
Abstract
Objective: To compare the pharmacokinetics (PK), safety and tolerability of biosimilar infliximab (CT-P13 [Remsima®, Inflectra®]) with two formulations of the reference medicinal product (RMP) (Remicade®) from either Europe (EU-RMP) or the USA (US-RMP). Methods: This was a double-blind, three-arm, parallel-group study (EudraCT number: 2013–003173-10). Healthy subjects received single doses (5 mg/kg) of CT-P13 (n = 71), EU-RMP (n = 71) or US-RMP (n = 71). The primary objective was to compare the PK profiles for the three formulations. Assessments of comparative safety and tolerability were secondary objectives. Results: Baseline demographics were well balanced across the three groups. Primary end points (Cmax, AUClast and AUCinf) were equivalent between all formulations (CT-P13 vs EU-RMP; CT-P13 vs US-RMP; EU-RMP vs US-RMP). All other PK end points supported the high similarity of the three treatments. Tolerability profiles of the formulations were similar. Conclusion: The PK profile of CT-P13 is highly similar to EU-RMP and US-RMP. All three formulations were equally well tolerated.
A biosimilar is a biologic drug that is highly similar to an already approved biologic, or ‘reference medicinal product (RMP)’. However, because biologics are produced in living systems, have complex structures and undergo various post-translational modifications, it is theoretically impossible for a biosimilar to be identical to its RMP, or for any RMP biologic product to be identical from batch to batch. With this in mind, regulatory authorities have defined strict criteria requiring a biosimilar to be sufficiently similar to its RMP to permit clinical use Citation[1–3]. One of these criteria is the demonstration of equivalence in clinical pharmacokinetics (PK).
Infliximab is a chimeric full-length bivalent IgG1 mAb that inhibits the proinflammatory cytokine, TNF. Neutralization of TNF signaling by infliximab and other TNF antagonists has emerged as a successful therapeutic strategy in many inflammatory diseases including rheumatoid arthritis (RA), ankylosing spondylitis (AS), Crohn’s disease, ulcerative colitis, psoriatic arthritis and psoriasis Citation[4,5].
CT-P13 (Remsima®, Inflectra®) is a biosimilar infliximab that was approved in Europe for the same indications as the infliximab RMP (Remicade®) in September 2013. PK equivalence of CT-P13 and RMP approved in the European Union (EU-RMP) was established in a randomized, parallel-group, Phase I study in patients with AS Citation[6]. The primary end points of the study were area under the serum concentration–time curve (AUC) and maximum serum concentration (Cmax) at steady state (Cmax,ss), assessed between week 22 and 30 of treatment. PK equivalence was demonstrated because the 90% CIs for the ratios of these parameters (CT-P13/EU-RMP) were within the predefined equivalence margins of 80–125%. Efficacy equivalence of CT-P13 and EU-RMP was demonstrated in a randomized, parallel-group, Phase III study in patients with RA Citation[7]. The primary end point was the American College of Rheumatology 20% (ACR20) response at week 30. Therapeutic equivalence of clinical response according to ACR20 criteria was demonstrated because the 95% CIs for the predefined treatment difference between CT-P13 and EU-RMP were within ±15%.
Biosimilar guidelines issued by the US FDA state that at least one clinical PK study should include an adequate comparison of the biosimilar with the RMP licensed in the USA (US-RMP), otherwise bridging data are required for justification to use data from a clinical study comparing the proposed biosimilar product with a non-US-licensed product Citation[2,3]. The guidelines further state that PK studies should be performed in healthy subjects, as long as the drugs can be safely administered to this population. This is because there is likely to be less PK variability in such subjects than in patient populations who are likely to have multiple confounding factors, including the presence of the underlying disease and concomitant medication use Citation[1,2]. To address this need, the current study compared the PK among CT-P13, EU-RMP and US-RMP in healthy volunteers. The safety and tolerability of these three infliximab formulations were also assessed.
Methods
Study subjects & design
Healthy male and female volunteers aged 18–55 years participated in this randomized, double-blind, three-arm, parallel-group, single-dose, Phase I study (EudraCT number: 2013-003173-10). A healthy state for study volunteers was defined as the absence of any clinically relevant abnormalities, as identified by a detailed medical history and full physical examination including blood pressure and pulse rate measurement, 12-lead ECG and clinical laboratory tests. Eligible subjects had a BMI of 18.0–29.9 kg/m2 and a body weight of 55–99.9 kg. Female subjects were of non-childbearing potential (either surgically sterile or ≥12 months post-menopausal). Male subjects and their female partners of child-bearing potential used suitable contraception.
Exclusion criteria included: recent surgical intervention; previous exposure to a mAb or current use of a biologic; evidence of drug abuse or excessive alcohol consumption; moderate/heavy smoking; recent live vaccination within 30 days prior to randomization; recent use of over-the-counter medications, use of prescription drug or herbal remedies that could affect study measurements; and active or latent tuberculosis (TB) or a history of TB. Eligible subjects were required to have a negative result on an IFN-γ release assay (IGRA) using Quantiferon TB Gold-IT test.
Subjects enrolled were randomized 1:1: to receive a single dose (5 mg/kg) of CT-P13 (Celltrion, Inc., Republic of Korea), EU-RMP (Schering-Plough, Ireland) or US-RMP (Janssen Biotech, Inc., PA, USA) by intravenous (IV) infusion over 120 min on day 1 of the study and were followed up for 57 days afterward. Randomization was stratified by gender in order to achieve a balance of males and females across the three treatment arms. Subjects were premedicated with IV hydrocortisone (100 mg), oral paracetamol (1000 mg) and oral loratadine (10 mg) 30–60 min before infusion with study medication.
Subjects were admitted to the study center (PAREXEL Early Phase Clinical Unit, Berlin, Germany) on day -1 and were discharged on day 4. Subsequent visits were held on an outpatient basis, up to and including the end-of-study visit on day 57.
The study was conducted in accordance with the principles of the Declaration of Helsinki and International Conference on Harmonisation good clinical practice guidelines. The study protocol and other documentation were approved by an independent ethics committee. All subjects provided written informed consent.
Study objectives
The primary objective of the study was to evaluate and compare the PK profiles of the three infliximab formulations (CT-P13 vs EU-RMP; CT-P13 vs US-RMP and EU-RMP vs US-RMP). Secondary objectives included assessment of the safety and tolerability of the three formulations.
Pharmacokinetic assessments & end points
Blood samples for PK analysis were obtained immediately before infusion of study drug (predose), at the end of infusion, and 1 h after the end of infusion. Further samples were obtained at 6, 12, 24, 48 and 72 h after the start of infusion and again on days 8, 15, 29, 43 and 57. Blood samples were processed and serum was then analyzed for concentrations of CT-P13, EU-RMP and US-RMP using a validated method on a flow-through immunoassay platform (GyrolabxP; Gyros AB, Sweden).
The primary objective was to evaluate whether the three infliximab formulations were equivalent for Cmax, AUC from time zero to last quantifiable concentration (AUClast) and from time zero to infinity (AUCinf). Secondary PK end points included volume of distribution during the terminal phase (Vz), terminal half-life (t1/2), total body clearance (CL) and mean residence time (MRT).
Safety & tolerability assessments
Safety and tolerability were assessed throughout the study. These assessments included adverse events (AEs), concomitant medications, clinical laboratory testing (including hematology, chemistry and urinalysis), vital signs (blood pressure, heart rate, body temperature and respiratory rate), 12-lead ECG, physical examination and signs and symptoms of TB infection.
Statistical analyses
A sample size of 201 (67 subjects per group) was calculated to provide 90% overall power to show similarity in PK among the three groups using a 90% CI approach, based on an 80–125% equivalence margin for a 20% difference. Since PK data for the RMP had not been reported in healthy subjects, an inter-subject coefficient of variation of 35% for Cmax and AUCinf was assumed based on data in patients with RA Citation[8,9]. A two one-sided t-test with 5% significance levels was applied using geometric mean ratio. A 5% dropout rate was anticipated and hence a study population of 213 subjects (71 in each group) was planned.
The PK analysis set included all subjects who received study treatment, provided at least one PK sample with a concentration above the lower limit of quantification, and had no major protocol violation that may have affected PK measurements. The safety analysis set included all subjects who received study treatment.
Statistical analysis of the log-transformed primary PK end points was based on an analysis of covariance (ANCOVA) model with treatment as a fixed effect and gender as covariate. The differences in least squares means and associated 90% CIs were determined for the following comparisons: CT-P13 versus EU-RMP, CT-P13 versus US-RMP and EU-RMP versus US-RMP. The ratio of geometric means and 90% CIs for these ratios was determined by back transformation. Similarity of systemic exposure was considered to be demonstrated if the 90% CI for the ratio of geometric means was within the interval of 80–125%.
All reported terms for AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA, version 16.1). Grading of AEs was recorded based on Common Terminology Criteria for Adverse Events (CTCAE v.4.0). A treatment-emergent AE (TEAE) was defined as an AE that began or worsened in severity after administration of study treatment.
Results
Subjects
A total of 213 subjects were randomized (n = 71 in each group). All subjects received a single dose of their allocated treatment and were included in the safety analysis set. Two subjects were excluded from the PK analysis set: one because of a protocol deviation (CT-P13 group; indeterminate IGRA test result) and one due to withdrawal of subject consent (US-RMP group). All other subjects completed the study .
Figure 1. Subject disposition.
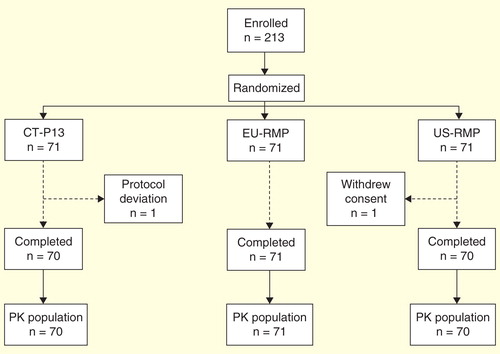
Baseline demographics were well balanced across the three groups . Subjects randomized in the study had a mean age of 40.7 years (range 18–55 years). The majority of patients in the study were male (85.9%) and Caucasian (98.6%).
Table 1. Baseline subject demographics (safety analysis set).
Pharmacokinetics
shows the mean serum concentration–time profiles for the three treatment groups. These profiles were highly similar to each other throughout the study. Geometric mean values and geometric mean ratios for the primary study end points (Cmax, AUClast and AUCinf) are shown by treatment group in .
Figure 2. Mean ± standard deviation serum concentrations of three formulations of infliximab over time.
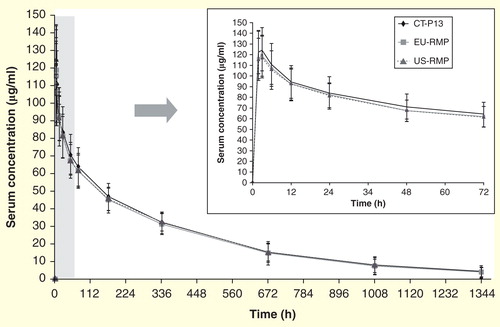
Table 2. Primary pharmacokinetic end points between three formulations of infliximab: CT-P13, EU-RMP and US-RMP.
The ratios (90% CI) of geometric means (CT-P13 to EU-RMP) were 105.03% (100.64–109.62%), 101.81% (96.36–107.56%) and 100.76% (94.37–107.59%) for Cmax, AUClast and AUCinf, respectively. For CT-P13 versus US-RMP, the ratios were 106.32% for Cmax (101.86–110.99%), 98.44% for AUClast (93.16–104.03%) and 96.48% for AUCinf (90.36–103.02%). Finally, the ratios for EU-RMP to US-RMP were 101.23% (96.99–105.65%), 96.70% (91.52–102.16%) and 95.75% (89.72–102.19%) for Cmax, AUClast and AUCinf, respectively. For all three primary end points and for each treatment comparison, the 90% CIs of the ratio of geometric means were entirely contained within the predefined equivalence limits of 80–125%.
All secondary PK end points were also similar among the three treatment groups .
Table 3. Mean (± standard deviation) values for the secondary pharmacokinetic end points (pharmacokinetic analysis set).
Safety & tolerability
All subjects were exposed to a single 5 mg/kg dose of their assigned study treatment. TEAEs were experienced by 37 (52.1%), 21 (29.6%) and 33 (46.5%) subjects in the CT-P13, EU-RMP and US-RMP groups, respectively. The most common TEAEs were nasopharyngitis, headache and rhinitis. No TEAEs led to study discontinuation. TEAEs considered by the study investigator to be related to study treatment were experienced by 28 (39.4%), 17 (23.9%) and 30 (42.3%) subjects, respectively. The common treatment-related TEAEs are shown in . Two serious AEs were reported (humerus fracture, CT-P13 group; acute cholecystitis, EU-RMP group), but neither was considered to be related to study treatment.
Table 4. Treatment-related TEAEs. Data are shown for TEAEs that occurred in ≥3% of subjects in any treatment group (safety analysis set).
Almost all treatment-related TEAEs were considered by the study investigator to be of CTCAE grade 1 or 2 in intensity. There was one treatment-related TEAE of CTCAE grade 3 (increased serum alanine transaminase, US-RMP group). No grade 4 or 5 TEAEs were reported. All treatment-related TEAEs resolved by the end of the study.
No infusion-related reaction, hypersensitivity or anaphylactic reaction TEAEs (either related or unrelated to treatment) were reported. There were no deaths and no cases of cardiovascular disorder, malignancy or lymphoma were reported. Vital sign measurements, 12-lead ECG and physical examination revealed no notable changes.
No signs or symptoms of TB were reported by any subjects. One subject in the CT-P13 group had an indeterminate IGRA test result at screening. A repeated IGRA test became positive 6 days later, and another test 33 days after screening rendered a negative result. The subject was withdrawn 7 days after dosing because of a major protocol violation (randomization with an indeterminate IGRA test result). The subject was followed-up for safety assessment including signs and symptoms of TB up to 8 weeks after dosing, with no specific finding except for an AE of nasopharyngitis (CTCAE grade 1) which was resolved after treatment.
Discussion
The primary objective of this double-blind, three-arm, parallel-group, single-dose, clinical study was to evaluate and compare the PK profiles of CT-P13, EU-RMP and US-RMP, all administered as a single dose of 5 mg/kg, to healthy subjects. Not only is this the first study to compare the PK of CT-P13 with its RMPs in healthy subjects, it is also, to our knowledge, the first to compare the PK of RMP manufactured in the EU and USA, and to report the PK profile of the infliximab RMP in this population.
The primary PK end points (Cmax, AUClast and AUCinf) were shown to be equivalent between all formulations studied (i.e., for CT-P13 vs EU-RMP; CT-P13 vs US-RMP and EU-RMP vs US-RMP). The statistical analysis was based on an ANCOVA model (treatment as a fixed effect and gender as a covariate). The geometric means for all primary PK end point comparisons fell within the predefined margins of 80–125%, thereby satisfying the criteria set for PK equivalence of the three pair-wise comparisons. This margin of equivalence is deemed clinically appropriate because of the wide therapeutic window and high variability of the infliximab RMP Citation[8,10]. All other PK end points, including tmax, t1/2 and CL were also similar among the three infliximab formulations tested.
To meet regulatory guidelines, the current study was conducted in healthy subjects. This is because administration of infliximab RMP to patients with RA can lead to high inter-patient variability in serum concentrations Citation[9,11]. Furthermore, concomitant therapy with methotrexate may also contribute to PK variability; higher infliximab serum concentrations have been reported in RA patients treated with methotrexate compared with those without methotrexate Citation[12–14].
The finding that the PK of CT-P13 and RMP were equivalent in healthy subjects is consistent with the results of a multiple-dose, Phase I study in AS and a multiple-dose, Phase III study in RA. In the Phase I study, equivalence of steady state PK was established between CT-P13 and EU-RMP (both at a dose of 5 mg/kg) from week 22 to 30 of treatment Citation[6]. Analysis up to the end of the parallel-group phase (week 54) confirmed that the PK profiles of CT-P13 and EU-RMP continued to be highly similar Citation[15]. Although the primary aim of the Phase III study was to assess efficacy equivalence between 3 mg/kg CT-P13 and the same dose of the EU-RMP, comparability of PK was also assessed. All PK end points were highly similar up to the time of the primary study analysis (week 30) and also to the end of the parallel-group phase (week 54) Citation[7,16].
Although the validity of cross-study comparisons of PK is inherently limited, geometric mean Cmax and AUClast values after a single 5 mg/kg dose of CT-P13 in healthy subjects (132 µg/ml and 32527 h*µg/ml, respectively) were similar to the values observed at steady state after a 5 mg/kg dose of CT-P13 in patients with AS (Cmax,ss and AUCτ 147 µg/ml and 32766 h*µg/ml, respectively) Citation[6]. Nevertheless, the values from the study in patients with AS are slightly higher than those from the current three-way PK study because the time point for PK assessment is different in each case. Patients in the AS study had already had drug administered on four occasions and PK parameters were measured at steady state. In contrast, the current PK study involved volunteers who had not previously received the study treatments. Even though same doses were used in this PK study, treatment had been started at a different time point and the population is different. Nevertheless, there were no differences in PK between both CT-P13 and RMP.
The safety profiles displayed by single-dose CT-P13, EU-RMP and US-RMP in the current study were similar. The common TEAEs included nasopharyngitis, headache and rhinitis. No TEAEs led to discontinuation and there were no serious AEs related to treatments.
Conclusions
This study compared the PK of CT-P13 with those of RMPs approved from Europe and licensed in the USA, thereby addressing important regulatory requirements for biosimilars. CT-P13 was shown to be equivalent to both formulations of the infliximab RMP. Furthermore, EU-RMP and US-RMP were also equivalent to each other with respect to PK. All three formulations of infliximab were generally well tolerated.
Biosimilar guidelines issued by the US FDA require at least one clinical pharmacokinetic (PK) study showing a comparison of the biosimilar with the reference medical product (RMP) licensed in the USA (US-RMP).
Such PK studies should be performed in healthy subjects because there is likely to be less variability than is seen in patient populations (who are likely to have multiple confounding factors due to the presence of the underlying disease and concomitant medication use).
The study enrolled 213 healthy subjects who were randomized 1:1:1 to receive a single dose (5 mg/kg) of CT-P13 (Celltrion, Inc., Republic of Korea), European RMP (EU-RMP; Schering-Plough, Ireland) or US-RMP (Janssen Biotech, Inc., PA, USA) by intravenous infusion.
The primary objective of the study was to compare the PK profiles of three infliximab formulations. Secondary objectives included assessment of the safety and tolerability of study treatments.
The 90% CIs of the ratio of geometric means for maximum serum concentration, area under the serum concentration–time curve (AUC) from time zero to last quantifiable concentration (AUClast) and AUC from time zero to infinity (AUCinf) – the primary study end points – were entirely contained within the predefined equivalence limits of 80–125% for all treatment comparisons, confirming that the PK profiles of CT-P13, EU-RMP and US-RMP are all highly similar to each other.
The safety profiles of CT-P13, EU-RMP and US-RMP were similar. No treatment-emergent adverse events led to discontinuation and there were no serious adverse events related to treatment.
This comparison of the PK of CT-P13 with those of RMP approved from Europe and RMP licensed from the USA further emphasize the high degree of clinical similarity between CT-P13 and both RMPs, even from different manufacturing sources.
Acknowledgements
Editorial support (writing assistance, assembling tables and figures, collating author comments, grammatical editing and referencing) was provided by Rick Flemming (Aspire Scientific Limited, Bollington, UK) and was funded by Celltrion Healthcare Co., Ltd (Incheon, Republic of Korea).
Financial & competing interests disclosure
This study was conducted by Celltrion. DH Yoo is a Scientific Consultant, and has received a research grant, which was not related to biosimilars from Celltrion. W Park has received research grants and consultancy fees from Celltrion. SJ Lee and J Yun are full-time employees of Celltrion. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Notes
References
- European Medicines Agency. Guideline on similar biological medicinal products 2014. Available from: www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf [Last accessed 21 July 2015]
- US Department of Health and Human Services; Food and drug administration; center for drug evaluation and research (CDER); Center for biologics evaluation and research (CBER). Guidance for Industry. Clinical pharmacology data to support a demonstration of biosimilarity to a reference product 2014. Available from: www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM397017.pdf [Last accessed 16 March 2015]
- US Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product: Guidance for Industry 2015. Available from: www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf [Last accessed 27 May 2015]
- Kuek A, Hazleman BL, Ostor AJ. Immune-mediated inflammatory diseases (IMIDs) and biologic therapy: a medical revolution. Postgrad Med J 2007;83:251-60
- Tracey D, Klareskog L, Sasso EH, et al. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther 2008;117:244-79
- Park W, Hrycaj P, Jeka S, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis 2013;72:1605-12
- Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis 2013;72:1613-20
- Maini R, St Clair EW, Breedveld F, et al. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised Phase III trial. ATTRACT Study Group. Lancet 1999;354:1932-9
- St Clair EW, Wagner CL, Fasanmade AA, et al. The relationship of serum infliximab concentrations to clinical improvement in rheumatoid arthritis: results from ATTRACT, a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2002;46:1451-9
- Fasanmade AA, Adedokun OJ, Ford J, et al. Population pharmacokinetic analysis of infliximab in patients with ulcerative colitis. Eur J Clin Pharmacol 2009;65:1211-28
- Wolbink GJ, Voskuyl AE, Lems WF, et al. Relationship between serum trough infliximab levels, pretreatment C reactive protein levels, and clinical response to infliximab treatment in patients with rheumatoid arthritis. Ann Rheum Dis 2005;64:704-7
- Maini RN, Breedveld FC, Kalden JR, et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheum 1998;41:1552-63
- Klotz U, Teml A, Schwab M. Clinical pharmacokinetics and use of infliximab. Clin Pharmacokinet 2007;46:645-60
- Mori S. A relationship between pharmacokinetics (PK) and the efficacy of infliximab for patients with rheumatoid arthritis: characterization of infliximab-resistant cases and PK-based modified therapy. Mod Rheumatol 2007;17:83-91
- Park W, Jaworski J, Brzezicki J, et al. A randomised, double-blind, parallel-group, Phase I study comparing the pharmacokinetics, safety and efficacy of CT-P13 and infliximab in patients with active ankylosing spondylitis: 54 week results from the PLANETAS study. Ann Rheum Dis 2013;72(Suppl 3):516 [FRI0421]
- Yoo DH, Racewicz A, Brzezicki J, et al. A Phase III randomised controlled trial to compare CT-P13 with infliximab in patients with active rheumatoid arthritis: 54 week results from the PLANETRA study. Ann Rheum Dis 2013;72(Suppl 3):[OP0068]