation[40]. As a consequence, routine phlebotomy is contraindicated as it impairs oxygen transport capacity, reduces exercise tolerance Citation[41], induces iron deficient anemia Citation[30] and augments the risk of stroke Citation[42]. If unavoidable due to clear hyperviscosity symptoms, phlebotomy should only be performed in the absence of iron deficiency and by avoiding dehydration Citation[40,43]. Especially, in patients with ES, there appears to be a high prevalence of pulmonary arterial thrombi of 20–29%. Abnormal flow properties secondary to PA dilatation and intimal damage may play a role in thrombogenesis. On the other hand, bleeding under oral anticoagulation or spontaneous bleeding are not uncommon and may have a significant impact on morbidity and mortality Citation[44]. Due to this therapeutic dilemma, routine anticoagulation is not recommended in these patients in the absence of additional indications such as atrial fibrillation, pulmonary arterial thrombus or mechanical prosthetic valves Citation[39]. Strenuous exercise or competitive sports are contraindicated in this population. Endocarditis prophylaxis continues to be recommended for relevant dental procedures by current guidelines. Furthermore, annual immunization against influenza and pneumococcal infections should be performed Citation[39]. Noncardiac surgery is known to be associated with significant mortality, therefore careful planning and intraoperative monitoring in this population is required Citation[45]. Overall, the underlying concept of care has to be not to disturb the delicate balance between systemic and pulmonary circulation Citation[46].
Medical treatment: targeted PAH therapy
Although calcium antagonists have been demonstrated to be beneficial in patients with iPAH and positive response to acute pulmonary vasodilator testing, this approach fails in the majority of patients with CHD and PAH and is contraindicated in the ES population due to the risk of systemic vasodilation and deterioration of cyanosis Citation[39].
Prostacyclins
The first available pulmonary vasodilators with antiproliferative action (DTT) were intravenous prostacyclins, which were shown in the 1990s to improve symptoms in patients with iPAH and other forms of PAH Citation[47]. The uptake of this form of therapy, however, was slow in CHD patients due to the short half life of the substances, demanding continuous intravenous infusion. Improvement of functional capacity, oxygen saturation and pulmonary hemodynamics have been reported in this cohort Citation[48,49]. Intravenous prostacyclin administration, however, is technically challenging and prolonged intravenous therapy is associated with frequent complications such as sepsis and line dislocation Citation[48]. Therefore, intravenous administration requires special expertise and extensive patient training, so it is avoided whenever possible.
Treprostinil, available for subcutaneous infusion, may represent an alternative. A double-blind, placebo-controlled multicenter trial in 470 patients patients with PAH (10% of whom had CHD) has shown that subcutaneous treprostinil has the potential to improve functional capacity and pulmonary hemodynamics and may represent an alternative in selected patients. However, problems with considerable pain at the infusion site (in up to 85% of the patients) requiring discontinuation of therapy in up to 8% of the patients may limit its use Citation[50]. A new approach of intravenous application with an implanted pump may become an alternative in the future.
Inhaled iloprost and inhaled treprostinil have been demonstrated to improve 6MWD and pulmonary hemodynamics and may represent an alternative to intravenous or subcutaneous prostacyclin analogs in selected patients Citation[51,52]; however, robust data are currently lacking in the CHD population. Despite these shortcomings, prostacyclins remain the therapy of choice in highly symptomatic CHD-PAH patients, with cardiac decompensation or presenting in WHO class IV patients Citation[53]. For most other patients, however, oral therapies are preferred. Two main groups of oral substances for the treatment of PAH are currently available: ERAs and PDEs.
ERAs & PDEs
ERAs include bosentan and ambrisentan, while the most commonly used PDEs include sildenafil and tadalafil. The landmark clinical trials to justify marketing authorization for these substances have been discussed in detail elsewhere Citation[54]. It is noteworthy, however, that only the minority of patients included in these trials were CHD patients and ES patients were generally excluded. Therefore, specific data on the CHD-PAH population are required. To date, the most robust evidence on the efficiency of DTT exists for bosentan. This includes a prospective, randomized, controlled, double-blind trial against placebo in 54 ES patients over a period of 16 weeks. The coprimary end points of the study were stability of peripheral oxygen saturation under treatment (i.e., a safety end point) and improvement in 6MWD. Both primary end points were met and an increase in 6MWD of 53 meters against placebo could be demonstrated. This was accompanied by an improvement in WHO functional class in patients on bosentan. Furthermore, a reduction of indexed PVR could be demonstrated Citation[55]. This study was followed by a 6- month open-label extension study, demonstrating a significant improvement in 6MWD in former placebo patients switched to open-label bosentan Citation[56]. Subsequent observational studies confirmed the beneficial effect on symptoms and exercise capacity and, with a few exceptions Citation[57,58], provided evidence that this symptomatic improvement is maintained mid-term. More recently, two studies have addressed the long-term impact of DTT in ES patients Citation[59,60], confirming a sustained positive impact on WHO class and 6MWD in the long term. Improvement in quality of life has also been demonstrated Citation[61]. Limited data are available on the effect of DTT in CHD patients with patent or previously closed left-to-right shunt lesions: Schulze-Neick and colleagues reported improved functional status as well as exercise capacity in 33 CHD-PAH patients treated with bosentan over a mean period of 2.1 years Citation[62]. In addition, these patients were (albeit in small numbers) included in some of the major PAH trials.
The evidence supporting the use of PDEs in CHD-PAH patients with closed defects stems from the SUPER-1 trial, which included 6.5% CHD-PAH patients Citation[22] with the assumption that these patients are, in pathophysiologic terms, very similar to iPAH patients. In contrast, the evidence in the ES population is less robust compared with bosentan. A number of observational studies and extended case reports exist, with the largest observation series published recently by Zhang et al. Citation[63]. The series included 168 ES patients, mostly with post-tricuspid defects (85%) treated with open-label sildenafil for 1 year. The authors were able to demonstrate significant improvements in pulmonary hemodynamics (including PVR and mean pulmonary arterial pressure) as well as in symptoms, peripheral oxygen saturations and 6MWD. In addition, Mukhopadhyay et al. Citation[64] reported data from 16 ES patients that showed that tadalafil reduces pulmonary vascular PVR, mean pulmonary arterial pressure and improves peripheral oxygen saturation after 12 weeks of therapy.
Current European guidelines recommend the use of bosentan in WHO class III patients (IB recommendation), while the use of PDEs can be considered (IIaC recommendation). Unfortunately, there are limited data on the use of combination therapy in this population. While patients with CHD-PAH and closed defects can be treated analogously to iPAH patients Citation[53], the use of combination therapy is less well founded on evidence in the ES population. A prospective, randomized double-blind crossover study in 21 ES patients, failed to show a significant improvement in exercise capacity after addition of sildenafil to bosentan monotherapy Citation[53,65]. However, these were stable ES patients and, therefore, the study does not adequately reflect routine clinical practice of initiating combination therapy in patients deteriorating on oral monotherapy. Two observational studies Citation[60,66] in contrast have demonstrated that initiation of dual or combination therapy in ES patients with a clinical indication improves 6MWD and hemodynamics. Therefore, in accordance to current guidelines, combination therapy may be considered in WHO class III ES patients. Eventually, specific goal-orientated treatment algorithms need to be defined for CHD-PAH patients or ES patients. Currently, however, concrete cut-off values for parameters of exercise capacity, neurohormone levels and echocardiographic measurements are lacking and further research is required to establish these values. It is currently also unclear whether an earlier start of therapy in WHO class II should be considered in CHD-PAH patients. The EARLY trial has demonstrated beneficial effects of bosentan in WHO functional class II patients with IPAH Citation[67]; however, such data are currently lacking for ES patients. Due to marked differences in physiology and the natural history between these groups, the results cannot simply be applied to CHD patients and further studies are required before targeted PAH therapy can be recommended at this early stage. The planned MAESTRO study is designed to also include ES patients in WHO class II and it is hoped that this will provide data on the effects of the early start of therapy (ClinicalTrials.gov identifier: NCT01743001 Citation[101]).
The impact of DTT on survival in CHD remains uncertain. In a retrospective analysis, Dimopoulos et al. reported a markedly better survival in DTT-treated ES patients compared with a historical untreated cohort Citation[68]. The difference remained significant after adjustment for known risk factors; however, such data can only be considered as a hypothesis and further studies are required.
New medical therapies
The cellular mechanisms of vascular remodeling in PAH show a certain similarity to tumor growth (proliferation, increased migration, resistance against apoptosis). New therapeutic options may arise with the advent of antiproliferative agents. Imatinib (a tyrosine kinase inhibitor used to treat chronic myeloid leukemia) has now been studied in experimental models of PAH with promising initial results Citation[69]. A recent randomized controlled study of imatinib as an add-on therapy in iPAH patients showed some encouraging results but serious adverse effects and discontinuation were common Citation[70]. In addition, experience with imatinib in the setting of CHD-PAH is limited. Currently available studies on DTT in CHD are summarized in .
Expert commentary
PH is present in a significant percentage of adults with CHD and is associated with markedly increased morbidity and mortality. Careful evaluation is required to select those patients who may still benefit from defect repair. DTT – in particular with ERAs and less well-documented PDEs – has been shown to improve hemodynamics, symptoms, exercise capacity and quality of life while the effect on survival remains still uncertain. Treatment should be started in WHO functional class III. Whether or not earlier treatment is beneficial, it requires further study. Better criteria to define the optimal timing of treatment initiations are also needed. Traditionally, the majority of clinical studies in PAH have relied on 6MWD as primary end points. It is increasingly recognized, however, that this may not be the best choice; it correlates poorly with morbidity and mortality and may be too insensitive in mildly symptomatic patients. Time to clinical worsening has been suggested as a superior alternative, better assessing the impact of treatment on disease progression and is therefore increasingly being used in current studies Citation[71].
Five-year view
CHD-PAH will continue to represent a major challenge. The mortality of CHD patients has been decreasing due to earlier and more complex operations. However, with more of these patients reaching adulthood, the morbidity – including PAH – is expected to increase. In addition, a growing portion of patients are thought to present with more complex CHD-PAH. With advances in medical therapy, especially the availability of new ERAs, oral prostacyclin analogs and soluble guanylatcyclase activators, it is hoped that symptoms can be further improved and survival benefits may eventually be provided.
Table 1. Currently available studies assessing disease targeting therapies in pulmonary arterial hypertension associated with congenital heart disease and selected studies of mixed pulmonary arterial hypertension populations.
Box 1. Clinical classification of pulmonary hypertension.
Pulmonary arterial hypertension
• Idiopathic pulmonary arterial hypertension
• Heritable
– BMPR2
– ALK1, endoglin (with or without hereditary hemorrhagic telangiectasia)
– Unknown
• Drug- and toxin-induced
• Associated with:
– Connective tissue diseases
– HIV infection
– Portal hypertension
– Congenital heart diseases
– Schistosomiasis
– Chronic hemolytic anemia
• Persistent pulmonary hypertension of the newborn
• Pulmonary veno-occlusive disease and/or pulmonary capillary hemangiomatosis
Pulmonary hypertension owing to left heart disease
• Systolic dysfunction
• Diastolic dysfunction
• Valvular disease
Pulmonary hypertension owing to lung diseases and/or hypoxia
• Chronic obstructive pulmonary disease
• Interstitial lung disease
• Other pulmonary diseases with mixed restrictive and obstructive pattern
• Sleep-disordered breathing
• Alveolar hypoventilation disorders
• Chronic exposure to high altitude
• Developmental abnormalities
Chronic thromboembolic pulmonary hypertensionPulmonary hypertension with unclear multifactorial mechanisms
• Hematologic disorders: myeloproliferative disorders, splenectomy
• Systemic disorders: sarcoidosis, pulmonary Langerhans cell histiocytosis: lymphangioleiomyomatosis, neurofibromatosis, vasculitis
• Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders
• Others: tumoral obstruction, fibrosing mediastinitis, chronic renal failure on dialysis
Box 2. Clinical classification of congenital systemic-to-pulmonary shunts associated with pulmonary arterial hypertension.
Eisenmenger syndrome
• Includes all systemic-to-pulmonary shunts resulting from large defects and leading to a severe increase in PVR and a reversed (pulmonary-to-systemic) or bidirectional shunt; cyanosis, erythrocytosis and multiple organ involvement are present
PAH associated with systemic-to-pulmonary shunts
• Includes moderate to large defects; PVR is mildly to moderately increased, systemic-to-pulmonary shunt is still prevalent and no cyanosis is present at rest
PAH with small defects
• Small defects (usually ventricular septal defects <1 cm and atrial septal defects <2 cm of effective diameter assessed by echocardiography); clinical picture is very similar to idiopathic PAH
PAH after corrective cardiac surgery
• Congenital heart disease has been corrected, but PAH is still present immediately after surgery or recurs several months or years after surgery in the absence of significant postoperative residual lesions
Key issues
• A significant number of adult patients with congenital heart disease suffer from pulmonary arterial hypertension (PAH).
• Pathological changes in the pulmonary vasculature include hypertrophy of vascular smooth muscle cells, plexiform lesions and displacement of smooth muscle cells distally into normally nonmuscular arterioles.
• The largest group of patients at risk consists of patients with septal defects.
• The most extreme form of PAH in this setting is Eisenmenger syndrome (ES) with pulmonary arterial pressures reaching systemic levels, leading to flow reversal and cyanosis at rest.
• PAH associated with congenital heart disease leads to an increased morbidity and mortality. The worst survival was shown for ES patients.
• Surgical repair is contraindicated in patients who have developed ES but patients with PAH and left-to-right shunt can be carefully evaluated for surgical or interventional options.
• Supportive therapy used to be the mainstay of management, including addressing infections, diuretics, treatment of arrhythmias, digoxin and oxygen therapy.
• Disease-targeting therapies using prostacyclin analogs, endothelin receptor antagonists and phosphodiesterase inhibitors have made some symptomatic and hemodynamic improvements possible.
Financial & competing interests disclosure
RM Radke, GP Diller and H Baumgartner have received educational and travel grants by Actelion Pharmaceuticals Deutschland GmbH. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Notes
Data taken from Citation[1].
PAH: Pulmonary arterial hypertension; PVR: Pulmonary vascular resistance.
Data taken from Citation[1].
References
- Simonneau G, Robbins IM, Beghetti M et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 54(Suppl. 1), S43–S54 (2009).
- Hoeper MM. Definition, classification, and epidemiology of pulmonary arterial hypertension. Semin. Respir. Crit. Care Med. 30(4), 369–375 (2009).
- Wood P. The Eisenmenger syndrome or pulmonary hypertension with reversed central shunt. Br. Med. J. 2(5099), 755–762 (1958).
- Engelfriet PM, Duffels MG, Möller T et al. Pulmonary arterial hypertension in adults born with a heart septal defect: the Euro Heart Survey on adult congenital heart disease. Heart 93(6), 682–687 (2007).
- Lowe BS, Therrien J, Ionescu-Ittu R, Pilote L, Martucci G, Marelli AJ. Diagnosis of pulmonary hypertension in the congenital heart disease adult population impact on outcomes. J. Am. Coll. Cardiol. 58(5), 538–546 (2011).
- Fuster V, Steele PM, Edwards WD, Gersh BJ, McGoon MD, Frye RL. Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation 70(4), 580–587 (1984).
- Beghetti M, Galiè N. Eisenmenger syndrome a clinical perspective in a new therapeutic era of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 53(9), 733–740 (2009).
- Duffels MG, Engelfriet PM, Berger RM et al. Pulmonary arterial hypertension in congenital heart disease: an epidemiologic perspective from a Dutch registry. Int. J. Cardiol. 120(2), 198–204 (2007).
- McLaughlin VV, Presberg KW, Doyle RL et al.; American College of Chest Physicians. Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest 126(Suppl. 1), 78S–92S (2004).
- Humenberger M, Rosenhek R, Gabriel H et al. Benefit of atrial septal defect closure in adults: impact of age. Eur. Heart J. 32(5), 553–560 (2011).
- Huang ZW, Fan ZX, Sun JT et al. The short- and medium-term results of transcatheter closure of atrial septal defect with severe pulmonary arterial hypertension. Heart Vessels 27(6), 603–609 (2012).
- Therrien J, Rambihar S, Newman B et al. Eisenmenger syndrome and atrial septal defect: nature or nurture? Can. J. Cardiol. 22(13), 1133–1136 (2006).
- Gatzoulis MA, Alonso-Gonzalez R, Beghetti M. Pulmonary arterial hypertension in paediatric and adult patients with congenital heart disease. Eur. Respir. Rev. 18(113), 154–161 (2009).
- Marelli AJ, Mackie AS, Ionescu-Ittu R, Rahme E, Pilote L. Congenital heart disease in the general population: changing prevalence and age distribution. Circulation 115(2), 163–172 (2007).
- Schuuring MJ, Bouma BJ, Cordina R et al. Treatment of segmental pulmonary artery hypertension in adults with congenital heart disease. Int. J. Cardiol. 164(1), 106–110 (2013).
- Heath D, Edwards JE. The pathology of hypertensive pulmonary vascular disease; a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects. Circulation 18(4 Pt 1), 533–547 (1958).
- Rabinovitch M, Haworth SG, Castaneda AR, Nadas AS, Reid LM. Lung biopsy in congenital heart disease: a morphometric approach to pulmonary vascular disease. Circulation 58(6), 1107–1122 (1978).
- Farber HW, Loscalzo J. Pulmonary arterial hypertension. N. Engl. J. Med. 351(16), 1655–1665 (2004).
- Haworth SG. Role of the endothelium in pulmonary arterial hypertension. Vascul. Pharmacol. 45(5), 317–325 (2006).
- Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Invest. 122(12), 4306–4313 (2012).
- Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 113(13), 1708–1714 (2006).
- Galiè N, Ghofrani HA, Torbicki A et al.; Sildenafil Use in Pulmonary Arterial Hypertension (SUPER) Study Group. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 353(20), 2148–2157 (2005).
- Schiffrin EL. Vascular endothelin in hypertension. Vascul. Pharmacol. 43(1), 19–29 (2005).
- Dupuis J, Hoeper MM. Endothelin receptor antagonists in pulmonary arterial hypertension. Eur. Respir. J. 31(2), 407–415 (2008).
- Falcetti E, Hall SM, Phillips PG et al. Smooth muscle proliferation and role of the prostacyclin (IP) receptor in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 182(9), 1161–1170 (2010).
- Diller GP, Dimopoulos K, Broberg CS et al. Presentation, survival prospects, and predictors of death in Eisenmenger syndrome: a combined retrospective and case-control study. Eur. Heart J. 27(14), 1737–1742 (2006).
- McLaughlin VV, Davis M, Cornwell W. Pulmonary arterial hypertension. Curr. Probl. Cardiol. 36(12), 461–517 (2011).
- Benza RL, Miller DP, Gomberg-Maitland M et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 122(2), 164–172 (2010).
- Mulder BJ. Changing demographics of pulmonary arterial hypertension in congenital heart disease. Eur. Respir. Rev. 19(118), 308–313 (2010).
- Daliento L, Somerville J, Presbitero P et al. Eisenmenger syndrome. Factors relating to deterioration and death. Eur. Heart J. 19(12), 1845–1855 (1998).
- Cantor WJ, Harrison DA, Moussadji JS et al. Determinants of survival and length of survival in adults with Eisenmenger syndrome. Am. J. Cardiol. 84(6), 677–681 (1999).
- Saha A, Balakrishnan KG, Jaiswal PK et al. Prognosis for patients with Eisenmenger syndrome of various aetiology. Int. J. Cardiol. 45(3), 199–207 (1994).
- Oya H, Nagaya N, Satoh T et al. Haemodynamic correlates and prognostic significance of serum uric acid in adult patients with Eisenmenger syndrome. Heart 84(1), 53–58 (2000).
- Clarkson PM, Frye RL, DuShane JW, Burchell HB, Wood EH, Weidman WH. Prognosis for patients with ventricular septal defect and severe pulmonary vascular obstructive disease. Circulation 38(1), 129–135 (1968).
- Moceri P, Dimopoulos K, Liodakis E et al. Echocardiographic predictors of outcome in Eisenmenger syndrome. Circulation 126(12), 1461–1468 (2012).
- Diller GP, Alonso-Gonzalez R, Kempny A et al. B-type natriuretic peptide concentrations in contemporary Eisenmenger syndrome patients: predictive value and response to disease targeting therapy. Heart 98(9), 736–742 (2012).
- Budts W, Van Pelt N, Gillyns H, Gewillig M, Van De Werf F, Janssens S. Residual pulmonary vasoreactivity to inhaled nitric oxide in patients with severe obstructive pulmonary hypertension and Eisenmenger syndrome. Heart 86(5), 553–558 (2001).
- Post MC, Janssens S, Van de Werf F, Budts W. Responsiveness to inhaled nitric oxide is a predictor for mid-term survival in adult patients with congenital heart defects and pulmonary arterial hypertension. Eur. Heart J. 25(18), 1651–1656 (2004).
- Baumgartner H, Bonhoeffer P, De Groot NM et al.; Task Force on the Management of Grown-up Congenital Heart Disease of the European Society of Cardiology (ESC); Association for European Paediatric Cardiology (AEPC); ESC Committee for Practice Guidelines (CPG). ESC Guidelines for the management of grown-up congenital heart disease (new version 2010). Eur. Heart J. 31(23), 2915–2957 (2010).
- Diller GP, Gatzoulis MA. Pulmonary vascular disease in adults with congenital heart disease. Circulation 115(8), 1039–1050 (2007).
- Broberg CS, Bax BE, Okonko DO et al. Blood viscosity and its relationship to iron deficiency, symptoms, and exercise capacity in adults with cyanotic congenital heart disease. J. Am. Coll. Cardiol. 48(2), 356–365 (2006).
- Ammash N, Warnes CA. Cerebrovascular events in adult patients with cyanotic congenital heart disease. J. Am. Coll. Cardiol. 28(3), 768–772 (1996).
- Perloff JK, Rosove MH, Child JS, Wright GB. Adults with cyanotic congenital heart disease: hematologic management. Ann. Intern. Med. 109(5), 406–413 (1988).
- Broberg CS, Ujita M, Prasad S et al. Pulmonary arterial thrombosis in Eisenmenger syndrome is associated with biventricular dysfunction and decreased pulmonary flow velocity. J. Am. Coll. Cardiol. 50(7), 634–642 (2007).
- Ammash NM, Connolly HM, Abel MD, Warnes CA. Noncardiac surgery in Eisenmenger syndrome. J. Am. Coll. Cardiol. 33(1), 222–227 (1999).
- Oechslin E. Eisenmenger’s syndrome. In: Adult Congenital Heart Disease. Gatzoulis M, Webb G, Daubeney P (Eds). Elsevier, PA, USA, 363–378 (2003).
- Rubin LJ, Mendoza J, Hood M et al. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trial. Ann. Intern. Med. 112(7), 485–491 (1990).
- Fernandes SM, Newburger JW, Lang P et al. Usefulness of epoprostenol therapy in the severely ill adolescent/adult with Eisenmenger physiology. Am. J. Cardiol. 91(5), 632–635 (2003).
- Rosenzweig EB, Kerstein D, Barst RJ. Long-term prostacyclin for pulmonary hypertension with associated congenital heart defects. Circulation 99(14), 1858–1865 (1999).
- Simonneau G, Barst RJ, Galie N et al.; Treprostinil Study Group. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a doubleblind, randomized, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 165(6), 800–804 (2002).
- Olschewski H, Simonneau G, Galiè N et al.; Aerosolized Iloprost Randomized Study Group. Inhaled iloprost for severe pulmonary hypertension. N. Engl. J. Med. 347(5), 322–329 (2002).
- Channick RN, Olschewski H, Seeger W, Staub T, Voswinckel R, Rubin LJ. Safety and efficacy of inhaled treprostinil as add-on therapy to bosentan in pulmonary arterial hypertension. J. Am. Coll. Cardiol. 48(7), 1433–1437 (2006).
- Task Force for Diagnosis and Treatment of Pulmonary Hypertension of European Society of Cardiology (ESC), European Respiratory Society (ERS), International Society of Heart and Lung Transplantation (ISHLT) et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J. 34(6), 1219–1263 (2009).
- Galiè N, Palazzini M, Manes A. Pulmonary arterial hypertension: from the kingdom of the near-dead to multiple clinical trial meta-analyses. Eur. Heart J. 31(17), 2080–2086 (2010).
- Galiè N, Beghetti M, Gatzoulis MA et al.; Bosentan Randomized Trial of Endothelin Antagonist Therapy-5 (BREATHE-5) Investigators. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled study. Circulation 114(1), 48–54 (2006).
- Gatzoulis MA, Beghetti M, Galiè N et al.; BREATHE-5 Investigators. Longer-term bosentan therapy improves functional capacity in Eisenmenger syndrome: results of the BREATHE-5 open-label extension study. Int. J. Cardiol. 127(1), 27–32 (2008).
- Apostolopoulou SC, Manginas A, Cokkinos DV, Rammos S. Effect of the oral endothelin antagonist bosentan on the clinical, exercise, and haemodynamic status of patients with pulmonary arterial hypertension related to congenital heart disease. Heart 91(11), 1447–1452 (2005).
- van Loon RL, Hoendermis ES, Duffels MG et al. Long-term effect of bosentan in adults versus children with pulmonary arterial hypertension associated with systemic-to-pulmonary shunt: does the beneficial effect persist? Am. Heart J. 154(4), 776–782 (2007).
- Vis JC, Duffels MG, Mulder P et al. Prolonged beneficial effect of bosentan treatment and 4-year survival rates in adult patients with pulmonary arterial hypertension associated with congenital heart disease. Int. J. Cardiol. 164(1), 64–69 (2013).
- Diller GP, Alonso-Gonzalez R, Dimopoulos K et al. Disease targeting therapies in patients with Eisenmenger syndrome: response to treatment and long-term efficiency. Int. J. Cardiol. doi:10.1016/j.ijcard.2012.02.007 (2012) (Epub ahead of print).
- Tay EL, Papaphylactou M, Diller GP et al. Quality of life and functional capacity can be improved in patients with Eisenmenger syndrome with oral sildenafil therapy. Int. J. Cardiol. 149(3), 372–376 (2011).
- Schulze-Neick I, Gilbert N, Ewert R et al. Adult patients with congenital heart disease and pulmonary arterial hypertension: first open prospective multicenter study of bosentan therapy. Am. Heart J. 150(4), 716 (2005).
- Zhang ZN, Jiang X, Zhang R et al. Oral sildenafil treatment for Eisenmenger syndrome: a prospective, open-label, multicentre study. Heart 97(22), 1876–1881 (2011).
- Mukhopadhyay S, Sharma M, Ramakrishnan S et al. Phosphodiesterase-5 inhibitor in Eisenmenger syndrome: a preliminary observational study. Circulation 114(17), 1807–1810 (2006).
- Iversen K, Jensen AS, Jensen TV, Vejlstrup NG, Søndergaard L. Combination therapy with bosentan and sildenafil in Eisenmenger syndrome: a randomized, placebo-controlled, double-blinded trial. Eur. Heart J. 31(9), 1124–1131 (2010).
- D’Alto M, Romeo E, Argiento P et al. Bosentan-sildenafil association in patients with congenital heart disease-related pulmonary arterial hypertension and Eisenmenger physiology. Int. J. Cardiol. 155(3), 378–382 (2012).
- Galiè N, Rubin Lj, Hoeper M et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet 371(9630), 2093–2100 (2008).
- Dimopoulos K, Inuzuka R, Goletto S et al. Improved survival among patients with Eisenmenger syndrome receiving advanced therapy for pulmonary arterial hypertension. Circulation 121(1), 20–25 (2010).
- Patterson KC, Weissmann A, Ahmadi T, Farber HW. Imatinib mesylate in the treatment of refractory idiopathic pulmonary arterial hypertension. Ann. Intern. Med. 145(2), 152–153 (2006).
- Hoeper MM, Barst RJ, Bourge RC et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation 127(10), 1128–1138 (2013).
- Peacock A, Keogh A, Humbert M. Endpoints in pulmonary arterial hypertension: the role of clinical worsening. Curr. Opin. Pulm. Med. 16(Suppl. 1), S1–S9 (2010).
- Rubin LJ, Badesch DB, Barst RJ et al. Bosentan therapy for pulmonary arterial hypertension. N. Engl. J. Med. 346(12), 896–903 (2002).
- Christensen DD, McConnell ME, Book WM, Mahle WT. Initial experience with bosentan therapy in patients with the Eisenmenger syndrome. Am. J. Cardiol. 94(2), 261–263 (2004).
- Barst RJ, Langleben D, Frost A et al.; STRIDE-1 Study Group. Sitaxsentan therapy for pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 169(4), 441–447 (2004).
- Gatzoulis MA, Rogers P, Li W et al. Safety and tolerability of bosentan in adults with Eisenmenger physiology. Int. J. Cardiol. 98(1), 147–151 (2005).
- Kotlyar E, Sy R, Keogh AM et al. Bosentan for the treatment of pulmonary arterial hypertension associated with congenital cardiac disease. Cardiol. Young 16(3), 268–274 (2006).
- Benza RL, Rayburn BK, Tallaj JA et al. Efficacy of bosentan in a small cohort of adult patients with pulmonary arterial hypertension related to congenital heart disease. Chest 129(4), 1009–1015 (2006).
- Sitbon O, Beghetti M, Petit J et al. Bosentan for the treatment of pulmonary arterial hypertension associated with congenital heart defects. Eur. J. Clin. Invest. 36(Suppl. 3), 25–31 (2006).
- Singh TP, Rohit M, Grover A, Malhotra S, Vijayvergiya R. A randomized, placebo-controlled, double-blind, crossover study to evaluate the efficacy of oral sildenafil therapy in severe pulmonary artery hypertension. Am. Heart J. 151(4), 851.e1–851.e5 (2006).
- Barst RJ, Langleben D, Badesch D et al.; STRIDE-2 Study Group. Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J. Am. Coll. Cardiol. 47(10), 2049–2056 (2006).
- D’Alto M, Vizza CD, Romeo E et al. Long term effects of bosentan treatment in adult patients with pulmonary arterial hypertension related to congenital heart disease (Eisenmenger physiology): safety, tolerability, clinical, and haemodynamic effect. Heart 93(5), 621–625 (2007).
- Apostolopoulou SC, Manginas A, Cokkinos DV, Rammos S. Long-term oral bosentan treatment in patients with pulmonary arterial hypertension related to congenital heart disease: a 2-year study. Heart 93(3), 350–354 (2007).
- Diller GP, Dimopoulos K, Kaya MG et al. Long-term safety, tolerability and efficacy of bosentan in adults with pulmonary arterial hypertension associated with congenital heart disease. Heart 93(8), 974–976 (2007).
- Chau EM, Fan KY, Chow WH. Effects of chronic sildenafil in patients with Eisenmenger syndrome versus idiopathic pulmonary arterial hypertension. Int. J. Cardiol. 120(3), 301–305 (2007).
- Galiè N, Olschewski H, Oudiz RJ et al.; Ambrisentan in Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled, Multicenter, Efficacy Studies (ARIES) Group. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 117(23), 3010–3019 (2008).
- Galiè N, Brundage BH, Ghofrani HA et al.; Pulmonary Arterial Hypertension and Response to Tadalafil (PHIRST) Study Group. Tadalafil therapy for pulmonary arterial hypertension. Circulation 119(22), 2894–2903 (2009).
- Duffels MG, Vis JC, van Loon RL et al. Effect of bosentan on exercise capacity and quality of life in adults with pulmonary arterial hypertension associated with congenital heart disease with and without Down’s syndrome. Am. J. Cardiol. 103(9), 1309–1315 (2009).
- Kermeen FD, Franks C, O’Brien K et al. Endothelin receptor antagonists are an effective long term treatment option in pulmonary arterial hypertension associated with congenital heart disease with or without trisomy 21. Heart. Lung Circ. 19(10), 595–600 (2010).
- Lu XL, Xiong CM, Shan GL et al. Impact of sildenafil therapy on pulmonary arterial hypertension in adults with congenital heart disease. Cardiovasc. Ther. 28(6), 350–355 (2010).
- D’Alto M, Romeo E, Argiento P et al. Therapy for pulmonary arterial hypertension due to congenital heart disease and Down’s syndrome. Int. J. Cardiol. 164(3), 323–326 (2013).
- Zeng WJ, Lu XL, Xiong CM et al.; Sildenafil Therapy on Pulmonary Arterial Hypertension Associated With Different Types of Congenital Heart Disease Study Group. The efficacy and safety of sildenafil in patients with pulmonary arterial hypertension associated with the different types of congenital heart disease. Clin. Cardiol. 34(8), 513–518 (2011).
- Monfredi O, Griffiths L, Clarke B, Mahadevan VS. Efficacy and safety of bosentan for pulmonary arterial hypertension in adults with congenital heart disease. Am. J. Cardiol. 108(10), 1483–1488 (2011).
- Dong MF, Ma ZS, Ma SJ et al. Effect of prostaglandin E1 on pulmonary arterial hypertension following corrective surgery for congenital heart disease. J. Cardiovasc. Pharmacol. Ther. 17(3), 303–307 (2012).
Website
- ClinicalTrials.gov database. www.clinicaltrials.gov.uk