
ABSTRACT
An extended series of studies indicate that endogenous phase shifts in circadian neuronal input signaling to the biological clock system centered within the hypothalamic suprachiasmatic nucleus (SCN) facilitates shifts in metabolic status. In particular, a diminution of the circadian peak in dopaminergic input to the peri-SCN facilitates the onset of fattening, insulin resistance and glucose intolerance while reversal of low circadian peak dopaminergic activity to the peri-SCN via direct timed dopamine administration to this area normalizes the obese, insulin resistant, glucose intolerant state in high fat fed animals. Systemic circadian-timed daily administration of a potent dopamine D2 receptor agonist, bromocriptine, to increase diminished circadian peak dopaminergic hypothalamic activity across a wide variety of animal models of metabolic syndrome and type 2 diabetes mellitus (T2DM) results in improvements in the obese, insulin resistant, glucose intolerant condition by improving hypothalamic fuel sensing and reducing insulin resistance, elevated sympathetic tone, and leptin resistance. A circadian-timed (within 2 hours of waking in the morning) once daily administration of a quick release formulation of bromocriptine (bromocriptine-QR) has been approved for the treatment of T2DM by the U.S. Food and Drug Administration. Clinical studies with such bromocriptine-QR therapy (1.6 to 4.8 mg/day) indicate that it improves glycemic control by reducing postprandial glucose levels without raising plasma insulin. Across studies of various T2DM populations, bromocriptine-QR has been demonstrated to reduce HbA1c by –0.5 to –1.7. The drug has a good safety profile with transient mild to moderate nausea, headache and dizziness as the most frequent adverse events noted with the medication. In a large randomized clinical study of T2DM subjects, bromocriptine-QR exposure was associated with a 42% hazard ratio reduction of a pre-specified adverse cardiovascular endpoint including myocardial infarction, stroke, hospitalization for congestive heart failure, revascularization surgery, or unstable angina. Bromocriptine-QR represents a novel method of treating T2DM that may have benefits for cardiovascular disease as well.
Introduction
Overall summary
Vertebrate species in the wild, from teleosts to mammals, within and outside of the temperate zones, exhibit marked annual cycles of metabolism, oscillating between obese and lean conditions at different seasons of the year. The seasonally obese condition is coupled with marked hyperinsulinemia, and insulin resistance, similar to the human metabolic syndrome phenotype. This evolutionarily preserved mechanism for the induction of the seasonal obese, insulin-resistant state imparts a survival advantage during long periods (a season) of low-food availability (e.g. winter [December to March] in northern temperate zones) as this condition facilitates increased hepatic glucose output to supply the brain with glucose when little to none is available in the environment while the insulin-resistant peripheral tissues increase utilization of the increased fat stores as an energy source. This annual cycle of metabolism is also observed in modern-day humans. Studies investigating this annual cycle of metabolism revealed that seasonal changes in metabolism were the manifestation of seasonal changes in circadian (24-h) phase relationships of neurotransmitter activities communicating with the biological clock pacemaker system in the brain. For example, the circadian peak in dopaminergic activity at the area juxtaposed to and communicating with the biological clock (the suprachiasmatic nucleus [SCN]) is reduced in seasonally obese, insulin-resistant animals. A reduction in the circadian peak of dopaminergic activity at this biological clock area is also observed in animals made insulin resistant by high-fat feeding. Subsequent studies established that a diminution in the circadian peak of dopaminergic activity at the area of the hypothalamic biological clock pacemaker, the SCN, actually contributes significantly to the development of insulin resistance syndrome (IRS). Emerging evidence suggests that in humans, chronic (year-long) exposure to the ‘western diet’ rich in saturated fat and simple sugars locks the individual into the insulin-resistant season circadian neuroendocrinology (e.g. diminution of the normal circadian peak of dopaminergic activity at the biological clock among other changes) all year long that over time facilitates development of the metabolic syndrome. Daily administration of bromocriptine, a potent dopamine D2 receptor agonist, timed to re-establish this circadian peak of dopaminergic activity in IRS improves multiple metabolic dysfunctions of the disorder (e.g. glucose intolerance, insulin resistance, dyslipidemia, hypertension) across a wide variety of animal models of this condition. In humans with type 2 diabetes mellitus (T2DM), daily morning administration of bromocriptine-QR, a quick release formulation of bromocriptine that provides a brief 4–5 h daily pulse of the agent to the body, improves glycemic control in such subjects particularly during the postprandial state across the three standard meals of the day without raising the plasma insulin level. Several studies indicate that when bromocriptine-QR is used as add-on therapy to sulfonylurea (SU), metformin plus SU, or thiazolidinedione (TZD), it reduces percent glycated hemoglobin A1c (HbA1c) by −0.6 to −0.9 over a 24-week exposure period to the drug. Pilot studies demonstrate that as add-on to insulin, bromocriptine-QR reduces percent-glycated HbA1c by −0.7 to −1.7 in different studies. Preclinical and clinical studies indicate that timed bromocriptine-QR therapy improves glucose disposal and insulin action in insulin-resistant states by correcting hypothalamic aberrations that predispose to decreased hypothalamic fuel-sensing-driven peripheral insulin action, increased sympathetic tone, and leptin resistance. Such circadian dopaminergic mechanisms may also potentiate cardiovascular health and clinical studies of T2DM subjects indicate that bromocriptine-QR therapy is associated with a significant 40–50% hazard rate reduction (HRR) of adverse cardiovascular events within 1 year of time. Given available evidence suggesting that bromocriptine-QR improves postprandial insulin sensitivity, the agent’s utility in treating T2DM may best be maximized in patients with sufficient postprandial insulin levels as a consequence of early disease state or treatment with insulin secretion enhancers such as dipeptidyl peptidase-4 inhibitors (DPP4Is) and glucagon like peptide-1 (GLP-1) analogs, or insulin itself.
Evolution of insulin resistance and its potentiation by the westernized diet
Insulin resistance and decreased beta cell insulin secretory capacity are the major pathological events predisposing to the decreased glucose disposal and increased hepatic glucose output characteristic of T2DM [Citation1–Citation6]. It is now evident that environmentally induced alterations of multiple organ systems, most prominently the central nervous system (CNS), contribute to these predisposing pathologies (reviewed in [Citation7–Citation17]). Respecting fuel metabolism, insulin resistance is generally characterized by a diminished ability of insulin to (1) stimulate glucose uptake in tissues responsive to such insulin action, (2) inhibit glucose production and stimulate glucose storage in liver, (3) inhibit lipolysis in adipose tissue, and (4) promote vascular relaxation and health [Citation1–Citation6,Citation18,Citation19]. In insulin-resistant states, the ability of insulin to stimulate lipogenesis however is not diminished and may actually be increased in absolute terms due to compensatory hyperinsulinemia associated with the insulin resistance [Citation20]. Such hyperinsulinemia therefore facilitates fattening in insulin-resistant conditions. While obesity is a common correlate of insulin resistance, it is clear that exposure to high-saturated fat diets can induce insulin resistance long before obesity ensues and that such diet-precipitated insulin resistance actually facilitates further fattening [Citation21–Citation24]. How a westernized diet high in saturated fats (and simple sugars) induces insulin resistance has been the focus of much investigation in the past few decades during which time the prevalence of obesity and T2DM has risen dramatically worldwide. The fact that the recent rise of IRS (characterized by hyperinsulinemia, insulin resistance, increased fat stores, dyslipidemia, low grade systemic inflammation, and hypertension), obesity, and T2DM are rapid onset, common occurrences across ethnic lines and genetic backgrounds without geographic borders argues strongly for an environmental factor versus genetic defect causative in the widespread rapid rise in the disease across the planet. In fact, the rise of the disease in specific geographic areas of the world correlates well with the introduction of the westernized diet in those areas [Citation25–Citation27]. Human and animal studies have clearly demonstrated the impact of high-saturated fat diets to rapidly induce resistance to insulin-stimulated glucose disposal in muscle and to insulin inhibition of hepatic glucose output, while facilitating increased lipogenic responsiveness to insulin, the most potent lipogenic hormone known [Citation28,Citation29]. However, the story is not quite so simple. Not all humans or animals exposed to high-fat diets actually develop insulin resistance or obesity [Citation30,Citation31]. Rather, it is now becoming increasingly evident that the CNS exerts control over responsiveness to a multitude of environmental input signals including the high-fat diet such that specific CNS neural circuit activities either predispose or prevent the insulin resistance-inducing effects of such environmental inputs including the westernized diet (and stress – see below) depending upon the specific functional organization of such CNS neurochemistry and circuit activities controlling metabolism. That is to say, it is the brain’s response to the environment, more so that the environment itself, that dictates the full metabolic outcome of environmental changes such as the presentation of the westernized diet. The neurophysiological literature is now replete with studies demonstrating the important roles of several neural circuits that impact peripheral fuel metabolism (reviewed in [Citation32]) including hypothalamic arcuate-alpha melanocortin-stimulating hormone (αMSH) [Citation33–Citation35] and arcuate neuropeptide Y (NPY) – paraventricular nuclei (PVN) circuits [Citation36–Citation38], the lateral hypothalamic (LH) leptin – orexin mesolimbic circuit [Citation39–Citation42], ventromedial hypothalamic and arcuate leptin autonomic/neuroendocrine circuits [Citation43–Citation47], and the insulin arcuate [Citation48] and dopamine mesolimbic circuits [Citation49,Citation50] to name just a few. Emerging experimental evidence suggests important roles for the biological clock system residing within and around the SCN of the hypothalamus in the integration of these CNS circuits to create an orchestrated organized CNS output signal (program) to the peripheral organs respecting fuel metabolism (see discussion below) [Citation51–Citation54].
Role of the biological clock (SCN) and circadian rhythms in the regulation of metabolism
Due to the earth’s rotation on its axis and to its revolution around the sun, life evolved on this planet in a cyclic environment of periods of daylight and darkness that summate to 24 h of time (1 day) and a longer cyclic environment of changing ratio of daily periods of daylight and darkness (e.g. seasons) that summate to 365 days (1 year). Coincident with these cyclic patterns of photoperiod, are cyclic changes in several other environmental conditions such as temperature, barometric pressure, rain fall, and the presence or absence of food supply for organisms of all types. Biological systems, from single-celled eukaryotes to multicellular organisms, have evolved biochemical mechanisms to perceive the environment and importantly to anticipate the future occurrence of such (daily and annual) cyclic events in the environment.
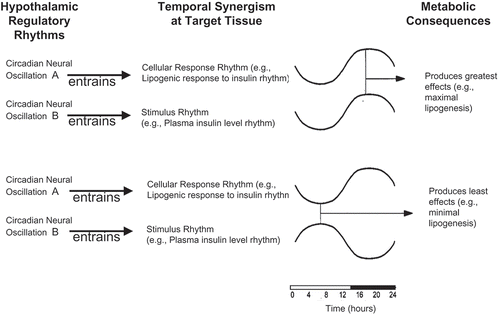
Circadian rhythms are cycles of biological events that persist in constant environmental conditions with periods that approximate 24 h. The basic biological unit of circadian rhythm is the cell and circadian rhythms of biological activities are ubiquitously expressed in the biome. In a multicellular organism, it is the role of the hypothalamic SCN, including its local neural connections (collectively – the seat of the endogenous circadian pacemaker system in vertebrates), to function via its direct and indirect efferent connections to other CNS centers including several hypothalamic centers regulating physiology (e.g. the ventromedial hypothalamus [VMH], PVN, arcuate nucleus, LH, dorsomedial nucleus, etc.), the autonomic nervous system, and the endocrine system, to synchronize cellular circadian activities for the production of tissue and organ level biological rhythms that temporally organize the organism internally and synchronize it with its cyclic environment [Citation51–Citation54]. Simple examples of such SCN-driven organization of metabolism include the synchronization of the circadian rhythms of locomotor activity (wakefulness), feeding, glucose disposal, and lipogenesis during one portion of the day and of sleep, fasting, increased hepatic glucose output, and lipolysis during another portion of the day that coordinate with the animal’s cyclic environment (e.g. daily active metabolic/feeding period when the prey/predator ratio is greatest) among vertebrates in the wild (reviewed in [51,52]). Daily rhythms of cellular biochemical activities, such as lipogenesis, are orchestrated via the SCN by coordinating circadian rhythms of stimulus (neuroendocrine factors) and response (cellular neuroendocrine factor receptor/signaling) rhythms. Preclinical studies indicate that the circadian rhythm of lipogenesis is manifested by a temporal interaction of the phase in the circadian peak in hepatic lipogenic responsiveness to insulin (response rhythm) with the phase in the circadian peak in plasma insulin level (secretory response to glucose and other metabolites) (stimulus rhythm) [Citation51,Citation52,Citation55–Citation59]. The greatest biological effect (in this case lipogenic output) occurs when the circadian peak in the stimulus level (insulin) and cellular response activity (insulin receptor-signal transduction pathway) rhythms are ‘in phase’ (both rhythms peak at the same time of day). A gradation of lesser effects occurs as these two rhythms move out-of-phase from each other. A multitude of such circadian stimulus and cellular response rhythms regulate the biochemistry of the cell ().
Figure 1. Temporal synergism of circadian rhythms of biological activities at the target tissue level regulate metabolism.
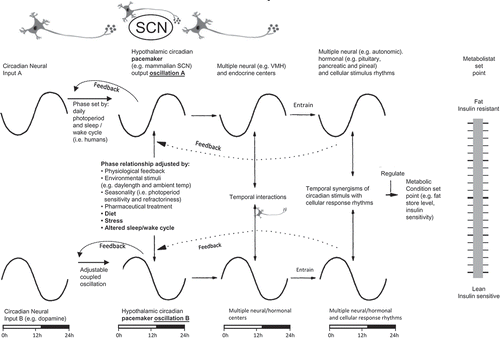
Moreover, a multitude of such SCN-orchestrated circadian stimulus and cellular response rhythms throughout the tissues/organs of the body regulating multiple metabolic functions such as lipid mobilization, glucose production and storage, and protein turnover, and so on are ultimately integrated to produce an overall organismal level metabolic status (e.g. insulin-sensitive or resistant state) (reviewed in detail in [Citation51,Citation52,Citation60–Citation62]). Circadian activities regulate metabolic states that are not generally viewed in circadian terms, such as level of obesity, degree of insulin resistance or glucose intolerance, magnitude of hyperlipidemia, and so on. Furthermore, the SCN receive input from and respond to circadian and noncircadian stimuli from numerous photic (eye) and nonphotic centers throughout the CNS, other hypothalamic nuclei, the peripheral neuroendocrine system and humoral factors, including metabolites (and therefore food itself). As such, physiologic (or pharmacologic) modulation of this circadian metabolic control center (and therefore its output control of metabolism) need not necessarily itself be circadian for example food quality stimuli or stress inputs (see discussion below). Ultimately, it is the preprogrammed, integrated response of the SCN circuit to its endogenous and environmental circadian and noncircadian input signals that modulates peripheral fuel metabolism via SCN-directed changes in the autonomic and neuroendocrine axes [Citation51–Citation56] ().
Figure 2. Circadian metabolistat and blueprint for regulation of metabolism.
The CNS controls metabolic responsiveness to high-fat diet – evolutionary aspects
Such CNS programming of metabolism is a prominent feature of metabolic control among vertebrate animals in the wild representing hundreds of millions of years of evolution. Among vertebrate species in the wild, from teleosts to mammals (spanning some 425 million years of evolutionary time), animals express the seasonal ability to fatten in anticipation of an ensuing season of low/no food availability which metabolic state is characterized by hyperinsulinemia, insulin resistance, and resultant increased fat stores. The insulin-resistant state that evolved in a 24-h-day, 365-day-year cyclic environment imparted a survival strategy against predictable ensuing adverse environmental conditions wherein food availability was/is low or nonexistent for long periods of time (i.e. a season). In such seasonal circumstances, hyperinsulinemia supports increased lipogenesis and fattening, while concurrent insulin resistance provides for an endogenous source of glucose (liver gluconeogenesis) when little to none is environmentally available. Increased hepatic glucose output supplies the CNS (that has an absolute requirement for glucose as an energy source) with its obligatory glucose fuel, and this supply is augmented by peripheral insulin resistance, where insulin-sensitive tissues (e.g. muscle) are preferentially fueled by adipose and intracellular fat stores (further maintaining the tissue insulin resistance). Liver-derived glucose may thus be ‘shunted’ to a certain extent to the brain by the coexistent peripheral lipid-induced insulin resistance, and the animal is better able to survive long periods of low/no food (glucose) availability. The annual cycle of metabolism among vertebrates in the wild evolved in synchrony with the annual cycle of food quality supply. Hints of an annual cycle of fuel metabolism exist even in prehistoric man exposed to the Paleolithic diet (reviewed in [Citation50]), which is known to support insulin sensitivity [Citation63–Citation65]. The high protein, omega-3 fatty acid, and flavonoid content of the Paleolithic diet was only available seasonally as a result of seasonal changes in the availability of flora and fauna [Citation51]. Most importantly however, while this seasonal programming ‘switch’ to the insulin-resistant state can be coupled to seasonal increases in environmental food quality (e.g. lipid, protein quality) supply, no such environmental food alteration is necessary for its expression (reviewed in [51]). That is to say, the induction (and the reversal) of this evolutionarily well-preserved insulin-resistant state is endogenously programmed and only supported by, not driven by, seasonal changes in food quality (e.g. lipid content). For example, the fattening effects of a high-fat diet are marked in winter but minimal or nonexistent in summer in many seasonal mammalian species under natural conditions in temperate zones [Citation51]. Likewise, the annual expression of the obese, insulin-resistant state persists in animals maintained under natural conditions on low-fat diets throughout the year. This evolutionarily preserved mechanism for regulating seasonal metabolism including metabolic responsiveness to dietary constituents offers major clues into the nature of CNS regulation of metabolism. Inasmuch as this seasonal survival mechanism (anthropomorphically termed strategy) is not merely the ability to develop insulin resistance but rather to develop insulin resistance at precisely the appropriate time of year, it follows that the construct of this endogenous CNS mechanism for regulating metabolism comprises clock/timing components. Importantly, the CNS control system for metabolism is malleable and able to shift back and forth from one metabolic regulating organization (e.g. potentiating insulin sensitivity) to another (e.g. potentiating insulin resistance). Moreover, it must be appreciated that this seasonal mechanism for regulating metabolism that has been preserved over hundreds of millions of years of evolution is also manifested in man, as multiple studies have clearly demonstrated [Citation66–Citation75].
What neurophysiological framework may provide for the biological convergence of seasonal-induced insulin resistance preserved over millions of years of evolution as well as western diet-induced sustained insulin resistance observed in so many humans across the globe irrespective of genetic background and within a very short period of time of exposure to the diet? Such information may likely lead to greater insights of the regulation of metabolism and treatment strategies for metabolic diseases such as IRS and T2DM.
Involvement of the biological clock and circadian rhythms in the manifestation of seasonal metabolism and IRS
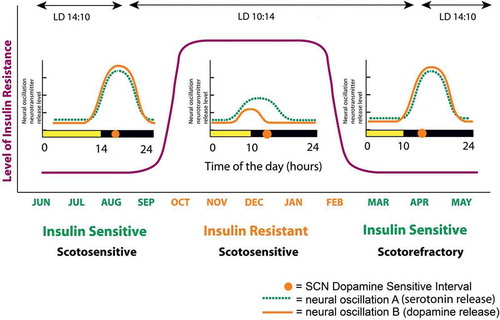
Studies, initiated by Albert H. Meier’s laboratory at Louisiana State University over a half-century ago, provided the original, foundational, and seminal research of biological clock mechanisms operative in the expression of the annual cycle of physiological events, including metabolism, among species from all the major vertebrate classes and this work has been reviewed in detail elsewhere [Citation51,Citation76,Citation77]. To summarize this work succinctly, it was concluded that the annual cycle of metabolism (oscillation from the lean/insulin-sensitive state to the obese/insulin-resistant state) among vertebrates is the result of seasonally changing phase relationships of circadian rhythms of multiple neural input signals to and within the biological clock (SCN) and that the amplitude and phase relationship of circadian dopamine and serotonin neuronal input activities to the SCN area are particularly dominant among such inputs in this role (discussed below). One particular circadian phase relationship of dopaminergic and serotonergic (5HT) neuronal peak input activities at the SCN area potentiated the seasonal lean/insulin-sensitive state while another circadian phase relationship of these neurotransmitter peak activities at the SCN potentiated the seasonal obese/insulin-resistant state. Merely mimicking the circadian phase relationships of these neurotransmitter peaks of a particular season by appropriately timed daily injections of their precursors (3,4-dihydroxy-L-phenylalanine (L-DOPA) and 5-hydroxytryptophan, respectively) was able to produce that seasonal condition irrespective of the actual time of year across studies of species from the major vertebrate classes from fish to mammals [Citation77–Citation79]. Changes in the phase relationships of these circadian input signals to the SCN area can be influenced by the environment (e.g. photoperiod, food quality – see discussion below) ().
Figure 3. Circadian organization of the annual cycle of metabolism of a representative mammalian species.
The circadian rhythm of dopamine input activity to the SCN is critical in the regulation of its output control of peripheral metabolism
Comparing seasonal animals from the glucose tolerant versus glucose intolerant periods of the year while they are maintained on the same photoperiod and low-fat diet in the laboratory throughout this time, it has been observed that the circadian peak in dopaminergic activity at the area of the SCN observed in glucose tolerant animals is diminished appreciably in glucose intolerant animals [Citation80]. Similarly, in animals sensitive to the fattening/insulin resistance inducing effects of a high-saturated fat diet (60% of total calories), the circadian peak in dopamine release at the SCN is markedly diminished [Citation81]. Moreover, at the site of the major dopaminergic neuronal projection to the SCN area, the supramammillary nucleus (SuMN), the circadian rhythm of tyrosine hydroxylase activity (the rate limiting enzyme for dopamine synthesis), that coincides with the circadian rhythm of dopamine release activity at the SCN, is also greatly diminished in animals sensitive to the fattening/insulin resistance-inducing effects of a high-fat diet [Citation81] ().
Figure 4. High saturated fat diet (HFD) reduces circadian peak SCN and SuMN dopaminergic neuronal activity.
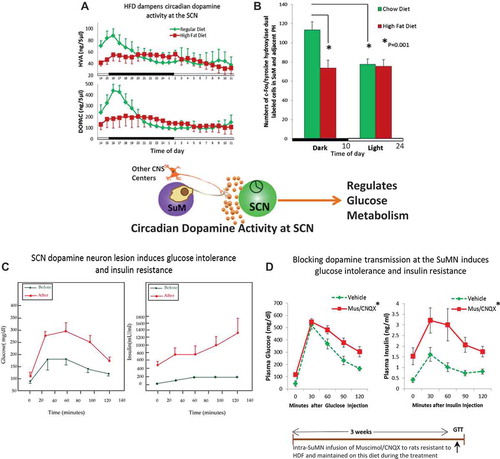
Importantly, several studies have provided evidence for a cause–effect relationship between circadian peak dopaminergic activity at the SuMN and SCN areas and insulin sensitivity as follows. In insulin-sensitive animals, specific lesion of dopaminergic neurons at the SCN area induces insulin resistance [Citation82] as does inhibition of dopamine release from the SuMN [Citation83]. Moreover, administration of dopamine to the SCN area or activation of SuMN dopaminergic projections to the SCN in obese, insulin-resistant animals maintained on a high-fat diet each ameliorate IRS in these animals ([Citation84,Citation85], A.H. Cincotta, unpublished data). An important question is ‘How does this circadian peak in hypothalamic dopaminergic activity regulate peripheral fuel metabolism and insulin sensitivity?’
Diminution of the circadian peak in hypothalamic dopaminergic activity characteristic of insulin-resistant states is coupled to marked increases in VMH noradrenergic (NE), and 5HT input activities to the VMH in seasonal animals, and the IRS is coupled to such VMH changes across the spectrum of seasonal, diet-induced, and genetic models of the syndrome (reviewed in [52,86]). It has been demonstrated in insulin-sensitive animals that induction of increased VMH NE and 5HT activities by direct VMH infusion of these neurotransmitters induces the full IRS, including obesity, insulin resistance, glucose intolerance, and hypertension. Such animals exhibit hyperleptinemia characteristic of leptin resistance, increased sympathetic tone, hyperinsulinemia [Citation52,Citation86], and a loss of appropriate hypothalamic fuel sensing [Citation87]. In insulin sensitive states, the hypothalamus senses meal-related increases in fuels such as glucose and fatty acids and generates signals to the periphery that promote postmeal muscle and hepatic insulin sensitivity [Citation88–Citation92], and this normal hypothalamic response to the meal is markedly attenuated by increased VMH NE activity thus potentiating postprandial insulin resistance [Citation87]. In parallel, increased VMH NE and 5HT activities also induce increases in sympathetic tone and hyperleptinemia/leptin resistance [Citation93], which also contribute to postprandial insulin resistance [Citation54,Citation94–Citation97]. The VMH NE/5HT activity-induced hyperinsulinemia potentiates lipid synthesis and increased fat stores while these same VMH hypothalamic activities stimulate lipid/free fatty acid (FFA) mobilization from adipose potentiating lipotoxicity in muscle, liver, and the pancreatic islet beta cell [Citation93,Citation98,Citation99]. This hyperinsulinemia also feeds back on the CNS to reduce dopaminergic activity and increase VMH NE activity [Citation52,Citation100], thereby locking the organism in a vicious cycle of CNS-driven peripheral insulin resistance. It was initially postulated that loss of appropriate hypothalamic fuel sensing was a causative factor leading to glucose intolerance with brain-fuel-sensing neurons inappropriately sensing and responding to local hyperglycemia as if they were sensing local hypoglycemia [Citation52]. This hypothesis was subsequently supported by results of studies of VMH regulation of fuel-sensing activities as described above [Citation52,Citation87]. Increases in VMH NE activity initiate the acute counter-regulatory response to hypoglycemia [Citation101–Citation103], but this increased VMH NE activity is also a chronic phenomenon present in insulin-resistant states, altering the appropriate response to hyperglycemia and/or hyperFFAemia from stimulation of insulin sensitivity to induction of neuroendocrine events (including counter-regulatory type events including increased sympathetic tone and hyperglucagonemia) that facilitate insulin resistance [Citation52,Citation86,Citation87]. Available evidence suggests that a reduction in the circadian dopaminergic input to the clock allows for this altered VMH NE/5HT state to arise.
Additionally, low hypothalamic dopaminergic tone precipitates increases in PVN NPY and corticotrophin-releasing hormone (CRH) levels [Citation104]. The concurrent increases in NPY and CRH activities stimulate hyperinsulinemia and fattening concurrent with increased sympathetic tone in the periphery again potentiating increased lipid supply to the muscle, liver, and beta cell (lipotoxicity) and potentiating insulin resistance (reviewed in [Citation104]).
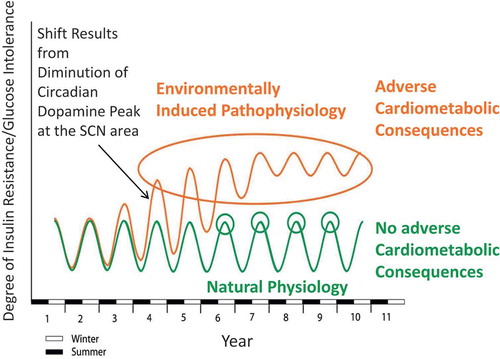
The diminution of the circadian peak in dopaminergic activity at the SCN area coupled to/facilitating the increase in VMH NE and 5HT activities and PVN NPY and CRH activities forms a ‘treacherous triad’ of hypothalamic circuitry that orchestrates a self-sustaining insulin-resistant state (). While such a circadian-based hypothalamic neurochemistry provides for survival against low/no food availability in a seasonal natural environment setting via CNS seasonal control mechanisms, the annual persistence of this neurochemical organization precipitated by the persistent presence of the westernized diet (as discussed above) (or westernized lifestyle – including stress and/or altered sleep/wake cycle) as in the human condition potentiates metabolic disease. Several studies have now documented that alterations to daily sleep (either shortened sleep [<5.5 h] or lengthened sleep [≥9 h]) as well as misalignment of sleep with the daily photoperiod (as with shift work during the daily scotophase) augment insulin resistance and IRS (postprandial hyperglycemia, increased sympathetic tone, inflammation, and reactive oxygen species generation) and are associated with increased risk for cardiovascular disease (CVD) [Citation105–Citation108]. Moreover, stress identified by hypercortisolemia is also coupled with such increased risk for metabolic and CVD [Citation109–Citation111]. In this regard, it is worth noting that sleep/wake disruptions and stress each are known to affect (reduce) CNS dopamine function and CNS dopamine function is a regulator of sleep and mood [Citation112–Citation115]. A working hypothesis for sleep/wake and stress-induced cardiometabolic disease is that such chronic disturbances reduce the circadian peak of dopaminergic activity at the biological clock and/or alter other circadian input signals to the clock to manifest a clock output that in part constitutes the treacherous triad and potentiates IRS. In essence, the clock is responding to a ‘stress’ (a very abnormal persistent environmental condition) by organizing the body in a default survival mode (the insulin-resistant state). This organization maintained chronically however becomes maladaptive leading to cardiometabolic disease. Our laboratory is currently investigating the circadian neurophysiological dimensions of such environment–clock interactions in the regulation of metabolism.
Figure 5. Schematic of dopamine—clock interactions in the regulation of peripheral fuel metabolism.
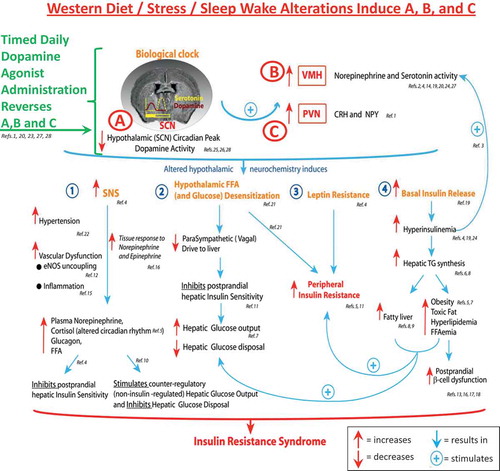
In conclusion, the acrophase and amplitude of circadian-timed dopaminergic input activity to the SCN (e.g. from the SuMN and other CNS areas) are critical in the maintenance of insulin sensitivity via its modulation of SCN communication with other hypothalamic centers that regulate neuroendocrine functions and autonomic balance at the liver, adipose, and beta cell and peripheral leptin sensitivity, and tissue glucose/lipid utilization to thereby regulate insulin sensitivity/action at the organismal level. A diminution of the natural circadian peak of dopaminergic input to the SCN, as occurs naturally in seasonal insulin-resistant animals, precipitates insulin resistance and glucose intolerance, and this diminution of dopaminergic activity can be manifested/accelerated in response to a high-saturated fat diet. That is, the annual cycle of metabolism, although regulated by circadian rhythms and endogenous in nature, can be influenced by the quality of foods consumed at different seasons. Food quality (molecular constituents of food) for foraging animals changes seasonally with the life cycles of flora and fauna prey. Among vertebrate species in the wild, this annual cycle of food quality interacts with and upon the above-described circadian neural oscillators to modulate the CNS regulation of the annual cycle of metabolism [Citation51,Citation52]. The expression of the insulin-resistant state evolved under such circumstances and is regulated by this interaction of food quality with an endogenous circadian neuroendocrine control system for metabolism. A high-saturated fat diet functions to induce insulin resistance in part by reducing the circadian peak in dopaminergic activity at the SCN area. The degree of responsiveness to the ability of the high-saturated fat diet to reduce SCN circadian dopaminergic peak activity contributes significantly to the degree of the insulin resistance-inducing effect of the high-saturated fat diet. Persistence of the high-saturated fat diet (or westernized diet, lifestyle) over a longer period of time (multiple seasons) may lead to an annual seasonal insulin-resistant state much like locking the individual in the ‘winter’ metabolic condition all year long as a result of feedback mechanisms from the periphery (e.g. hyperinsulinemia, hyperleptinemia) upon this clock regulatory system ([Citation52], see ).
Figure 6. Postulate for clock involvement in Insulin Resistance Syndrome (IRS): modern insulin resistant man “locked” into seasonal insulin resistance syndrome physiology (neuroendocrinology) (winter condition all year long).
Non-SCN CNS dopaminergic activities regulating peripheral metabolism
Besides the major impact of circadian dopaminergic activity at the SCN in the regulation of metabolism reviewed above, several studies have identified other CNS sites wherein decreased dopaminergic activity is coupled to the insulin-resistant state. High-fat feeding of rodents leads to decreased dopaminergic function in the mesolimbic system, a main center for reward enforcement and feeding and glucose tolerance regulation [Citation116–Citation118]. Dopamine transmission at this center provides the molecular reward cue that both initiates and terminates feeding. It has been postulated that decreased dopaminergic signaling within the mesolimbic system allows for overfeeding and glucose intolerance in the absence of a sufficient reward signal [Citation116–Citation121]. Moreover, leptin activation of dopaminergic neurons within the mesolimbic system is believed to account in part for its actions to maintain body weight and normal insulin sensitivity, and this leptin effect on dopamine function is diminished in obesity [Citation41,Citation47,Citation122]. Although not well recognized, dopamine acts within the arcuate hypothalamus to stimulate proopiomelanocortin (POMC) – α-MSH synthesis, a major contributor to feeding inhibition and insulin sensitivity (reviewed in [52]). Dopamine also functions to inhibit feeding in the perifornical area of the hypothalamus and to inhibit sympathetic nervous system (SNS) activation via its actions at the PVN [Citation52,Citation123]. Moreover, positron emission topography scan studies with radiolabeled dopaminergic receptor ligands in humans have identified a decreased dopaminergic function in obese and insulin-resistant individuals [Citation49,Citation50]. However, our recent studies suggest that these non-SCN CNS functions of dopamine that collectively tend to maintain the lean/insulin-sensitive state are actually regulated in part by circadian SNC output signals that, as described herein, are modulated by circadian dopaminergic input signals to the SCN.
To summarize, animals resistant to the fattening/insulin resistance-inducing effects of a high-saturated fat diet can be made sensitive to this dietary effect by reducing the circadian peak in dopaminergic input signaling to the SCN and animals sensitive to the fattening/insulin resistance-inducing effects of a high-saturated fat diet can be made resistant to such dietary effects merely by providing dopamine to reinstate the circadian peak in dopaminergic activity at the SCN area (even while maintained on a high-saturated fat diet). Other non-SCN CNS dopaminergic activities also align increased dopaminergic function with increased insulin sensitivity, and these activities are also likely regulated by the SCN. Such studies suggest that treatment of insulin-resistant states with a systemic dopamine agonist administered at the appropriate time of day to reinstate the normal circadian peak of dopaminergic activity at the clock area may be an effective means to improve insulin resistance and dysglycemia.
Preclinical studies evaluating the use of a dopamine agonist to treat IRS
Several preclinical studies in various animal models of IRS including seasonal insulin-resistant hamsters, genetically leptin-deficient ob/ob mice, and spontaneously hypertensive rats (SHRs) have demonstrated the ability of circadian-timed bromocriptine (a potent dopamine D2 receptor agonist) administration timed so as to reinstate the normal circadian CNS/SCN peak in dopaminergic activity to normalize both the elevated VMH NE/5HT activities and PVN NPY/CRH levels described above that markedly potentiate the loss of appropriate hypothalamic fuel sensing and the IRS [Citation86,Citation104,Citation124]. Moreover, numerous studies have consistently demonstrated the ability of such circadian-timed daily administration of bromocriptine (systemic or intracerebroventricular) to markedly reduce insulin resistance (particularly during the postprandial state [Citation125,Citation126], in agreement with its ability to improve VMH hypothalamic fuel-sensing mechanisms as described above), hyperinsulinemia and/or glucose intolerance without raising the plasma insulin level, in a variety of animal models of IRS including seasonal insulin-resistant hamsters, SHRs, high-fat fed rats, genetically leptin-deficient ob/ob mice, fattened pigs, and high-fat fed dogs [Citation86,Citation124,Citation126–Citation132]. As a composite, these animal studies provide evidence that timed bromocriptine treatment improves dysglycemia by improving (postprandial) insulin action in the liver and/or peripheral insulin-sensitive tissues (e.g. muscle). As such, animal studies were undertaken to investigate the interactive effects of the insulin-sensitizing impact of chronic timed bromocriptine treatment with the acute insulin secretory effects of glucagon like peptide-1 (GLP-1) analog (exendin-4) or exogenous insulin administration just prior to the administration of a glucose tolerance test (GTT) [Citation133]. It was found that chronic timed bromocriptine treatment plus either acute GLP-1 analog or insulin administered just prior to the GTT produced synergistic effects on improving glucose intolerance (i.e. the improvement in glucose intolerance was greater than the additive effects of the individual bromocriptine plus GLP-1/insulin administrations) in an animal model of T2DM. These findings suggest that combining bromocriptine-QR therapy with a postprandial insulin secretion enhancer or insulin itself may provide a physiological-based, complementary mechanism approach to uniquely improve glycemic control in T2DM patients.
Such timed bromocriptine treatment also markedly reduced basal and insulin-stimulated hepatic lipogenesis, hepatic triglyceride (TG) secretion, and plasma TG level during the circadian peak of hepatic lipogenic responsiveness to insulin in insulin-resistant hamsters [Citation134,Citation135]. Bromocriptine therapy also reduced total body fat stores and hepatic lipid content in animal models of obesity and fatty liver disease (high-saturated fat diet fed rats, SHRs) [Citation86,Citation131]. Bromocriptine-induced reductions in hepatic lipid levels and insulin resistance were coupled to reductions in hyperleptinemia and liver protein content of transcription factors and enzymes for lipogenesis and gluconeogenesis, and transcription factors for activation of parallel pathways of hepatic inflammation (nuclear factor-kappa B (NFkB), suppressor of cytokine signaling 3 (SOCS3), and Jun N-terminal kinase (JNK)) [Citation86]. Moreover, consistent with its effects to reduce sympathetic tone via the above hypothalamic mechanisms, such bromocriptine therapy also reduced hypertension and whole-body adipose lipolysis and plasma FFA levels in insulin-resistant animals [Citation86,Citation128,Citation136]. Bromocriptine-induced decreases in body fat stores were not associated with any change in food consumption but were coupled to substantial increases in whole-body protein turnover rate and whole-body protein content [Citation128,Citation137]. In total, these studies indicate that such bromocriptine treatment of obese, insulin-resistant animals results in a shift of metabolism from a lipid accretion, glucose producing, insulin-resistant state to a protein accretion, insulin-sensitive condition.
Respecting the cardiovascular system, in line with the bromocriptine effect to reduce insulin resistance, leptin resistance, hyperFFAemia, elevated sympathetic tone, and inflammation, bromocriptine therapy of SHRs has been observed to reduce markers of endothelial nitric oxide synthase uncoupling [Citation138], an event that is a major early contributor to CVD by way of its ability to reduce nitric oxide and generate reactive oxygen species at the vascular endothelium [Citation139–Citation141]. Bromocriptine therapy has also been observed to reduce arterial stiffness in SHRs [Citation138]. Bromocriptine treatment has also been demonstrated to reduce ischemic reperfusion injury in rat kidney and heart [Citation142–Citation144]. Most importantly, the effects of bromocriptine to improve IRS and cardiovascular pathology were time of day dependent on SHRs (Cincotta et al., unpublished data) with the maximal effect observed when administered at the peak of the circadian rhythm of SCN area dopaminergic activity and of little impact when administered several hours after this sensitive interval. Likewise, dopamine administration directly to the SCN area of high-fat fed, insulin-resistant, rats improved insulin sensitivity when administered at the time of day such endogenous dopamine activity peaks in insulin-sensitive animals but had no effect when administered several hours after this sensitive interval of the day ([Citation84], Cincotta et al. unpublished data). Timed bromocriptine treatment of insulin-resistant animals was demonstrated to reset toward normal the circadian rhythm of cortisol secretion as well as the circadian expression of the circadian clock gene, per1 in the SCN of animals held on a photoperiod thus demonstrating its ability to reset circadian clock systems in the animal [Citation52,Citation93]. These resetting effects of timed bromocriptine treatment on the clock system are consistent with a mechanism of circadian-dependent timed dopaminergic activity at the clock to reset clock regulation of metabolism. Such neuroendocrine and metabolic studies of circadian CNS dopaminergic regulation of metabolism suggest that similar improvements in metabolism via timed bromocriptine therapy may be manifested in man.
Clinical effects of bromocriptine-QR therapy in the treatment of metabolic disease and T2DM
Bromocriptine-QR is a unique, quick release, high absorbing formulation of bromocriptine mesylate, a potent dopamine D2 receptor agonist. Bromocriptine-QR therapy for the treatment of T2DM is a circadian-timed therapy that is administered in the morning within 2 h of waking to provide a discrete and brief daily interval of circulating bromocriptine [Citation145], thereby providing a timed pulse of increased dopaminergic activity centrally at this early morning time of day that studies suggest is the natural daily peak of central dopaminergic activity in healthy individuals [Citation115] and diminished in insulin-resistant states [Citation52].
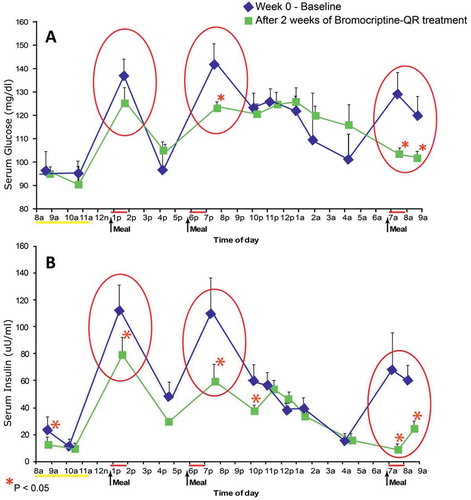
In normal healthy humans, plasma prolactin concentrations peak at night during sleep and are relatively low during the wakeful daylight period of the day (reviewed in [Citation50]), and animal studies indicate this inhibition is due in large part to inhibition from hypothalamic dopaminergic tone [Citation146]. Generally, a large fraction of obese, insulin-resistant humans have diurnal (day-time) plasma prolactin levels that are elevated relative to lean, insulin-sensitive subjects [Citation147–Citation151]. These elevated plasma prolactin levels are generally 2–4-fold greater than normal and do not constitute frank clinical hyperprolactinemia and would not generally even be assessed as elevated by clinical laboratory standards. However, this relatively elevated plasma prolactin level has been taken as an index of reduced dopaminergic tone at this time of day and several studies have associated high plasma prolactin levels with obesity and insulin resistance in man [Citation150–Citation159]. Therefore, available evidence in humans supports the tenet that a decreased circadian peak in central (hypothalamic) dopaminergic activity is coupled to the insulin-resistant state, much the same as is observed in other vertebrate animals. Consequently, clinical studies were undertaken to investigate the influence of morning (within 2 h of waking) bromocriptine-QR dosing upon glycemic control and insulin sensitivity. Initial Phase 2 studies evaluated the effect of bromocriptine-QR on postprandial glycemic control in obese, hyperinsulinemic nondiabetic subjects [Citation160]. Study subjects with fasting serum insulin >20 μU/ml (N = 12) were admitted into a clinical research center (CRC) for a 24-h period and fed standardized meals at 0700, 1200, and 1800 hours. Fasting and postprandial blood samples were drawn throughout the day for analyses of serum insulin and glucose levels. Subjects were then discharged on bromocriptine-QR (1.6 mg/day, administered at 0800) for 2 weeks and instructed to consume an ad libitum diet. After 2 weeks, subjects were readmitted to the CRC for blood sampling under the same initial dietary conditions. Bromocriptine-QR therapy reduced mean postprandial glucose levels from 135 ± 5 mg/dl to 117 ± 3 mg/dl (P < 0.05) and fasting and postprandial plasma insulin levels from 27 ± 6 μU/ml and 99 ± 13 μU/ml to 15 ± 2 μU/ml and 50 ± 7 μU/ml, respectively (P < 0.05) ().
Figure 7. Effect of bromocriptine-QR on postprandial serum glucose and insulin levels of obese hyperinsulinemic subjects after 2 weeks of treatment.
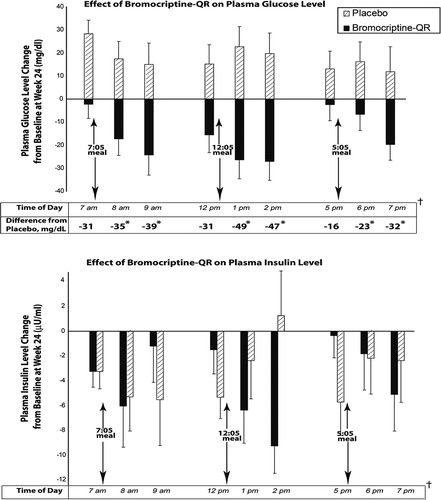
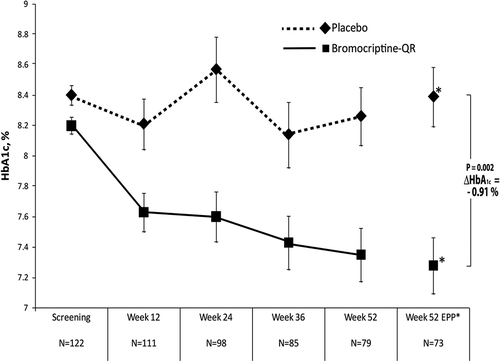
The above study of bromocriptine-QR impact on mealtime glucose and insulin was reconducted in T2DM subjects naïve to any anti-diabetes drug treatment [Citation161,Citation162]. Average study population baseline demographics included a percent-glycated HbA1c of 8.9, BMI of 31.5, duration of diabetes of 3.9 years (however, the study was conducted at a time when a fasting glucose level of 140 mg/dl was used as a diagnosis of T2DM so dysglycemia likely was present for a longer period of time), and age of 55 years. Such T2DM subjects were randomized to treatment with bromocriptine-QR (l.6–4.8 mg/day; titrated up at a rate of 0.8 mg/week until a maximum tolerated dose of 1.6–4.8 mg/day was achieved; N = 80) or placebo (N = 79) therapy (administered within 2 h of waking in the morning) for 24 weeks. Study subjects were administered standardized meals at breakfast, lunch, and dinner for the analyses of premeal and 1- and 2-h postmeal plasma insulin and glucose levels at baseline and again at week 24 for comparison. Relative to placebo, bromocriptine-QR therapy resulted in a significant decrease from baseline in postprandial glucose after each meal of the day without altering plasma insulin levels (37 mg/dl average decline of postprandial glucose of all three meals, P < 0.002; ). Given that the drug is substantially cleared from the circulation by lunch time, yet postprandial effects are observed at lunch and dinner, these results (and those described above in insulin-resistant, nondiabetes subjects) support preclinical data concluding a ‘resetting’ of hypothalamic nutrient-sensing responsiveness to a meal to improve postprandial insulin sensitivity with this therapy [Citation125].
Figure 8. Effect of once daily-morning administration of bromocriptine-QR vs. Placebo on pre- and post meal plasma glucose and insulin levels in drug naïve T2DM subjects following 24 Weeks of therapy.
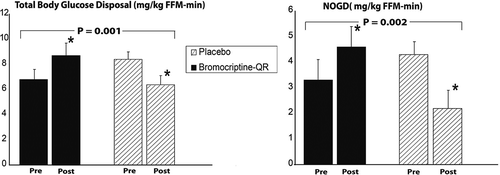
Based upon these findings, a subsequent study investigated the impact of once-daily morning administration of bromocriptine-QR (1.6–4.8 mg/day; titrated up at a rate of 0.8 mg/week until a maximum tolerated dose of 1.6–4.8 mg/day was achieved) versus placebo on insulin-stimulated glucose disposal in T2DM subjects on diet with or without SU therapy [Citation163]. Average study population baseline demographics included a percent-glycated HbA1c of 8.6, BMI of 34.7, duration of diabetes of 3.4 years (the study was conducted at a time when a fasting glucose level of 140 mg/dl was used as a diagnosis of T2DM so dysglycemia likely was present for a longer period of time), and age of 53 years. The study demonstrated a notable effect of such bromocriptine-QR therapy on postprandial (oral glucose tolerance test [OGTT]) glucose, insulin sensitivity, and glycemic control (as measured by fasting and HbAlc change from baseline) versus placebo over a 16-week-treatment period. The between-group difference in change from baseline area under the glucose curve from an OGTT was −46 mg/dl (P = 0.02), in fasting glucose was −54 mg/dl (P < 0.01) and HbAlc was −1.2% (P = 0.0l). The between-group difference in change from baseline total and nonoxidative glucose disposal rates were improved by 51% and 84%, respectively, during the second step of a hyperinsulinemic–euglycemic clamp among bromocriptine-QR versus placebo treated subjects (P < 0.002) ( and ) and a subsequent analysis of the metabolic clearance rate (MCR) (an index of insulin action adjusted for plasma glucose level) from this study demonstrated a similar magnitude of effect on between group-difference in MCR during the first and second steps of the clamp (44% and 56% improvements in MCR, P ≤ 0.01) (A.H. Cincotta, unpublished data).
Figure 9. Effect of 16 weeks treatment with morning Bromocriptine-QR or placebo therapy upon HbA1c, and fasting and mean OGTT plasma glucose in T2DM subjects on diet with or without sulfonylurea therapy.
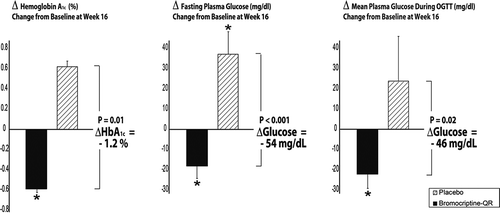
Figure 10. Effect of 16 weeks treatment with morning Bromocriptine-QR or placebo therapy upon insulin-stimulated glucose disposal in T2DM subjects on diet with or without sulfonylurea therapy.
Subsequent clinical studies investigated the possible beneficial effects of bromocriptine-QR on glycemic control in obese T2DM subjects whose dysglycemia was poorly controlled on SU and/or metformin therapies. One such study investigated the impact of morning administration (8 a.m.) of bromocriptine-QR (1.6–4.8 mg/day; titrated up at a rate of 0.8 mg/week until a maximum tolerated dose of 1.6–4.8 mg/day was achieved) or placebo upon glycemic control in obese T2DM subjects very poorly controlled on baseline SU therapy (485 subjects) while held on a weight maintaining diet for 24 weeks (the SU add-on study) [Citation161]. Average population baseline demographics included a percent-glycated HbA1c level of 9.4, BMI of 32.1, duration of disease of 6.3 years (the study was conducted at a time when a fasting glucose level of 140 mg/dl was used as a diagnosis of T2DM so dysglycemia likely was present for a longer period of time) and age of 55 years. Bromocriptine-QR therapy over the treatment period reduced percent-glycated HbA1c by 0.56 (P < 0.0001). However, further analyses of this SU add-on study data revealed salient features of this therapy that allow for the expression of a greater impact on glycemic control in specific T2DM subjects as follows. First, as reviewed above, available evidence suggests that timed bromocriptine-QR therapy improves glycemic control by improving insulin sensitivity, particularly during the postprandial state. As such, effectiveness of the agent to reduce postprandial glucose levels should improve with greater circulating insulin levels during the postprandial state. However, the majority of these study subjects had a poor postprandial insulin secretory response to a standardized meal (<50 μU/ml plasma insulin level at 1-h-post meal). An analysis of the effect of bromocriptine-QR on glycemic control as a function of the postprandial insulin level was therefore conducted in this SU add-on study group. The between-group difference in impact of bromocriptine-QR versus placebo on decrease in percent-glycated HbA1c was the greatest in study subjects whose postprandial insulin levels were at least moderate (≥50 μU/ml) (HbA1c Δ = −0.79, P < 0.05) and not present in those few subjects without any clinically relevant postprandial insulin release (insulin levels <30 μU/ml) (HbA1c Δ = −0.11, not significant) (A.H. Cincotta, unpublished data). Second, a major mechanism of improving glycemic control with bromocriptine-QR therapy appears to be an increased hypothalamic dopaminergic tone at the clock, a reduction of elevated sympathetic tone, and an improved circadian hypothalamic–pituitary–adrenal (HPA) axis function as described above (amelioration of the treacherous triad). Mounting converging evidence from multiple preclinical and clinical studies indicates important contributory roles for increased SNS tone and a disrupted HPA axis (inducing hypercortisolemia) in the genesis and maintenance of IRS [Citation51,Citation52,Citation95–Citation97,Citation109,Citation164,Citation165]. These SNS and HPA aberrations potentiate increased peripheral and hepatic insulin resistance, adipose lipolytic activity, dyslipidemia, hypertension, and low grade systemic inflammation and oxidative stress that all contribute to dysglycemia and increased cardiovascular risk. Also, this neuroendocrine phenotype is in fact often associated with IRS and increased cardiovascular risk in humans [Citation95–Citation97,Citation109,Citation164,Citation165]. Timed daily administration of bromocriptine to obese insulin-resistant animals reduces hypothalamic drive for increased SNS tone and HPA axis activity while alleviating insulin resistance, glucose intolerance, dyslipidemia, hypertension, and obesity [Citation84,Citation122,Citation126,Citation128] and available evidence suggests that a large component to this effect is via resetting toward ‘normal’ central circadian systems that control autonomic balance (to reduce elevated sympathetic tone) and the neuroendocrine axis (to improve circadian HPA axis function) [Citation52,Citation86,Citation166]. It follows therefore that T2DM subjects with elevated SNS and HPA axis activity may be particularly suitable for and responsive to treatment with bromocriptine to improve their glycemic control. Inasmuch as it is not currently practical to assess patients’ SNS tone or HPA axis activity in a clinical setting, a simple plausible surrogate for such aberrant neuroendocrine activity may simply be elevation of both the plasma TG and blood pressure (BP) levels [Citation167,Citation168]. As defined by NCEP ATP III criteria, elevated levels of plasma TG for inclusion in metabolic syndrome (IRS) are ≥150 mg/dl and elevated BP levels for such inclusion are systolic ≥130 and diastolic ≥85, based upon epidemiological data [Citation169]. Therefore, we examined the effects of bromocriptine-QR on glycemic control in the SU add-on study population, stratifying the results by baseline plasma TG and BP levels [Citation170]. Study subjects with plasma TG and BP measures that met the NCP APT III criteria for metabolic syndrome (TG ≥ 150 mg/dl and BP ≥ 130/85), taken as a surrogate measure of elevated sympathetic tone [Citation167,Citation168], were analyzed for impact of bromocriptine-QR upon glycemic control. It was observed that in those subjects whose TG and BP measures met the metabolic syndrome definition criteria, bromocriptine-QR reduced percent-glycated HbA1c by −0.76 versus placebo (N = 75) (P < 0.02) and as the plasma TG level increased to ≥200 mg/dl, this effect increased to a delta of −1.22 versus placebo (N = 52) (P < 0.005). These preliminary findings suggest that bromocriptine-QR therapy may be particularly effective in improving glycemic control in T2DM subjects with elevated TG and BP levels.
Effect of bromocriptine-QR on dyslipidemia in T2DM subjects
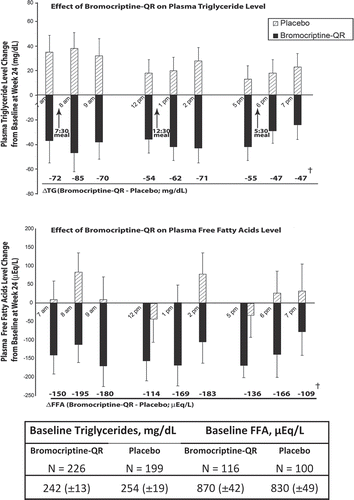
Preclinical studies have repeatedly demonstrated the ability of timed bromocriptine therapy to reduce hepatic lipogenesis and plasma TG and FFA levels in obese, insulin-resistant states as described above [Citation86,Citation127,Citation128,Citation131,Citation134,Citation135,Citation171]. These studies indicate that such bromocriptine administration reduces plasma TG and FFA levels by reducing TG synthesis and secretion in liver and TG mobilization from adipose tissue. Therefore, the SU add-on study also investigated the impact of bromocriptine-QR therapy upon diurnal plasma TG and FFA levels [Citation161,Citation162]. Diurnal TG levels were assessed from 0700 to 1900 hours including 1 h before and 1 and 2 h after each of breakfast, lunch, and dinner standardized meals, before and 24 weeks after treatment. Study subjects had elevated levels of plasma TG (248 mg/dl) and FFA (850 μEq/l) at baseline. Bromocriptine-QR significantly reduced plasma TGs by 29% and free fatty acid levels by 19% throughout the diurnal portion of the day relative to placebo treatment (P < 0.0001 and P < 0.02, respectively) (). Such findings are consistent with those observed in hyperlipidemic animal models, where timed bromocriptine therapy has been demonstrated to reduce hepatic lipogenesis and adipose lipolysis thus reducing circulating TG and FFA levels, respectively [Citation86,Citation127,Citation128,Citation131,Citation134,Citation135,Citation171].
Figure 11. Effect of 24 weeks treatment with bromocriptine-QR or placebo on diurnal plasma TG and FFA levels in T2DM subjects on baseline sulfonylurea therapy.
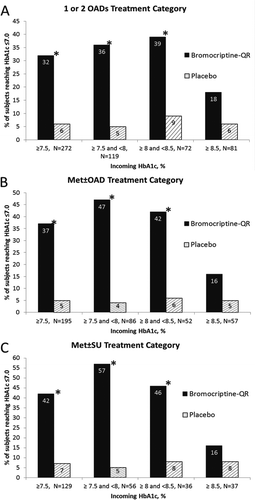
Based upon the findings described above of bromocriptine-QR therapy-induced improvement in glycemic control in T2DM subjects whose dysglycemia was very poorly controlled on SU therapy, additional studies were undertaken to investigate the effect of timed bromocriptine-QR therapy on glycemic control in a broad population of T2DM subjects whose hyperglycemia was poorly controlled with one or two commonly used oral anti-diabetes drugs (OADs), including metformin with or without another OAD (the metformin add-on study) [Citation172]. Five-hundred fifteen T2DM subjects representing a cohort from the Cycloset Safety Trial (CST) [Citation173] (ages 18–80 and average BMI of 32.7) with baseline HbA1c ≥ 7.5 and on one or two OADs (metformin, SU, and/or TZDs) were randomized 2:1 to bromocriptine-QR (1.6–4.8 mg/day; titrated up at a rate of 0.8 mg/week until a maximum tolerated dose of 1.6–4.8 mg/day was achieved) or placebo for a 24 week-treatment period. Study investigators were allowed to adjust, if necessary, subject concomitant anti-diabetes medications during the study to attempt to achieve glycemic control in the case of glycemic deterioration. The impact of bromocriptine-QR treatment intervention on glycemic control was assessed in subjects on any one or two OADs (ALL treatment category) (N = 515), or on metformin with or without another OAD (Met ± OAD treatment category) (N = 356), or on metformin plus a SU (Met ± SU treatment category) (N = 245) (1) by examining the between-group difference in change from baseline (a) concomitant OAD medication changes during the study and (b) HbA1c and (2) by determining the odds of reaching HbA1c of ≤7.0% on bromocriptine-QR versus placebo in the evaluable per protocol (EPP) and intention to treat (ITT) populations. The prespecified EPP population was defined as those subjects who were at least 80% compliant with prescribed dosing of study drug and completed treatment through the 24-week study without any major protocol violations during the trial. Respecting the EPP population, significantly more people (approximately 1.5–2-fold more; P < 0.05) intensified concomitant anti-diabetes medication therapy during the study in the placebo versus the bromocriptine-QR arm () after 24 weeks on therapy. In subjects that did not change the intensity of the baseline diabetes therapy (72% of the study population), and that were on ALL, or on metformin with or without another OAD (Met ± OAD), or on metformin plus SU (Met ± SU), the HbA1c change for bromocriptine-QR versus placebo was −0.69 (P < 0.0001), −0.81 (P < 0001), and −0.83 (P < 0.0001), respectively, after 24 weeks on therapy (). The odds ratio of reaching HbA1c of ≤7.0% on bromocriptine-QR versus placebo therapy for the ALL, Met ± OAD, and Met ± SU groups was 6.50, 12.03, and 11.45 for these three groups, respectively (P < 0.0002). For the ALL, Met ± OAD, and Met ± SU groups, the percent of bromocriptine-QR versus placebo subjects reaching an HbA1c goal of ≤7.0% was 32%, 37%, and 42% versus 6%, 5%, and 7%, respectively (P < 0.0001), representing a 5–7-fold increase in the percent of subjects on bromocriptine-QR versus placebo reaching this goal across all treatment categories. On closer inspection, for the baseline metformin-treated subjects (Met ± OAD and Met ± SU) with baseline HbA1c of between 7.5 and 8.5, it was observed that 43% and 52% of those randomized to bromocriptine-QR achieved a goal of ≤7.0 versus 5% and 6% for placebo, respectively (P < 0.0001) () [Citation172].
Table 1. Concomitant diabetes medication regimen change for the EPP population during the study among ALL, Met/OAD, and Met/SU treatment categories.
Figure 12. Effect of 24 weeks therapy with bromocriptine-QR versus placebo on change from baseline HbA1c (%) among T2DM subjects whose glycemia was poorly controlled (HbA1c ≥ 7.5) on any 1 or 2 oral antidiabetic drugs (OADs—Metformin, sulfonylurea, TZD), Met±OAD, and Met±SU treatment regimens at baseline and whose OAD medications intensification did not change during this period.
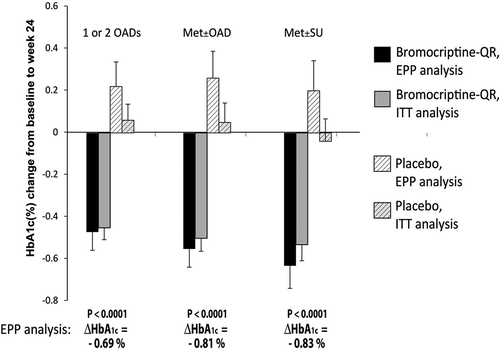
Figure 13. Effect of 24 weeks therapy with bromocriptine-QR on glycemic control (% to goal of ≤ 7.0) in T2DM subjects whose glycemia was poorly controlled on any 1 or 2 OADs, Met±OAD, and Met±SU Treatment Regimens at Baseline.
As described above, preclinical and clinical studies indicate that bromocriptine-QR therapy functions as a centrally acting insulin sensitizer, particularly during the postprandial state to improve dysglycemia. Therefore, the possibility that bromocriptine-QR may have additive/synergistic effects when added to beta cell preserving agents/peripherally acting insulin sensitizers such as TZDs was explored in a specific cohort of subjects from the CST [Citation174]. A subgroup from the CST who were taking a TZD (rosiglitazone or pioglitazone-normalized average dose = 15 mg/day pioglitazone) at baseline with or without another OAD (90% with metformin or SU) and having an HbA1c level of ≥7.5 (N = 122) were randomized 2:1 to receive additional once daily (morning) bromocriptine-QR (1.6–4.8 mg/day) or placebo for up to 52 weeks. Basic inclusion/exclusion criteria were as described above for the CST participants. Glycemic efficacy analyses of between-group difference in change from baseline HbA1c and fasting glucose were based on both the intent to treat-modified (ITTm) population (all randomized subjects with a least one post-baseline visit) and EPP population (all subjects completing 52 weeks of therapy and 80% compliant with study drug dosing) using general linear model analyses after adjusting for baseline covariates. The odds ratio of participants achieving HbA1c ≤ 7% were also calculated. Bromocriptine-QR treatment led to a significant reduction in HbA1c (ITTm −0.81%, P = 0.001 and EPP −0.91%, P = 0.002) (), fasting plasma glucose (ITTm −21.5 mg/dl, P = 0.03 and EPP −20.5 mg/dl, P = 0.05), and higher frequency achieving an HbA1c ≤ 7% (32.1% vs. 15.9%, P = 0.05) when compared with placebo. Three times as many subjects reached a goal HbA1c of ≤7.0, after accounting for various baseline covariates and adjusting for the greater tendency of the placebo arm to intensify their diabetes regimen. Treatment with bromocriptine-QR had no adverse impact on weight or risk of hypoglycemia.
Figure 14. Effect of 52 week therapy with bromocriptine-QR upon HbA1c level in T2DM subjects whose dysglycemia was poorly controlled (baseline HbA1c ≥ 7.5) on TZD therapy.
There was no statistically significant increase in weight among participants who took a TZD and bromocriptine-QR and this combination was well tolerated. TZDs reduce insulin resistance by activating the nuclear receptor PPAR gamma to improve insulin-signal transduction [Citation175]; however, such agonist use also improves pancreatic beta cell function in T2DM subjects [Citation176,Citation177]. Bromocriptine-QR has been shown to reduce postprandial hyperglycemia without raising insulin levels in T2DM subjects and to enhance tissue sensitivity to insulin [Citation161–Citation163]. The current findings suggest that the combination of a TZD (with or without another OAA) plus bromocriptine-QR may provide a therapeutic strategy that confers complementary mechanisms of action to establish long-lasting improvements in glycemic control in patients with T2DM.
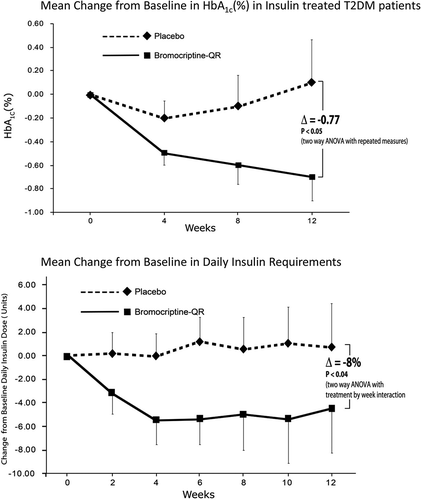
Glycemic control in T2DM patients on moderate-to-high-dose insulin therapy is difficult to manage. The concurrent use of a postprandial insulin-sensitizing agent, such as bromocriptine-QR, a quick release formulation of bromocriptine, a dopamine D2 receptor agonist, may offer a strategy to improve glycemic control and limit/reduce insulin requirement in such patients. Based upon the findings of the above-described preclinical and clinical investigations of bromocriptine-QR, further studies were undertaken to investigate the impact of this therapy upon glycemic control in T2DM subjects in poor glycemic control on insulin therapy. Two such pilot studies were conducted, one in subjects with moderate daily insulin requirement and one in subjects with high daily insulin requirement. In the first study, T2DM subjects receiving a minimum daily insulin dose of 25 U/day (subject population average 67 U/day) with baseline HbA1c average of 9.35% and average duration of disease of 13 years were randomized to bromocriptine-QR (1.6–4.8 mg/day; titrated up at a rate of 0.8 mg/week over a 6 weeks period to maximum tolerated dose) (N = 21) or placebo (N = 11) for a 12 week-treatment period [Citation178]. Following 12 weeks on therapy, bromocriptine-QR treatment reduced percent-glycated HbA1c level by 0.77 (P < 0.05) and the daily insulin requirement by 8% (P < 0.05) relative to placebo (). There were no differences in reported adverse events between the treatment groups.
Figure 15. Effect of bromocriptine-QR (N = 21) or placebo (N = 11) therapy on HbA1c and Daily Insulin Requirement in Type 2 Diabetes Mellitus subjects whose dysglycemia was poorly controlled (average baseline HbA1c = 9.2% for placebo and 9.5% for Bromocriptine-QR) on insulin therapy (average baseline insulin dose = 67 units/day).
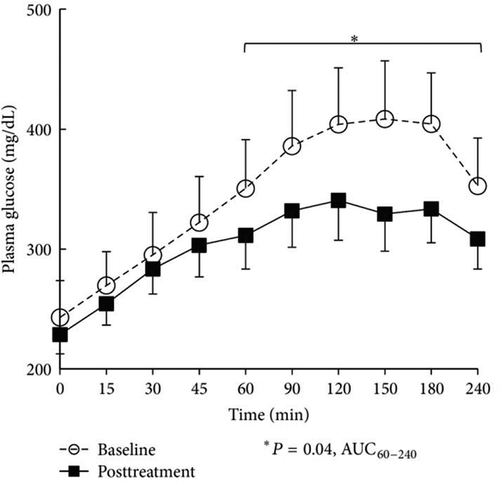
In the second open-label, single arm pilot study, 10 T2DM subjects on metformin (1–2 g/day) plus high dose basal-bolus insulin (minimum 65 U/day, subject population average daily dose = 190 U/day) were assigned treatment to bromocriptine-QR (1.6–4.8 mg/day, titrated up at a rate of 0.8 mg/week for a 6 weeks period to the maximum tolerated dose) for a 24 weeks period [Citation179]. Changes from baseline in HbA1c, total daily insulin requirement, and area under the glucose curve from a 4 h mixed meal tolerance test (MMTT) (in the absence of any anti-diabetes therapy) were monitored over the study period while glycemic control was treated with standard clinical practice (adjustment of insulin dose for hypo- or hyperglycemia). Compared to the baseline, average HbA1c decreased 1.76% (9.74 ± 0.56–7.98 ± 0.36, P = 0.01), average total daily insulin requirement decreased 27% (from 199 ± 33 to 147 ± 31, P = 0.009) and MMTT area under the curve (AUC)60–240 decreased 32% (P = 0.04) over the 24-week bromocriptine-QR treatment period. The decline in HbA1c and total daily insulin requirement were observed at 8 weeks and sustained over the remaining 16-week study duration [Citation179] ( and ). The more robust anti-hyperglycemic responses to bromocriptine-QR in T2DM subjects on insulin therapy versus those on SU therapy (−0.7 to −1.76 HbA1c reductions versus −0.6 to −0.83 HbA1c reductions, respectively) may reflect the better ability of exogenous insulin versus SU to raise the plasma insulin level that in turn interacts (additively or synergistically) with the insulin-sensitizing effects of bromocriptine-QR [Citation163] to better improve glycemic control. Additional studies are planned by our group to further substantiate these findings.
Figure 16. Effect of bromocriptine-QR therapy on HbA1c (Panel a) and total daily insulin dose (TDID) (Panel b) over 24 weeks in subjects (N = 8) on metformin plus high-dose basal-bolus insulin therapy at baseline.
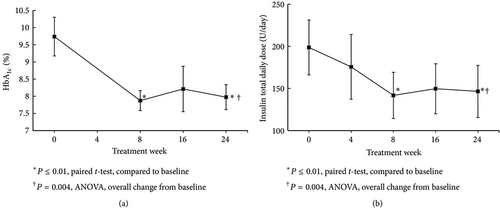
Figure 17. Effect of bromocriptine-QR therapy on mixed meal tolerance test postprandial glucose area under the curve (AUC) over 24 weeks in subjects (N = 7) on metformin plus high-dose basal-bolus insulin therapy at baseline.
Cardiovascular effects of bromocriptine-QR therapy
CVD is the leading cause of death among patients with T2DM, accounting for at least 65% of deaths among these patients even with earlier detection and treatment of T2DM as has occurred during the past decade. This patient population has a 2–4-fold higher risk of CVD relative to their nondiabetic counterparts [Citation180]. Why this is the case is still poorly understood. Several major clinical studies have demonstrated that the ability of glucose lowering with a wide variety of anti-diabetes medications in T2DM subjects to reduce CVD events is modest at best [Citation181], even though cellular and molecular studies of glucose impact upon the vasculature clearly indicate that elevated glucose levels induce vasculature pathologies leading to CVD [Citation182]. It is becoming increasingly clear that progression of the multifactorial pathological components of the IRS that precede and continue in T2DM, including loss of frank glycemic control, predisposes to increased incidence of CVD that cannot then be easily, quickly, and largely reversed by merely reversing the hyperglycemia of T2DM. Among such multifactorial systemic pathologies are insulin resistance, hyperinsulinemia, elevated sympathetic tone, altered/increased renin-angiotensin activity, leptin resistance, and lipotoxicity. These pathologies promote increased vascular cell (endothelium and smooth muscle cell) reactive oxygen species generation, in part by inducing endothelium nitric oxide synthase uncoupling, a self-sustaining process that converts this enzyme from generating the vascular protective agent, nitric oxide to producing the potent vascular damaging agents of reactive oxygen and nitrogen species [Citation139–Citation141]. Such multiple systemic and vascular events lead to systemic and local immune inflammatory activation precipitating arteriosclerosis and vascular damage. The increase in SNS activity as part of the IRS also promotes hypertension and a hypercoagulative state in the circulation, thereby exacerbating the local vascular pathology to induce CVD [Citation183–Citation185]. Such factors help explain why reducing hyperglycemia in and of itself has failed to produce dramatic improvements in CVD outcomes of T2DM patients.
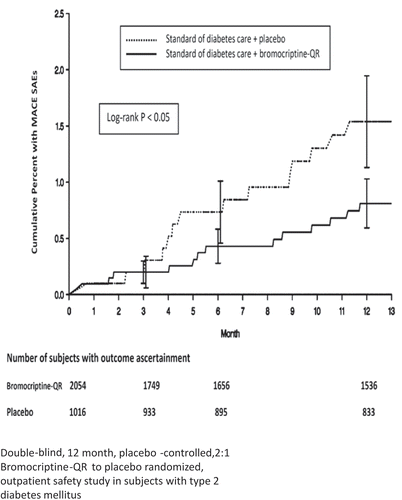
As described at the outset of this review article, clock mechanisms under dopaminergic control within the CNS regulate the onset (and reversal) of the IRS via the autonomic/neuroendocrine axes. Such wide-spread regulatory influences of central circadian dopaminergic activities on peripheral metabolism may be a part of a broader control system for overall physiology, including cardiovascular biology. Inasmuch as IRS, not merely hyperglycemia, appears to be a major contributor to cardiovascular pathology, it follows that an intervention that targets IRS via the ultimate regulator of physiology, the brain, may have the potential to improve cardiovascular pathology in IRS animals and human subjects. Such was the rationale for investigating the potential cardiovascular benefits of bromocriptine-QR therapy upon cardiovascular outcomes in T2DM subjects in the CST [Citation173]. The CST was designed to investigate the influence of bromocriptine-QR therapy upon overall and cardiovascular safety across a wide spectrum of T2DM subjects. The CST was a large (3070 subject), randomized, double blind, placebo controlled 1-year study of T2DM subjects across a wide range of glycemic control, and anti-diabetes therapy regimens [Citation186]. Inclusion criteria were the diagnosis of type 2 diabetes, age between 30 and 80 years, body mass index <43 kg/m2, stable anti-diabetes regimen consisting of either diet, oral hypoglycemic agents (no more than two), or insulin (alone or with no more than one oral hypoglycemic agent) for at least 30 days prior to randomization. Exclusion criteria were current chronic use of prescription sympathomimetic drugs, ergot alkaloid derivatives, clinically significant comorbid conditions such as uncontrolled hypertension, renal failure, or cancer. Subjects exhibited a wide range of glycemic control with percent-glycated HbA1c values ranging from 5.5 to 10.5 and held multiple risk factors for CVD, including history of CVD, being a prior or current smoker, as well as dyslipidemia and hypertension (but that were each well controlled by pharmacotherapy). All serious adverse events were adjudicated by an independent adverse event adjudication committee consisting of two cardiologists and an endocrinologist, blinded to treatment assignment, made the final classification for all cardiovascular adverse events as meeting or not the prespecified definition of the adverse cardiovascular study endpoint. Cycloset therapy resulted in a 40% HRR for the prespecified endpoint of myocardial infarction, stroke, or hospitalization for unstable angina, congestive heart failure, or revascularization surgery [Citation186] and a 52% HRR for the major adverse cardiovascular event (MACE) endpoint [Citation187] ( and ). Furthermore, this effect on the prespecified CVD endpoint was also manifested in a subset of this CST population with well-controlled glycemia (average HbA1c: 6.3; 48% HRR, P = 0.041) [Citation188]. These findings highlight a potent and unique potential of bromocriptine-QR therapy to reduce CVD event rate in T2DM subjects and suggest that treatment with bromocriptine-QR at any time in the course of the disease and preferably early may provide substantive CV benefit and further studies are warranted to confirm this postulate.
Figure 18. Effect of bromocriptine-QR on time to first prespecified composite CVD serious adverse event (myocardial infarction, stroke, hospitalization for congestive heart failure, unstable angina, or revascularization surgery) in T2DM subjects from the Cycloset Safety Trial on any 1 or 2 antidiabetes medications (including insulin) at baseline.
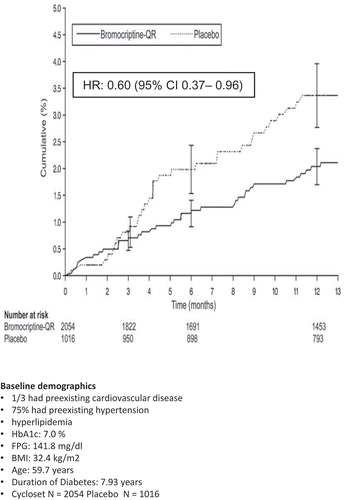
Figure 19. Effect of bromocriptine-QR on time to first major adverse cardiovascular event (MACE—myocardial infarction, stroke, and CV death) in T2DM subjects from the Cycloset Safety Trial on any 1 or 2 antidiabetes medications (including insulin) at baseline.
Preclinical studies suggest that mechanisms involved in this marked response to bromocriptine-QR are likely to include a reduction of elevated sympathetic drive to the peripheral adipose and liver that facilitates insulin resistance and inflammatory cytokine secretion, a reduction of elevated sympathetic drive to the vasculature itself that facilitates vasoconstriction, ROS generation, inflammation and eNOS uncoupling, and a reduction in the hypercoagulative state of the circulation. In hypertensive, insulin-resistant SHRs, timed bromocriptine therapy corrected the elevated levels of VMH NE and 5HT activities that predispose to IRS, attenuated the syndrome, corrected aortic endothelial dysfunction, and ameliorated arteriosclerosis [Citation86,Citation138]. Similar CVD pathologies to those of the SHR are operative in human CVD of T2DM and therefore are potentially similarly impacted by bromocriptine-QR to manifest its effects on CVD outcomes observed in these study subjects.
Expert commentary
Clinical use and treatment considerations summary
Bromocriptine-QR is approved by the US FDA as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. It may be used as monotherapy or in combination with other glucose-lowering agents. Bromocriptine-QR is administered once daily within 2 h after waking in the morning. The initial dose is one tablet (0.8 mg) daily, increased weekly by one tablet until a maximum daily dose of six tablets (4.8 mg) or until the maximal tolerated dose of between two and six tablets (1.6–4.8 mg) per day is achieved. Taking bromocriptine-QR with food is recommended to potentially reduce gastrointestinal side effects such as nausea.
After a 24-week-treatment period with bromocriptine-QR therapy of T2DM subjects whose dysglycemia was poorly controlled on metformin plus SU, among subjects within the prespecified EPP analysis, the therapy induced an HbA1c reduction of −0.83 versus placebo and 42% of the bromocriptine-QR study population reached an HbA1c goal of ≤7.0 versus only 7% for placebo from a baseline average of 8.2 [Citation172]. As add-on therapy to TZD plus OAD, among subjects within the prespecified EPP analysis, bromocriptine-QR therapy for 52 weeks resulted in a −0.91 reduction in HbA1c versus placebo in T2DM subjects with baseline HbA1c of 8.3 [Citation174]. In pilot studies, as add-on to insulin therapy, bromocriptine-QR treatment resulted in a −0.77 and −1.76 HbA1c reduction with concurrent insulin dose reductions of 8% and 25% in T2DM subjects with baseline HbA1c of 9.35 and 9.4, respectively [Citation178,Citation179]. Once-daily morning administration of bromocriptine-QR to T2DM subjects reduces postprandial hyperglycemia following each of the three meals of the day [Citation162]. The breadth of preclinical and clinical mechanistic studies conducted on bromocriptine-QR suggest that this therapy improves glycemic control in T2DM subjects by improving glucose disposal and insulin sensitivity, without increasing plasma insulin levels. These drug effects predominate during the postprandial state, theoretically owing in large part to the therapy’s influence (1) to improve attenuated brain (hypothalamic) fuel-sensing mechanisms that thus allows for improved postmeal hypothalamic-directed insulin action in muscle and liver, (2) to reduce elevated sympathetic tone and HPA axis activity that contribute to insulin resistance and glucose intolerance, and (3) to reduce leptin resistance that potentiates postprandial dysglycemia. Therefore, bromocriptine-QR may be best utilized either in early diagnosed T2DM subjects with good postprandial beta cell insulin secretory capacity or in conjunction with postprandial insulin secretion enhancing agents (e.g. DPP4 Is, GLP-1 analogs, meglitinides, prandial insulin) to achieve complementary (additive or synergistic) effects on glycemic control (vis-à-vis insulin secretory stimulus plus insulin tissue response interactions) [Citation133], though certain of these particular combination studies have yet to be conducted. In addition, results from pilot studies suggest that concurrent use of bromocriptine-QR in individuals with T2DM requiring moderate-to-high doses of insulin may be an effective strategy to improve glycemic control while limiting insulin dose in these individuals [Citation179]. Moreover, a substantive portion of the metabolic benefit from bromocriptine-QR therapy may be the result of its ability to reduce elevated sympathetic tone, a major potentiator of metabolic syndrome. Therefore, its utility may be particularly beneficial in treating hyperglycemia (and dyslipidemia) in metabolic syndrome T2DM patients identified using elevated plasma TG and BP levels as a surrogate marker for elevated sympathetic tone as preliminary studies suggest [Citation170].
Besides its role as a glucose-lowering agent as described above, other important considerations for use of bromocriptine-QR therapy in type 2 diabetes include the potential for CVD risk reduction associated with this therapy (40–50% HRR within 1 year). While the mechanisms by which timed bromocriptine-QR therapy produces the observed effects on CVD risk reduction are yet to be fully delineated, available evidence suggest important roles for its modulation of CNS and circadian hypothalamic functions to reduce elevated SNS activities that directly and indirectly potentiate vasoconstriction, vascular inflammation, and endothelial dysfunction, which in turn lead to increased CVD risk. Endothelial dysfunction, vascular inflammation, and elevated sympathetic tone are early pathological events in the progression of CVD that precede the onset of T2DM and are present at all stages of the disease process. Therefore, the possibility exists that bromocriptine-QR therapy may produce cardiovascular benefit across the continuum of T2DM disease course, from initial diagnosis to late stage disease and clinical studies to investigate this possibility are warranted. As an extension to this postulate, timed bromocriptine-QR therapy may have utility in treating CVD associated with other disorders such as obesity, hypertension, metabolic syndrome, prediabetes, chronic kidney disease, congestive heart failure, and/or diabetic cardiomyopathy. Though speculative at the present time, available evidence suggests that bromocriptine-QR may also have utility in treating prediabetes, elements of IRS, and end organ damage due to IRS or elevated sympathetic tone [Citation86,Citation131,Citation138,Citation142–Citation144,Citation170]. It is worth noting that the time of day that bromocriptine-QR is administered to restore the normal circadian peak in CNS dopaminergic activity (within 2 h of waking in the morning) to treat cardiometabolic disease coincides with the time of day of increased cardiovascular events and elevated sympathetic tone in metabolic syndrome (i.e. early morning hours of the day near awaking from sleep, nondipper pathophysiology) [Citation189–Citation194]. It may be interpreted that morning administration of bromocriptine-QR is attenuating the early morning abnormal elevation in sympathetic tone, insulin resistance, and inflammatory activity, and thus reducing increased morning risk of CV events due to its immediate inhibition of sympathetic tone and inflammation in the periphery. However, there are a few very important points to understand regarding the cardiometabolic benefits of this timed bromocriptine therapy as follows. First, it should be recalled that the metabolic effects of bromocriptine upon IRS can be produced with intracerebroventricular administration of 1/1000th the effective systemic dose and that systemic administration of bromocriptine corrects major hypothalamic aberrations that initiate and potentiate the IRS. These findings indicate a central target for such bromocriptine action. Second, it should be understood that while the effects of the morning administration of bromocriptine-QR upon dopaminergic receptor sites in the CNS (and periphery) can be rapid, it appears that it is the ‘resetting’ effect of this timed administration that ultimately lowers the elevated CVD risk factors at this critical time of day over the succeeding days [Citation86]. That is, it appears that it is the timed bromocriptine action to reprogram SCN output control of cardiometabolic functions that mostly impacts these circadian peripheral immune and autonomic activities (i.e. elevated morning inflammation, insulin resistance, and sympathetic tone) more so than does the immediate and direct effect of the bromocriptine itself on the CNS and peripheral tissues [Citation84,Citation130]. Lastly, administration of bromocriptine at a time of day out of phase from the natural circadian peak in CNS clock area dopaminergic activity is not only much less effective or ineffective but can actually be detrimental to normal physiology much the same as shift work influences or stress stimuli are (A.H. Cincotta, unpublished data). Much more work is needed to be done here to delineate the specific biochemical and physiological dimensions of these initial findings. The CST evaluated the overall safety of bromocriptine-QR (N = 2054) versus placebo (N = 1016), and the event rate for serious adverse events was 8.5% for bromocriptine-QR and 9.6% for placebo groups. None of the serious adverse events grouped by System–Organ–Class occurred more than 0.3 percentage points higher with bromocriptine-QR than with placebo. In controlled clinical trials, adverse effects reported in ≥5% of patients treated with bromocriptine-QR and reported more commonly than in patients treated with placebo, included mild–moderate nausea, fatigue, dizziness, vomiting, and headache. These adverse events were more likely to occur during the initial titration of bromocriptine-QR and lasted a median of 14 days. None of the reports of nausea or vomiting were described as serious. Risk of hypoglycemia with bromocriptine-QR therapy is very low and this therapy does not cause weight gain. Hypotension, including orthostatic hypotension, can occur infrequently, particularly upon initiation of bromocriptine-QR therapy and with dose escalation. Bromocriptine-QR may cause somnolence in some patients, which resolves over time in most. Bromocriptine-QR is not recommended for use in patients with severe psychotic disorder, syncopal migraines or women who are nursing. Bromocriptine-QR therapy is not recommended in patients who are taking dopamine antagonists or other dopamine agonists.
Although use of other dopamine agonists such as cabergoline and pergolide may have an increased risk of fibrotic valvular disease [Citation195–Citation197], in Parkinson’s patients, results of studies that evaluated for a similar association in patients with Parkinson’s disease treated with bromocriptine have been inconsistent with these findings [Citation195,Citation196,Citation198,Citation199]. Moreover, none of the studies evaluating a relationship between bromocriptine exposure and valvular heart disease in prolactinoma subjects demonstrated any clinically significant valvular disease [Citation200–Citation202]. In hyperprolactinemia patients treated with bromocriptine up to a cumulative dose of 40 g, there was no increased relative risk of valvular heart disease [Citation203]. The therapeutic dose of bromocriptine-QR used in treatment of type 2 diabetes is between 1.6 and 4.8 mg/day and is lower than bromocriptine doses typically used to treat hyperprolactinemia (5–15 mg/day) or Parkinson’s disease (10–50 mg/day). Lastly, it should be appreciated that bromocriptine-QR is administered once daily in the morning and provides only a brief 4–5-h rise in plasma bromocriptine level [Citation162] whereas multiple daily doses of bromocriptine are typically used to treat prolactinoma and Parkinson’s disease that consequently may raise the plasma bromocriptine level chronically over the entire day each day. No cases of cardiac valvulopathy have been reported in any clinical studies to date with bromocriptine-QR therapy [Citation204].
In summary, circadian timed bromocriptine-QR therapy is a safe and effective glucose lowering agent with unique postprandial insulin-sensitizing properties and potential for CVD event rate reduction. The mechanism of action of the therapy appears to target etiological factors in the genesis of IRS that reside within the biological clock system of the CNS.
Five-year view
Circadian-timed bromocriptine-QR therapy for the treatment of type 2 diabetes represents a novel approach in treating metabolic disorders that targets the circadian neural circuitry within the brain that controls peripheral fuel metabolism. This strategy represents the first application of a biomedical approach to treat biological disorders termed ‘circadian neuroendocrine resetting therapy™’. An appreciation of its developmental history gives unique insights into its potential future applications. Biological clocks evolved early in the development of cellular life on earth and persist in multicellular organisms. In multicellular organisms, circadian neural circuits within the CNS function to temporally organize the activities of organ systems of the body so their biochemical activities are synchronized internally and synchronized with the cyclic environment in a manner that optimizes survival of the organism. Due to the cyclicity of the earth’s rotation on its axis and its revolution around the sun, life equipped with sensory mechanisms evolved in a predictable (cyclic) environment. Evolution selected for biological systems capable of anticipating and coping with ensuing adverse environmental conditions such as low-food availability so as to enhance survival. The seasonal CNS-directed induction of insulin resistance provides a means to survive an ensuing season of low-food (glucose) availability and is the manifestation of seasonal alterations in circadian neural activities whose seasonally changing temporal interactions modulate autonomic and endocrine regulation of peripheral fuel metabolism as a function of their circadian phase relations. Studies indicate that circadian dopaminergic neuronal input to this CNS circadian pacemaker center, the SCN, is a prime regulator of clock output function regulating metabolism. A diminution of the circadian dopaminergic input signal to the clock neurons precipitates insulin resistance and glucose intolerance. Exposure to high-saturated fat diets is able to induce such a diminution in this circadian dopaminergic input to the clock and dopamine administration to the clock reverses the saturated fat diet-induced insulin resistance. The effect of saturated fat to reduce dopaminergic activity at the clock to initiate insulin resistance may itself be part of the evolutionarily derived mechanism by which seasonal insulin resistance, though endogenous, is facilitated by seasonal environmental increases in saturated fat in the food supply (e.g. ground squirrels fatten in winter irrespective of diet however acorns in autumn support seasonal fattening of ground squirrels for winter, and the CNS has evolved to take advantage of predicting such seasonal environmental changes in the food supply). Understanding how the brain functions to interpret food supply and the environment in general is critical to developing new treatment strategies for metabolic (and other) diseases. For example, determining the molecular constituents of the seasonal Paleolithic diet that couple with and/or support endogenous seasonal circadian neuroendocrine mechanisms to facilitate enhanced insulin sensitivity may likely lead to improved methods of treating metabolic diseases in modern man.
An important extension of this circadian neuroendocrine organization system is that this ancient brain system also appears to respond to other clock-perceived stresses common within humans of the twenty-first century including alteration of sleep/wake organization, altered photoperiod exposure, and societal stress by shifting metabolism to the survival mode (i.e. insulin resistance) [Citation51,Citation52,Citation205–Citation208]. That is to say, it appears that desynchronization of external cyclic cues (altered sleep/wake organization and photoperiod exposure) from the internal clock, loss of circadian feedback to the clock (e.g. societal stress induced changes to the circadian organization of plasma cortisol and prolactin rhythms), pharmaceutical interventions that affect clock organization (e.g. anti-psychotic dopamine antagonists and serotonin/norepinephrine modulators) [Citation209–Citation212], or even inherited malfunctions of the clock itself result in a ‘clock response’ that drives the survival output of insulin resistance induction. If these stresses persist for long periods of time (e.g. years) the clock response for survival transforms to a clock response that is detrimental (suicidal). In fact, the scientific literature is now replete with observations of reduced sleep, ‘shift worker’ sleep/wake activity, generalized societal stress, and pharmaceutical agents that impact dopaminergic and/or 5HT systems to induce insulin resistance and CVD (reviewed in [51,52,205–212]). Yet, the clock can be appropriately ‘reset’ to the insulin-sensitive condition (as with timed dopaminergic stimulation) even in the face of such environmental stress cues or resetting signals such as a high-saturated fat diet driving the clock output toward the insulin-resistant state [Citation138], and this resetting therapy may be augmented by simple circadian lifestyle adjustments to appropriately realign the body with its cyclic environment (e.g. aligning sleep/wakefulness with the dark/light cycle, dietary modification). The application of circadian dopaminergic resetting therapy, as with timed bromocriptine-QR therapy, to treating metabolic disease extends beyond the treatment of T2DM and may have potential clinical value in possible prevention of T2DM via treatment of metabolic syndrome or prediabetes, as preclinical and clinical studies reviewed herein suggest. Resetting toward normal those altered circadian neuroendocrine activities that initiate and potentiate the IRS early on in its genesis (or even preventatively before signs of insulin resistance) to thwart progression of the disorder seems a worthy and plausible research endeavor. Moreover, using the approach of timed dopaminergic resetting of circadian neuroendocrine regulation of metabolism, we have been able to markedly attenuate the onset of obesity induced by high-fat diets or genetic loss of leptin function ([Citation104,Citation129,Citation138], Cincotta et al., unpublished data). In a new world increasingly becoming ‘westernized’, an old brain’s misguided response for survival results in a maladaptive suicidal directive. Utilizing circadian neuroendocrine resetting therapy, as with timed bromocriptine-QR to treat metabolic disease, appears to be a potent method of ‘regulating the regulator’ (the brain) to maintain metabolic and physiological health and future scientific research in this area is certain to provide simple effective and safe ways to improve metabolic disease and the human condition. A final note is this regard. This ancient earth-clock brain system for regulating physiology needs to be addressed when considering not only long-term space travel [Citation213] but far more importantly when contemplating the futuristic possibility of life long habitation on other planets with varying daily and seasonal photoperiods from those on earth.
Financial and competing interests disclosure
The authors were supported by VeroScience LLC. AH. Cincotta serves as the President and Chief Scientific Officer and is a shareholder of VeroScience, LLC, the company that owns Cycloset (bromocriptine-QR). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Key Issues
Among vertebrate species, evolution selected for a circadian neuroendocrine system whose organization and function can oscillate seasonally to facilitate the seasonal onset and reversal of the obese, insulin-resistant state which enhances survival during periods (seasons) of low-food (glucose) availability.
The seasonal induction of insulin resistance is driven in large part by a diminution of the circadian dopaminergic input signaling to the hypothalamic biological clock that alters clock output signaling via the autonomic/neuroendocrine axes to potentiate postprandial insulin resistance.
A high-saturated fat diet reduces CNS dopaminergic activity, including a marked reduction in the circadian peak of dopaminergic input to the clock, thereby inducing insulin resistance which can be reversed by administering dopamine directly to the clock center or by systemic administration of bromocriptine at the appropriate time of day to re-establish the circadian peak of dopaminergic activity at the clock.
Clinical studies indicate that timed daily morning administration of bromocriptine-QR, a quick release formulation of bromocriptine that provides a 4–5 h morning pulse of the agent to the body, can improve glycemic control in T2DM subjects (average observed HbA1c reductions of −0.6 to −1.7 across studies) primarily via reducing postprandial hyperglycemia across the meals of the day without increasing plasma insulin levels.
Given available evidence suggesting that bromocriptine-QR improves postprandial insulin sensitivity, the agent’s utility in treating T2DM is best maximized in patients with sufficient postprandial insulin levels as a consequence of early disease state or treatment with insulin secretion enhancers such as DPP4 inhibitors and GLP-1 analogs, or insulin itself. In this regard, preclinical studies indicate and clinical studies suggest that the complementary mechanisms of action of the timed bromocriptine-QR therapy plus the insulin secretory effect of the DPP4 inhibitors or GLP-1 analogs (or plus prandial insulin administration) can provide for synergistic improvement of dysglycemia.
Acting as a sympatholytic agent, time bromocriptine-QR therapy effectiveness in treating hyperglycemia (and dyslipidemia) in T2DM may be enhanced in metabolic syndrome T2DM patients whose sympathetic tone is particularly elevated.
In a large 1-year, randomized clinical study of T2DM subjects, bromocriptine-QR therapy was associated with a significant 40–50% HRR for a prespecified and independently adjudicated adverse cardiovascular event endpoint, including MACE.
Timed bromocriptine-QR therapy is a unique treatment for T2DM that has potential to improve glycemic control and CVD in patients across a broad range of dysglycemia.
Supplemental_material_for_Figure_5.pdf
Download PDF (493.4 KB)References
- Ferrannini E, Gastaldelli A, Iozzo P. Pathophysiology of prediabetes. Med Clin North Am. 2011;95:327–339.
- DeFronzo RA. Pathogenesis of type 2 diabetes mellitus. Med Clin North Am. 2004;88:787–835.
- Ferrannini E, Mari A. β-Cell function in type 2 diabetes. Metabolism. 2014;63:1217–1227.
- Chang-Chen KJ, Mullur R, Bernal-Mizrachi E. Beta-cell failure as a complication of diabetes. Rev Endocr Metab Disord. 2008;9:329–343.
- Abdul-Ghani MA, Williams K, DeFronzo R, et al. Risk of progression to type 2 diabetes based on relationship between postload plasma glucose and fasting plasma glucose. Diabetes Care. 2006;29:1613–1618.
- Abdul-Ghani MA, Tripathy D, DeFronzo RA. Contributions of beta-cell dysfunction and insulin resistance to the pathogenesis of impaired glucose tolerance and impaired fasting glucose. Diabetes Care. 2006;29:1130–1139.
- Townsend KL, Lorenzi MM, Widmaier EP. High-fat diet-induced changes in body mass and hypothalamic gene expression in wild-type and leptin-deficient mice. Endocrine. 2008;33:176–188.
- Thaler JP, Schwartz MW. Minireview: inflammation and obesity pathogenesis: the hypothalamus heats up. Endocrinology. 2010;151:4109–4115.
- Chari M, Lam CKL, Lam TKT. Hypothalamic fatty acid sensing in the normal and disease states. In: Montmayeur JP, Le Coutre J, editors. Frontiers in neuroscience. Fat detection: taste, texture, and post ingestive effects. Boca Raton (FL): CRC Press; 2010. p. Ch20.
- Demuro G, Obici S. Central nervous system and control of endogenous glucose production. Curr Diab Rep. 2006;6:188–193.
- Martínez De Morentin PB, Varela L, Fernø J, et al. Hypothalamic lipotoxicity and the metabolic syndrome. Biochim Biophys Acta. 2010;1801:350–361.
- Parlevliet ET, Coomans CP, Rensen PC, et al. The brain modulates insulin sensitivity in multiple tissues. Front Horm Res. 2014;42:50–58.
- Routh VH, Hao L, Santiago AM, et al. Hypothalamic glucose sensing: making ends meet. Front Syst Neurosci. 2014;8:236.
- Picard A, Moullé VS, Le Foll C, et al. Physiological and pathophysiological implications of lipid sensing in the brain. Diabetes Obes Metab. 2014;16(Suppl1):49–55.
- Martinac M, Pehar D, Karlović D, et al. Metabolic syndrome, activity of the hypothalamic-pituitary-adrenal axis and inflammatory mediators in depressive disorder. Acta Clin Croat. 2014;53:55–71.
- Bisschop PH, Fliers E, Kalsbeek A. Autonomic regulation of hepatic glucose production. Compr Physiol. 2015;5:147–165.
- Jordan SD, Könner AC, Brüning JC. Sensing the fuels: glucose and lipid signaling in the CNS controlling energy homeostasis. Cell Mol Life Sci. 2010;67:3255–3273.
- Cersosimo E, DeFronzo RA. Insulin resistance and endothelial dysfunction: the road map to cardiovascular diseases. Diabetes Metab Res Rev. 2006;22:423–436.
- Younk LM, Lamos EM, Davis SN. The cardiovascular effects of insulin. Expert Opin Drug Saf. 2014;13:955–966.
- Otero YF, Stafford JM, McGuinness OP. Pathway-selective insulin resistance and metabolic disease: the importance of nutrient flux. J Biol Chem. 2014;289:20462–20469.
- Boden G, Carnell LH. Nutritional effects of fat on carbohydrate metabolism. Best Pract Res Clin Endocrinol Metab. 2003;17:399–410.
- Morrison CD, Huypens P, Stewart LK, et al. Implications of crosstalk between leptin and insulin signaling during the development of diet-induced obesity. Biochim Biophys Acta. 2009;1792:409–416.
- Martins AR, Nachbar RT, Gorjao R, et al. Mechanisms underlying skeletal muscle insulin resistance induced by fatty acids: importance of the mitochondrial function. Lipids Health Dis. 2012;11:30.
- Levin BE, Magnan C, Migrenne S, et al. F-DIO obesity-prone rat is insulin resistant before obesity onset. Am J Physiol Regul Integr Comp Physiol. 2005;289:R704–11.
- Zimmet PZ. Challenges in diabetes epidemiology – from west to the rest. Diabetes Care. 1992;232:252.
- Dowse G, Zimmet P. The thrifty genotype in non-insulin dependent diabetes. BMJ. 1993;306:532–533.
- Popkin BM, Slining MM. New dynamics in global obesity facing low- and middle-income countries. Obes Rev. 2013;14(Suppl2):11–20.
- Rivellese AA, De Natale C, Lilli S. Type of dietary fat and insulin resistance. Ann N Y Acad Sci. 2002;967:329–335.
- Riccardi G, Giacco R, Rivellese AA. Dietary fat, insulin sensitivity and the metabolic syndrome. Clin Nutr. 2004;23:447–456.
- Levin BE, Dunn-Meynell AA, Balkan B, et al. Selective breeding for diet-induced obesity and resistance in Sprague-Dawley rats. Am J Physiol. 1997;273:R725–30.
- Heitmann BL, Westerterp KR, Loos RJ, et al. Obesity: lessons from evolution and the environment. Obes Rev. 2012;13:910–922.
- Singh RB, Gupta S, Dherange P, et al. Metabolic syndrome: a brain disease. Can J Physiol Pharmacol. 2012;90:1171–1183.
- Biebermann H, Kühnen P, Kleinau G, et al. The neuroendocrine circuitry controlled by POMC, MSH, and AGRP. Handb Exp Pharmacol. 2012;209:47–75.
- Ellacott KL, Cone RD. The central melanocortin system and the integration of short- and long-term regulators of energy homeostasis. Recent Prog Horm Res. 2004;59:395–408.
- Fani L, Bak S, Delhanty P, et al. The melanocortin-4 receptor as target for obesity treatment: a systematic review of emerging pharmacological therapeutic options. Int J Obes (Lond). 2014;38:163–169.
- Baltatzi M, Hatzitolios A, Tziomalos K, et al. Neuropeptide Y and alpha-melanocyte-stimulating hormone: interaction in obesity and possible role in the development of hypertension. Int J Clin Pract. 2008;62:1432–1440.
- Cone RD, Cowley MA, Butler AA, et al. The arcuate nucleus as a conduit for diverse signals relevant to energy homeostasis. Int J Obes Relat Metab Disord. 2001;25(Suppl5):S63–7.
- Belgardt BF, Okamura T, Brüning JC. Hormone and glucose signalling in POMC and AgRP neurons. J Physiol. 2009;587:5305–5314.
- Niswender KD, Baskin DG, Schwartz MW. Insulin and its evolving partnership with leptin in the hypothalamic control of energy homeostasis. Trends Endocrinol Metab. 2004;15:362–369.
- Leinninger GM. Lateral thinking about leptin: a review of leptin action via the lateral hypothalamus. Physiol Behav. 2011;104:572–581.
- Opland DM, Leinninger GM, Myers MG Jr. Modulation of the mesolimbic dopamine system by leptin. Brain Res. 2010;1350:65–70.
- Van Zessen R, Van Der Plasse G, Adan RA. Contribution of the mesolimbic dopamine system in mediating the effects of leptin and ghrelin on feeding. Proc Nutr Soc. 2012;71:435–445.
- Bingham NC, Anderson KK, Reuter AL, et al. Selective loss of leptin receptors in the ventromedial hypothalamic nucleus results in increased adiposity and a metabolic syndrome. Endocrinology. 2008;149:2138–2148
- Irani BG, Le Foll C, Dunn-Meynell AA, et al. Ventromedial nucleus neurons are less sensitive to leptin excitation in rats bred to develop diet-induced obesity. Am J Physiol Regul Integr Comp Physiol. 2009;296:R521–7.
- Toda C, Shiuchi T, Kageyama H, et al. Extracellular signal-regulated kinase in the ventromedial hypothalamus mediates leptin-induced glucose uptake in red-type skeletal muscle. Diabetes. 2013;62:2295–2307.
- Toda C, Shiuchi T, Lee S, et al. Distinct effects of leptin and a melanocortin receptor agonist injected into medial hypothalamic nuclei on glucose uptake in peripheral tissues. Diabetes. 2009;58:2757–2765.
- Pfaffly J, Michaelides M, Wang GJ, et al. Leptin increases striatal dopamine D2 receptor binding in leptin-deficient obese (ob/ob) mice. Synapse. 2010;64:503–510.
- Marino JS, Xu Y, Hill JW. Central insulin and leptin-mediated autonomic control of glucose homeostasis. Trends Endocrinol Metab. 2011;22:275–285.
- Dunn JP, Kessler RM, Feurer ID, et al. Relationship of dopamine type 2 receptor binding potential with fasting neuroendocrine hormones and insulin sensitivity in human obesity. Diabetes Care. 2012;35:1105–1111.
- Caravaggio F, Borlido C, Hahn M, et al. Reduced insulin sensitivity is related to less endogenous dopamine at D2/3 receptors in the ventral striatum of healthy non-obese humans. Int J Neuropsychopharmacol. 2015;pii:pyv014.
- Meier A, Cincotta A. Circadian rhythms regulate the expression of the thrifty genotype/phenotype. Diabetes Rev. 1996;4:464–487.
- Cincotta A. Hypothalamic role in the insulin resistance syndrome. In: Hansen B, Shaffrir E, editors. Resistance and insulin resistance syndrome. London: Taylor and Francis; 2002. p. 271–312.
- La Fleur SE. Daily rhythms in glucose metabolism: suprachiasmatic nucleus output to peripheral tissue. J Neuroendocrinol. 2003;15:315–322.
- Kalsbeek A, Bruinstroop E, Yi CX, et al. Hypothalamic control of energy metabolism via the autonomic nervous system. Ann N Y Acad Sci. 2010;1212:114–129.
- Bailey SM, Udoh US, Young ME. Circadian regulation of metabolism. J Endocrinol. 2014;222:R75–96.
- Mazzoccoli G, Pazienza V, Vinciguerra M. Clock genes and clock-controlled genes in the regulation of metabolic rhythms. Chronobiol Int. 2012;29:227–251.
- Cincotta AH, Meier AH. Circadian rhythms of lipogenic and hypoglycaemic responses to insulin in the golden hamster (Mesocricetus auratus). J Endocrinol. 1984;103:141–146.
- Cincotta AH, Meier AH. Prolactin permits the expression of a circadian variation in lipogenic responsiveness to insulin in hepatocytes of the golden hamster (Mesocricetus auratus). J Endocrinol. 1985;106:173–176.
- Cincotta AH, Meier AH. Prolactin permits the expression of a circadian variation in insulin receptor profile in hepatocytes of the golden hamster (Mesocricetus auratus). J Endocrinol. 1985;106:177–181.
- Froy O. The relationship between nutrition and circadian rhythms in mammals. Front Neuroendocrinol. 2007;28:61–71.
- Kalsbeek A, La Fleur S, Fliers E. Circadian control of glucose metabolism. Mol Metab. 2014;3:372–383.
- Ramsey KM, Bass J. Circadian clocks in fuel harvesting and energy homeostasis. Cold Spring Harb Symp Quant Biol. 2011;76:63–72.
- Boers I, Muskiet FA, Berkelaar E, et al. Favourable effects of consuming a Palaeolithic-type diet on characteristics of the metabolic syndrome: a randomized controlled pilot-study. Lipids Health Dis. 2014;13:160.
- Manheimer EW, Van Zuuren EJ, Fedorowicz Z, et al. Paleolithic nutrition for metabolic syndrome: systematic review and meta-analysis. Am J Clin Nutr. 2015;102:922–932.
- Spreadbury I. Comparison with ancestral diets suggests dense acellular carbohydrates promote an inflammatory microbiota, and may be the primary dietary cause of leptin resistance and obesity. Diabetes Metab Syndr Obes. 2012;5:175–189.
- Tsujimoto T, Yamamoto-Honda R, Kajio H, et al. Seasonal variations of severe hypoglycemia in patients with type 1 diabetes mellitus, type 2 diabetes mellitus, and non-diabetes mellitus: clinical analysis of 578 hypoglycemia cases. Medicine (Baltimore). 2014;93:e148.
- Nordfeldt S, Ludvigsson J. Seasonal variation of HbA1c in intensive treatment of children with type 1 diabetes. J Pediatr Endocrinol Metab. 2000;13:529–535.
- Sakura H, Tanaka Y, Iwamoto Y. Seasonal fluctuations of glycated hemoglobin levels in Japanese diabetic patients. Diabetes Res Clin Pract. 2010;88:65–70.
- Liang WW. Seasonal changes in preprandial glucose, A1C, and blood pressure in diabetic patients. Diabetes Care. 2007;30:2501–2502.
- Ishii H, Suzuki H, Baba T, et al. Seasonal variation of glycemic control in type 2 diabetic patients. Diabetes Care. 2001;24:1503.
- Sohmiya M, Kanazawa I, Kato Y. Seasonal changes in body composition and blood HbA1c levels without weight change in male patients with type 2 diabetes treated with insulin. Diabetes Care. 2004;27:1238–1239.
- Gikas A, Sotiropoulos A, Pastromas V, et al. Seasonal variation in fasting glucose and HbA1c in patients with type 2 diabetes. Prim Care Diabetes. 2009;3:111–114.
- Partonen T. During winter the body resists insulin. Hypertens Res. 2013;36:390–391.
- Kamezaki F, Sonoda S, Nakata S, et al. Association of seasonal variation in the prevalence of metabolic syndrome with insulin resistance. Hypertens Res. 2013;36:398–402.
- Kamezaki F, Sonoda S, Tomotsune Y, et al., et al. Seasonal variation in metabolic syndrome prevalence. Hypertens Res. 2010;33:568–572.
- Meier AH. Temporal synergism of circadian neuroendocrine oscillations regulates seasonal conditions in the gulf killifish. Trans Am Fish Soc. 1984;113:422–431.
- Meier AH, Russo AC. Circadian organization of the avian annual cycle. In: Johnston RE, editor. Current ornithology. Vol. 2. New York (NY): Plenum; 1984. p. 303–343.
- Emata AC, Meier AH, Spieler RE. Temporal variations in gonadal and body fat responses to daily injections of a 5-hydroxytryptophan (5-HTP) and dihydroxyphenylalanine (DOPA) in the gulf killifish, Fundulus grandis. J Exp Zool. 1985;233:29–34.
- Wilson JM, Meier AH. Resetting the annual cycle with timed daily injections of 5-hydroxytryptophan and L-dihydroxyphenylalanine in Syrian hamsters. Chronobiol Int. 1989;6:113–121.
- Luo S, Luo J, Cincotta AH. Suprachiasmatic nuclei monoamine metabolism of glucose tolerant versus intolerant hamsters. Neuroreport. 1999;10:2073–2077.
- Luo S, Zhang Y, Ezrokhi M, et al. High-fat feeding abolishes the insulin-sensitizing peak in circadian dopamine activity at the biological clock. Diabetes. 2014;63(Suppl1):A470.
- Luo S, Luo J, Meier A. Dopaminergic neurotoxin administration to the area of the suprachiasmatic nuclei induces insulin resistance. Neuroreport. 1997;8:3495–3499.
- Zhang Y, Luo S, Ezrokhi M, et al. Increasing GABAa/AMPA receptor ratio stimulation at the supramammillary nucleus (SuMN) induces insulin resistance (IR) without weight gain in rats. Diabetes. 2014;63(Suppl1):A494.
- Luo S, Ezrokhi M, Trubitsyna Y, et al. One minute of circadian-timed daily dopamine (DA) administration at the biological clock for 2 weeks ameliorates metabolic syndrome in spontaneously hypertensive rats (SHR) held on a high fat diet (HFD). Diabetes. 2015;64(Suppl1):A523.
- Zhang Y, Luo S, Ezrokhi M, et al. Circadian-timed daily activation of supramammillary nucleus (SuMN) ameliorates obesity/insulin resistance in high fat diet-induced insulin resistant rats. Diabetes. 2015;64(Suppl1):A536.
- Ezrokhi M, Luo S, Trubitsyna Y, et al. Neuroendocrine and metabolic components of dopamine agonist amelioration of metabolic syndrome in SHR rats. Diabetol Metab Syndr. 2014;6:104.
- Luo S, Ezrokhi M, Trubitsyna Y, et al. Intrahypothalamic circuitry regulating hypothalamic fuel sensing to induce insulin sensitivity or insulin resistance. Diabetologia. 2008;51(Suppl1):S1–S588.
- Pocai A, Lam TK, Obici S, et al. Restoration of hypothalamic lipid sensing normalizes energy and glucose homeostasis in overfed rats. J Clin Invest. 2006;116:1081–1091.
- Lam TK. Neuronal regulation of homeostasis by nutrient sensing. Nat Med. 2010;16:392–395.
- Breen DM, Yang CS, Lam TK. Gut-brain signaling: how lipids can trigger the gut. Diabetes Metab Res Rev. 2011;27:113–119.
- Jordan SD, Könner AC, Brüning JC. Sensing the fuels: glucose and lipid signaling in the CNS controlling energy homeostasis. Cell Mol Life Sci. 2010;67:3255–3273.
- Morgan K, Obici S, Rossetti L. Hypothalamic responses to long-chain fatty acids are nutritionally regulated. J Biol Chem. 2004;279:31139–31148.
- Cincotta AH, Luo S, Zhang Y, et al. Chronic infusion of norepinephrine into the VMH of normal rats induces the obese glucose-intolerant state. Am J Physiol Regulatory Integrative Comp Physiol. 2000;278:R435–R444.
- Dicostanzo CA, Dardevet DP, Neal DW, et al. Role of the hepatic sympathetic nerves in the regulation of net hepatic glucose uptake and the mediation of the portal glucose signal. Am J Physiol Endocrinol Metab. 2006;290:E9–E16.
- Lambert GW, Straznicky NE, Lambert EA, at al Sympathetic nervous activation in obesity and the metabolic syndrome–causes, consequences and therapeutic implications. Pharmacol Ther. 2010;126:159–172.
- Grassi G. Sympathetic overdrive and cardiovascular risk in the metabolic syndrome. Hypertens Res. 2006;29:839–847.
- Tentolouris N, Liatis S, Katsilambros N. Sympathetic system activity in obesity and metabolic syndrome. Ann N Y Acad Sci. 2006;1083:129–152.
- Luo S, Luo J, Cincotta AH. Chronic ventromedial hypothalamic infusion of norepinephrine and serotonin promotes insulin resistance and glucose intolerance. Neuroendocrinology. 1999;70:460–465.
- Liang Y, Luo S, Cincotta AH. Long-term infusion of norepinephrine plus serotonin into the ventromedial hypothalamus impairs pancreatic islet function. Metabolism. 1999;48:1287–1289.
- Cincotta AH, Luo S, Liang Y. Hyperinsulinemia increases norepinephrine metabolism in the ventromedial hypothalamus of rats. Neuroreport. 2000;11:383–387.
- Shimazu T. Neuronal regulation of hepatic glucose metabolism in mammals. Diabetes Metab Rev. 1987;3:185–206.
- Steffens AB, Damsma G, Vasnder Gugten J, et al. Circulating fatty acids, insulin, and glucose during chemical stimulation of hypothalamus in rats. Am J Physiol. 1984;247:E765–71.
- Borg WP, Sherwin RS, During MJ, et al. Local ventromedial hypothalamus glucopenia triggers counterregulatory hormone release. Diabetes. 1995;44:180–184.
- Bina KG, Cincotta AH. Dopaminergic agonists normalize elevated hypothalamic neuropeptide Y and corticotropin-releasing hormone, body weight gain, and hyperglycemia in ob/ob mice. Neuroendocrinology. 2000;71:68–78.
- Ferrie JE, Kivimäki M, Akbaraly TN, et al. Change in sleep duration and type 2 diabetes: the Whitehall II Study. Diabetes Care. 2015;38:1467–1472.
- Vetter C, Devore EE, Ramin CA, et al. Mismatch of sleep and work timing and risk of type 2 diabetes. Diabetes Care. 2015;38:1707–1713.
- Scheer FA, Hilton MF, Mantzoros CS, et al. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc Natl Acad Sci USA. 2009;106:4453–4458.
- Leproult R, Holmback U, Van Cauter E. Circadian misalignment augments markers of insulin resistance and inflammation, independently of sleep loss. Diabetes. 2014;63:1860–1869.
- Chiodini I, Adda G, Scillitani A, et al. Cortisol secretion in patients with type 2 diabetes: relationship with chronic complications. Diabetes Care. 2007;30:83–88.
- Farag NH, Moore WE, Lovallo WR, et al. Hypothalamic-pituitary-adrenal axis function: relative contributions of perceived stress and obesity in women. J Womens Health (Larchmt). 2008;17:1647–1655.
- Jokinen J, Nordström P. HPA axis hyperactivity and attempted suicide in young adult mood disorder inpatients. J Affect Disord. 2009;116:117–120.
- Mendoza J, Challet E. Circadian insights into dopamine mechanisms. Neuroscience. 2014;282C:230–242.
- Argyropoulos SV, Nutt DJ. Anhedonia revisited: is there a role for dopamine-targeting drugs for depression? J Psychopharmacol. 2013;27:869–877.
- Lim BK, Huang KW, Grueter BA, et al. Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature. 2012 Jul 11;487(7406):183–189.
- Monti JM, Monti D. The involvement of dopamine in the modulation of sleep and waking. Sleep Med Rev. 2007;11:113–133.
- Geiger BM, Haburcak M, Avena NM, et al. Deficits of mesolimbic dopamine neurotransmission in rat dietary obesity. Neuroscience. 2009;159:1193–1199.
- Davis JF, Tracy AL, Schurdak JD, et al. Exposure to elevated levels of dietary fat attenuates psychostimulant reward and mesolimbic dopamine turnover in the rat. Behav Neurosci. 2008;122:1257–1263.
- Rada P, Bocarsly ME, Barson JR, et al. Reduced accumbens dopamine in Sprague-Dawley rats prone to overeating a fat-rich diet. Physiol Behav. 2010;101:394–400.
- Geiger BM, Behr GG, Frank LE, et al. Evidence for defective mesolimbic dopamine exocytosis in obesity-prone rats. FASEB J. 2008;22:2740–2746.
- Blum K, Chen TJ, Downs BW, et al. Neurogenetics of dopaminergic receptor supersensitivity in activation of brain reward circuitry and relapse: proposing “deprivation-amplification relapse therapy” (DART). Postgrad Med. 2009;121:176–196.
- Noble EP. Addiction and its reward process through polymorphisms of the D2 dopamine receptor gene: a review. Eur Psychiatry. 2000;15:79–89.
- Billes SK, Simonds SE, Cowley MA. Leptin reduces food intake via a dopamine D2 receptor-dependent mechanism. Mol Metab. 2012;1:86–93.
- Zheng H, Liu X, Li Y, et al. Attenuated dopaminergic tone in the paraventricular nucleus contributing to sympathoexcitation in rats with Type 2 diabetes. Am J Physiol Regul Integr Comp Physiol. 2014;306:R138–48.
- Luo S, Meier AH, Cincotta AH. Bromocriptine reduces obesity, glucose intolerance and extracellular monoamine metabolite levels in the ventromedial hypothalamus of Syrian hamsters. Neuroendocrinology. 1998;68:1–10.
- Ezrokhi M, Luo S, Trubitsyna Y, et al. Weighted effects of bromocriptine treatment on glucose homeostasis during hyperglycemic versus euglycemic clamp conditions in insulin resistant hamsters: bromocriptine as a unique postprandial insulin sensitizer. J Diabetes Metab. 2012;S2:7.
- Moore MC, Smith M, Farmer B, et al. Timed daily bromocriptine mesylate (BC) administration improves glucose disposal in a canine diet-induced model of impaired glucose tolerance. Diabetologia. 2014;57(Suppl1):S350.
- Cincotta AH, Schiller BC, Meier AH. Bromocriptine inhibits the seasonally occurring obesity, hyperinsulinemia, insulin resistance, and impaired glucose tolerance in the Syrian hamster, Mesocricetus auratus. Metabolism. 1991;40:639–644.
- Cincotta AH, MacEachern TA, Meier AH. Bromocriptine redirects metabolism and prevents seasonal onset of obese hyperinsulinemic state in Syrian hamsters. Am J Physiol. 1993;264:E285–93.
- Scislowski PW, Tozzo E, Zhang Y, et al. Biochemical mechanisms responsible for the attenuation of diabetic and obese conditions in ob/ob mice treated with dopaminergic agonists. Int J Obes Relat Metab Disord. 1999;23:425–431.
- Luo S, Liang Y, Cincotta AH. Intracerebroventricular administration of bromocriptine ameliorates the insulin-resistant/glucose-intolerant state in hamsters. Neuroendocrinology. 1999;69:160–166.
- Davis LM, Pei Z, Trush MA, et al. Bromocriptine reduces steatosis in obese rodent models. J Hepatol. 2006;45:439–444.
- Cincotta AH, Meier AH, Southern LL. Bromocriptine alters hormone rhythms and lipid metabolism in swine. Ann Nutr Metab. 1989;33:305–314.
- Ezrokhi M, Luo S, Trubitsyna T, et al. Synergism of dopamine agonist plus GLP-1 analog therapy on improvement of glucose intolerance in Syrian hamsters. Diabetes. 2011;60(Suppl1):A445.
- Cincotta AH, Schiller BC, Meier AH. Bromocriptine inhibits the seasonally occurring obesity, hyperinsulinemia, insulin resistance, and impaired glucose tolerance in the Syrian hamster, Mesocricetus auratus. Metabolism. 1991;40:639–644.
- Cincotta AH, Meier AH. Prolactin permits the expression of a circadian variation in lipogenic responsiveness to insulin in hepatocytes of the golden hamster (Mesocricetus auratus). J Endocrinol. 1985;106:173–176.
- Cincotta AH, Meier AH. Bromocriptine inhibits in vivo free fatty acid oxidation and hepatic glucose output in seasonally obese hamsters (Mesocricetus auratus). Metabolism. 1995;44:1349–1355.
- Liang Y, Jetton TL, Lubkin M, et al. Bromocriptine/SKF38393 ameliorates islet dysfunction in the diabetic (db/db) mouse. Cell Mol Life Sci. 1998;54:703–711.
- Ezrokhi M, Trubitsyna Y, Luo S, et al. Timed dopamine agonist treatment ameliorates both vascular nitrosative/oxidative stress pathology and aortic stiffness in arteriosclerotic, hypertensive SHR rats. Diabetes. 2010;59(Suppl1):A67.
- Kietadisorn R, Juni RP, Moens AL. Tackling endothelial dysfunction by modulating NOS uncoupling: new insights into its pathogenesis and therapeutic possibilities. Am J Physiol Endocrinol Metab. 2012;302:E481–95.
- Santilli F, Cipollone F, Mezzetti A, et al. The role of nitric oxide in the development of diabetic angiopathy. Horm Metab Res. 2004;36:319–335.
- Magenta A, Greco S, Capogrossi MC, et al. Nitric oxide, oxidative stress, and p66Shc interplay in diabetic endothelial dysfunction. Biomed Res Int. 2014;2014:193095.
- Narkar V, Kunduzova O, Hussain T, et al. Dopamine D2-like receptor agonist bromocriptine protects against ischemia/reperfusion injury in rat kidney. Kidney Int. 2004;66:633–640.
- Gao J, Guo J, Li H, et al. Involvement of dopamine D2 receptors activation in ischemic post-conditioning-induced cardioprotection through promoting PKC-ε particulate translocation in isolated rat hearts. Mol Cell Biochem. 2013;379:267–276.
- Li HZ, Guo J, Gao J, et al. Role of dopamine D2 receptors in ischemia/reperfusion induced apoptosis of cultured neonatal rat cardiomyocytes. J Biomed Sci. 2011;18:18.
- Cycloset® [package insert]. Tiverton (RI): VeroScience LLC;2010 9.
- Park S, Kang S, Lee HW, et al. Central prolactin modulates insulin sensitivity and insulin secretion in diabetic rats. Neuroendocrinology. 2012;95:332–343.
- Ferrari E, Bossolo P, Marelli G, et al. Hormonal circadian profiles in obesity. Annu Rev Chronopharmacol. 1986;3:99–402.
- Cupinschi G, DeLaet MH, Brion JP, et al. Simultaneous study of cortisol, growth hormone, and prolactin nyctohemeral variations in normal and obese subjects: influence of prolonged fasting in obesity. Clin Endocrinol. 1978;9:15–26.
- Ferrari E, Magri F, Dori D. Neuroendocrine abnormalities in primary obesity. In: Ferrari E, Brambilla F, Solerte SB, et al., editors. Primary and secondary eating disorders: a psychoneuroendocrine and metabolic approach. Vol. 90, Oxford (UK): Pergamon; 1993. p. 287.
- Kok P, Roelfsema F, Frölich M, et al. Prolactin release is enhanced in proportion to excess visceral fat in obese women. J Clin Endocrinol Metab. 2004;89:4445–4449.
- Kok P, Roelfsema F, Langendonk JG, et al. Increased circadian prolactin release is blunted after body weight loss in obese premenopausal women. Am J Physiol Endocrinol Metab. 2006;290:E218–24.
- Berinder K, Nyström T, Höybye C, et al. Insulin sensitivity and lipid profile in prolactinoma patients before and after normalization of prolactin by dopamine agonist therapy. Pituitary. 2011;14:199–207.
- Tuzcu A, Yalaki S, Arikan S, et al. Evaluation of insulin sensitivity in hyperprolactinemic subjects by euglycemic hyperinsulinemic clamp technique. Pituitary. 2009;12:330–334.
- Tuzcu A, Bahceci M, Dursun M, et al. Insulin sensitivity and hyperprolactinemia. J Endocrinol Invest. 2003;26:341–346.
- Foss MC, Paula FJ, Paccola GM, et al. Peripheral glucose metabolism in human hyperprolactinaemia. Clin Endocrinol (Oxf). 1995;43:721–726.
- Serri O, Beauregard H, Rasio E, et al. Decreased sensitivity to insulin in women with microprolactinomas. Fertil Steril. 1986;45:572–574.
- Dos Santos Silva CM, Barbosa FR, Lima GA, et al. BMI and metabolic profile in patients with prolactinoma before and after treatment with dopamine agonists. Obesity (Silver Spring). 2011;19:800–805.
- Landgraf R, Landraf-Leurs MM, Weissmann A, et al. Prolactin: a diabetogenic hormone. Diabetologia. 1977;13:99–104.
- Johnston DG, Alberti KG, Nattrass M, et al. Hyperinsulinaemia in hyperprolactinaemic women. Clin Endocrinol (Oxf). 1980;13:361–368.
- Cincotta AH, Meier AH, Taylor E, et al. Bromocriptine (Ergoset) reduces body fat, hyperinsulinemia and glucose intolerance in obese subjects. Diabetes. 1995;44(Suppl.1):168A.
- Cincotta AH, Meier AH, Cincotta M Jr. Bromocriptine improves glycaemic control and serum lipid profile in obese Type 2 diabetic subjects: a new approach in the treatment of diabetes. Expert Opin Investig Drugs. 1999;8:1683–1707.
- Scranton R, Cincotta A. Bromocriptine–unique formulation of a dopamine agonist for the treatment of type 2 diabetes. Expert Opin Pharmacother. 2010;11:269–279.
- Pijl H, Ohashi S, Matsuda M, et al. Bromocriptine: a novel approach to the treatment of type 2 diabetes. Diabetes Care. 2000;23:1154–1161.
- Mancia G, Bousquest P, Elghozi JL, et al. The sympathetic nervous system and the metabolic syndrome. J Hypertens. 2007;25:909–920.
- Roelfsema F, Kok P, Pereira AM, et al. Cortisol production rate is similarly elevated in obese women with or without the polycystic ovary syndrome. J Clin Endocrinol Metab. 2010;95:3318–3324.
- Cincotta AH, Ezrokhi M, Trubitsyna Y, et al. Lesion of dopaminergic afferent neurons communicating with the biological clock induces metabolic syndrome in rats. Diabetes. 2015;64(Suppl1):A540.
- Geerling JJ, Boon MR, Kooijman S, et al. Sympathetic nervous system control of triglyceride metabolism: novel concepts derived from recent studies. J Lipid Res. 2014;55:180–189.
- Straznicky NE, Lambert EA, Lambert GW, et al. Effects of dietary weight loss on sympathetic activity and cardiac risk factors associated with the metabolic syndrome. J Clin Endocrinol Metab. 2005;90:5998–6005.
- Grundy SM, Brewer HB Jr, Cleeman JI, et al. American Heart Association; National Heart, Lung, and Blood Institute. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation. 2004;109:433–438.
- Cincotta AH, Ezrokhi M. The anti-diabetes efficacy of Bromocriptine-QR in type 2 diabetes mellitus (T2DM) subjects is enhanced among those with elevated blood pressure and plasma triglyceride levels. Diabetes. 2013;62(Suppl1):A305.
- Cincotta AH, Meier AH. Reductions of body fat stores and total plasma cholesterol and triglyceride concentrations in several species by bromocriptine treatment. Life Sci. 1989;45:2247–2254.
- Vinik AI, Cincotta AH, Scranton RE, et al. Effect of bromocriptine-QR on glycemic control in subjects with uncontrolled hyperglycemia on one or two oral anti-diabetes agents. Endocr Pract. 2012;18:931–943.
- Scranton RE, Gaziano JM, Rutty D, et al. A randomized, double-blind, placebo-controlled trial to assess safety and tolerability during treatment of type 2 diabetes with usual diabetes therapy and either Cycloset or placebo. BMC Endocr Disord. 2007;7:3.
- Florez H, Scranton R, Farwell WR, et al. Randomized clinical trial assessing the efficacy and safety of bromocriptine-QR when added to ongoing thiazolidinedione therapy in patients with type 2 diabetes mellitus. J Diabetes Metab. 2011;2:142.
- Murphy GJ, Holder JC. PPAR-gamma agonists: therapeutic role in diabetes, inflammation and cancer. Trends Pharmacol Sci. 2000;21:469–474.
- Zeender E, Maedler K, Bosco D, et al. Pioglitazone and sodium salicylate protect human beta-cells against apoptosis and impaired function induced by glucose and interleukin-1beta. J Clin Endocrinol Metab. 2004;89:5059–5066.
- Diani AR, Sawada G, Wyse B, et al. Pioglitazone preserves pancreatic islet structure and insulin secretory function in three murine models of type 2 diabetes. Am J Physiol Endocrinol Metab. 2004;286:E116–22.
- Schwartz S. Bromocriptine (Ergoset) improves glycemic control in type 2 diabetics on insulin. Diabetes. 1999;48(Suppl1):A99.
- Roe ED, Chamarthi B, Raskin P. Impact of bromocriptine-QR therapy on glycemic control and daily insulin requirement in type 2 diabetes mellitus subjects whose dysglycemia is poorly controlled on high-dose insulin: a pilot study. J Diabetes Res. 2015;2015:834903.
- Zoungas S, Patel A. Cardiovascular outcomes in type 2 diabetes: the impact of preventative therapies. Ann N Y Acad Sci. 2010;1212:29–40.
- Holman RR, Paul SK, Bethel MA, et al. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359:1577–1589.
- Sheetz MJ, King GL. Molecular understanding of hyperglycemia’s adverse effects for diabetic complications. JAMA. 2002;288:2579–2588.
- Grassi G. Sympathetic overdrive and cardiovascular risk in the metabolic syndrome. Hypertens Res. 2006;29:839–847.
- Lambert GW, Straznicky NE, Lambert EA, et al. Sympathetic nervous activation in obesity and the metabolic syndrome–causes, consequences and therapeutic implications. Pharmacol Ther. 2010;126:159–172.
- Tentolouris N, Liatis S, Katsilambros N. Sympathetic system activity in obesity and metabolic syndrome. Ann N Y Acad Sci. 2006;1083:129–152.
- Gaziano JM, Cincotta AH, O’Connor CM, et al. Randomized clinical trial of quick-release bromocriptine among patients with type 2 diabetes on overall safety and cardiovascular outcomes. Diabetes Care. 2010;33:1503–1508.
- Gaziano JM, Cincotta AH, Vinik A, et al. Effect of bromocriptine-QR (a quick-release formulation of bromocriptine mesylate) on major adverse cardiovascular events in type 2 diabetes subjects. J Am Heart Assoc. 2012;1:e002279.
- Chamarthi B, Gaziano JM, Blonde L, et al. Timed bromocriptine-QR therapy reduces progression of cardiovascular disease and dysglycemia in subjects with well-controlled type 2 diabetes mellitus. J Diabetes Res. 2015;2015:157698.
- Halberg F, Powell D, Otsuka K, et al. Diagnosing vascular variability anomalies, not only MESOR-hypertension. Am J Physiol Heart Circ Physiol. 2013;305:H279–94.
- Hermida RC, Ayala DE, Fernandez JR, et al. Administration-time differences in effects of hypertension medications on ambulatory blood pressure regulation. Chronobiol Int. 2013;30:280–314.
- Verdecchia P, Angeli F, Mazzotta G, et al. Day-night dip and early-morning surge in blood pressure in hypertension: prognostic implications. Hypertension. 2012;60:34–42.
- Kanth R, Ittaman S, Rezkalla S. Circadian patterns of ST elevation myocardial infarction in the new millennium. Clin Med Res. 2013;11:66–72.
- Giles TD. Circadian rhythm of blood pressure and the relation to cardiovascular events. J Hypertens Suppl. 2006;24:S11–6.
- Muller JE, Tofler GH, Stone PH. Circadian variation and triggers of onset of acute cardiovascular disease. Circulation. 1989;79:733–743.
- Senard JM, Rai S, Lapeyre-Mestre M, et al. JL. Prevalence of orthostatic hypotension in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1997;63:584–589.
- Andersohn F, Garbe E. Cardiac and noncardiac fibrotic reactions caused by ergot-and nonergot-derived dopamine agonists. Mov Disord. 2009;24:129–133.
- Antonini A, Poewe W. Fibrotic heart-valve reactions to dopamine-agonist treatment in Parkinson’s disease. Lancet Neurol. 2007;6:826–829.
- Kim JY, Chung EJ, Park SW, et al. Valvular heart disease in Parkinson’s disease treated with ergot derivative dopamine agonists. Mov Disord. 2006;21:1261–1264.
- Tan LC, Ng KK, Au WL, et al. Bromocriptine use and the risk of valvular heart disease. Mov Disord. 2009;24:344–349.
- Halperin I, Aller J, Varela C, et al. No clinically significant valvular regurgitation in long-term cabergoline treatment for prolactinoma. Clin Endocrinol. 2012;77:275–280.
- Elenkova A, Shabani R, Kalinov K, et al. Increased prevalence of subclinical cardiac valve fibrosis in patients with prolactinomas on long-term bromocriptine and cabergoline treatment. Eur J Endocrinol. 2012;167:17–25.
- Boguszewski CL, Dos Santos CM, Sakamoto KS, et al. A comparison of cabergoline and bromocriptine on the risk of valvular heart disease in patients with prolactinomas. Pituitary. 2012;15:44–49.
- Drake WM, Stiles CE, Howlett TA, et al. A cross-sectional study of the prevalence of cardiac valvular abnormalities in hyperprolactinemic patients treated with ergot-derived dopamine agonists. J Clin Endocrinol Metab. 2014;99:90–96.
- Liang W, Gao L, Li N, et al. Efficacy and safety of bromocriptine-QR in type 2 diabetes: a systematic review and meta-analysis. Horm Metab Res. 2015;47:805–812.
- Kalsbeek A, Scheer FA, Perreau-Lenz S, et al. Circadian disruption and SCN control of energy metabolism. FEBS Lett. 2011;585:1412–1426.
- Reutrakul S, Van Cauter E. Interactions between sleep, circadian function, and glucose metabolism: implications for risk and severity of diabetes. Ann N Y Acad Sci. 2014;1311:151–173.
- Cipolla-Neto J, Amaral FG, Afeche SC, et al. Melatonin, energy metabolism, and obesity: a review. J Pineal Res. 2014;56:371–381.
- Antunes LC, Levandovski R, Dantas G, et al. Obesity and shift work: chronobiological aspects. Nutr Res Rev. 2010;23:155–168.
- Steylen PM, Van Der Heijden FM, Hoogendijk WJ, et al. Glycosylated hemoglobin as a screening test for hyperglycemia in antipsychotic-treated patients: a follow-up study. Diabetes Metab Syndr Obes. 2015;8:57–63.
- Musil R, Obermeier M, Russ P, et al. Weight gain and antipsychotics: a drug safety review. Expert Opin Drug Saf. 2015;14:73–96.
- Reynolds GP, Kirk SL. Metabolic side effects of antipsychotic drug treatment–pharmacological mechanisms. Pharmacol Ther. 2010;125:169–179.
- Newcomer JW. Second-generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs. 2005;19(Suppl1):1–93.
- Mallis MM, DeRoshia CW. Circadian rhythms, sleep, and performance in space. Aviat Space Environ Med. 2005;76(6Suppl):B94–107.