Abstract
Obesity is increasing at an epidemic rate in women of reproductive age. Not only does obesity during pregnancy lead to increased maternal health concerns, it is also linked to an increase in adiposity and components of the metabolic syndrome in the children and grandchildren of obese women. The potential transgenerational impact of maternal obesity on the health of future generations will undoubtedly result in increasing healthcare costs for society. This review will describe what is known about the specific impacts of maternal obesity on offspring in the human population as well as discuss how controlled animal experiments have shed light on the specific physiological mechanisms involved. Furthermore, preliminary experiments are presented describing potential dietary methods for preventing obesity-induced programming of offspring health concerns in postnatal life.
Medscape: Continuing Medical Education Online
This activity has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education through the joint sponsorship of Medscape, LLC and Expert Reviews Ltd. Medscape, LLC is accredited by the ACCME to provide continuing medical education for physicians.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 70% minimum passing score and complete the evaluation at www.medscape.org/journal/expertendo; (4) view/print certificate.
Release date: 29 April 2013; Expiration date: 29 April 2014
Learning objectives
Upon completion of this activity, participants will be able to:
• Evaluate the epidemiology and risks for offspring of maternal obesity
• Analyze the effect of maternal obesity on the placenta and fetal organs
• Distinguish the preferred means to treat obesity during pregnancy
• Assess the effects of maternal obesity on the metabolism of the infant after delivery
Financial & competing interests disclosure
EDITOR
Elisa Manzotti
Publisher, Future Science Group, London, UK.
Disclosure: E Manzotti has disclosed no relevant financial relationships.
CME AUTHOR
Charles Vega, MD
Associate Professor and Residency Director, Department of Family Medicine, University of California, Irvine, CA, USA.
Disclosure: CP Vega, MD, has disclosed no relevant financial relationships.
AUTHORS AND CREDENTIALS
Stephen P Ford
Department of Animal Science, Center for the Study of Fetal Programming, University of Wyoming, Laramie, WY, USA.
Disclosure: SP Ford has disclosed no relevant financial relationships.
Nuermaimaiti Tuersunjiang
Department of Animal Science, Center for the Study of Fetal Programming, University of Wyoming, Laramie, WY, USA.
Disclosure: N Tuersunjiang has disclosed no relevant financial relationships.
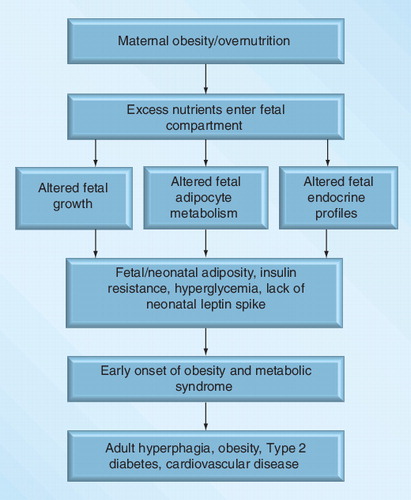
Both the developed and developing world are experiencing an epidemic within their populations that are overweight and obese in both sexes. This obesity is occurring earlier in life and affects women of childbearing age Citation[1–9]. The current global obesity epidemic, together with its associated chronic diseases, represent a major drain on healthcare resources. Estimates suggest that 18–35% of pregnant women in the USA are clinically obese Citation[10]. In the Western world, excess consumption of highly palatable diets combined with maternal obesity (MO) represent a significant problem, because of adverse effects on both maternal health and fetal development, which can result in harmful, persistent effects in offspring Citation[2,3]. It has been previously determined that being overweight is associated with poor maternal health and several chronic conditions, including Type II diabetes mellitus (T2DM), cardiovascular disease, hypertension and stroke Citation[11]. It is estimated that more than 42 million children under 5 years of age are overweight Citation[201] and childhood obesity is strongly associated with obesity in adolescence and adulthood Citation[12,13]. Furthermore, the prevalence of obesity is greatest among children of obese mothers Citation[14] and there is an independent association between maternal BMI and offspring adiposity and insulin resistance Citation[15–18]. Moreover, obesity in pregnancy is strongly associated with the development of gestational diabetes Citation[19,20]. Others report that maternal weight gain in pregnancy is independently associated with offspring adiposity Citation[21,22]. While these associations may be due to shared genetic traits that influence bodyweight or weight gain Citation[23,24], a more common interpretation is that susceptibility to obesity is partly programmed in the fetus in utero or in the neonate during early postnatal life Citation[25,26]. This is supported by the observation that birthweight, neonatal fat mass and BMI are significantly better correlated with maternal BMI during early pregnancy than with paternal BMI Citation[23,27–31]. Maternal diabetes and offspring insulin resistance and risk of T2DM are similarly linked and some authors also report an independent relationship between maternal diabetes and offspring obesity Citation[32]. Cardiovascular traits in children at the age of 9 years old have also been linked to maternal weight during pregnancy in a recent study Citation[33], where greater maternal weight was positively correlated with more adverse cardiovascular risk factors. Specifically, children who were born to overweight mothers showed higher systolic blood pressure and lower high-density lipoprotein cholesterol levels compared with children who were born to lean mothers Citation[33]. Therefore, there is strong evidence to suggest that the fetal/early postnatal environment associated with MO and diabetes leads to increased risk of obesity, T2DM and coronary heart disease in the offspring. This review will summarize our current knowledge of specific impacts of MO on offspring, using human epidemiological data as well as well-controlled animal studies to discern the mechanisms involved, as well as a discussion of potential strategies to ameliorate these effects.
Evidence that MO leads to offspring obesity
Two notable maternal conditions that predispose offspring to increased size and adiposity at birth are MO and maternal diabetes. Women with elevated pre-pregnancy maternal BMI, an indirect measure of fatness and plasma triglycerides tend to give birth to infants with higher birthweights Citation[34]. These heavier babies are not lean, having an increased bodyfat and fat mass in comparison with infants born to women of normal weight Citation[35]. In this regard, Pederson proposed a ‘hyperglycemia-hyperinsulinism’ pathway to explain the observation that offspring of diabetic mothers tend to exhibit higher birthweights Citation[36]. Furthermore, large-for-gestational-age fetuses have higher proportions of total bodyfat and relatively lower lean body mass than infants who are of an appropriate weight for gestational age Citation[37,38]. Thus, fetal hyperinsulinemia may exert lasting influences on body composition by increasing fat-cell size or number in early life leading to overweight and obesity in adolescence. Evidence for this hypothesis was presented by Silverman et al. who reported that amniotic fluid insulin levels, which reflected fetal pancreatic insulin production, correlated with obesity of offspring of diabetic mothers during adolescence Citation[39]. Among Pima Indians, siblings that were exposed to maternal diabetes in utero had a higher BMI from ages 9 to 24 years than unexposed siblings Citation[40], downplaying the roles of both shared genes and postnatal environment. Furthermore, direct associations have been reported between higher birthweight and the attainment of higher BMI in later life Citation[29,41]. In a study of 14,000 US adolescents, it was shown that for every 1 kg increase in birthweight among full term infants, there was a 50% increase in the risk of being overweight at ages 9–14 years Citation[42]. Children who were born to obese mothers (BMI ≥30 in first trimester) have been shown to develop obesity at twice the rate of children of nonobese mothers at the ages of 2-, 3- and 4-years in a retrospective cohort study Citation[15]. These data are consistent with the hypothesis that fetal exposure to maternal nutrient excess and obesity and to elevated insulin concentrations results in long-term impacts on bodyweight regulation.
Animal models of MO
Studies in animal models show strong parallels with the human observational studies as reviewed by Poston Citation[32]. They have also provided strong evidence that maternal overnutrition can contribute to this relationship Citation[43,44]. The association between maternal nutritional ‘excess’ and offspring obesity and insulin resistance, sometimes referred to as the ‘Developmental Overnutrition Hypothesis’, has focused on the programming role of ‘fuels’ that can pass directly from the mother to fetus. Maternal hyperglycemia stimulates fetal insulin synthesis and increases fetal adiposity, which may permanently influence fetal adipocyte mass Citation[45,46]. Maternal triglycerides, elevated in obese and insulin resistant women, do not cross the placenta but are hydrolyzed by placental lipases to fatty acids Citation[47] and may thereby affect fetal fuel supply. Indeed, several reports demonstrate independent associations between insulin resistance or plasma triglycerides and offspring adiposity Citation[48,49]. In this regard, Zhu et al. reported that MO in the sheep was associated with increased placental fatty acid transporter activity, mRNA expression and protein content, which was associated with marked elevations in cholesterol and fatty acids in fetal blood Citation[50]. Other studies in pregnant women and also in nonhuman primates have implicated the inflammatory state associated with MO, which may lead to activation of the hypothalamic–pituitary–adrenal pathway and persistent influences on offspring metabolic function Citation[26,51].
Animal models of maternal diabetes have focused on the role of neonatal hyperinsulinemia in the stimulation of pancreatic β-cell hyperplasia and eventual β-cell failure and also persistent effects on the developing hypothalamus leading to increased food intake and offspring obesity later in life Citation[52–55]. Although some human studies reported no differences in food intake of children born to obese mothers or nonobese mothers, the children of obese mothers had an increased percentage of abdominal fat compared with children of nonobese mothers Citation[56]. Gluck et al. concluded that children’s energy intake was closely associated with their mother’s energy intake, and thus was a strong predictor for their children’s energy intake Citation[57]. Furthermore, there are large bodies of data demonstrating that the food preference of children can be inherited from their mother as reviewed by Kral and Rauh Citation[58]. The relatively few animal studies addressing the consequences to offspring of MO combined with high caloric intakes during pregnancy and lactation have mostly been conducted in rodents. The data produced in rodents bolster the view that a diet-induced MO (either maternal cafeteria or high-fat diets) predisposes offspring to obesity and its related pathophysiology’s including obesity, insulin and leptin resistance, hypertension, fatty pancreatic disease, hepatic steatosis and nonalcoholic fatty liver disease, as reviewed by Li et al. Citation[59]. Samper et al. reported that feeding the cafeteria diet resulted in increased inflammation in white fat, brown fat and the liver compared with feeding the high fat diet or a control diet Citation[60].
The authors have used a sheep model to investigate the effects of maternal diet-induced obesity on fetal growth and development at mid-gestation, and metabolic health in their offspring. The authors have chosen to simply feed a highly palatable diet in excess of requirements to induce obesity, as ewes will eat in excess of requirements. The sheep is an appropriate animal model to study the impacts of maternal overnutrition/obesity on fetuses and offspring, as it exhibits many similarities with humans, including its willingness to overeat. The fetal sheep has a metabolism similar to the fetal human as shown by the large number of studies worldwide Citation[61–69]. The importance and relevance of all the metabolic studies is that the fetal sheep, like the fetal human, is dependent on glucose for its major source of energy. Furthermore, both the sheep and human produce well developed precocial offspring, exhibit the same newborn-to-maternal weight ratios, and exhibit the same temporal pattern of fetal tissue and organ development throughout pregnancy Citation[70]. Furthermore, investigators worldwide have utilized the sheep as a biomedical model to conduct controlled studies on human pregnancy. Our studies on the impacts of maternal overnutrition/obesity in the ewe on fetal growth and development and offspring health are conducted with animals from a closed flock at the Center for the Study of Fetal Programming, at the University of Wyoming (WY, USA). Ewes of similar breeding, size, age and weight are fed to National Research Council (NRC) dietary recommendation Citation[71] both before and throughout pregnancy and lactation, and female offspring to be used in ongoing studies are housed together and fed only to requirements from weaning to maturity. This provides assurance that animals have not been exposed to highly variable environments prior to use in ongoing studies and thus, limits the chance of markedly different environmental (epigenetic) influences on study results. Furthermore, all ewes in each experiment are bred to a single intact ram to minimize paternal effects.
Our approach to studying the impacts of MO on fetal growth and development and offspring health, was to feed a highly palatable diet at either 100% (control [CON]) or 150% (MO) of requirements from 60 days before conception through pregnancy Citation[72]. Composition of the experimental diet was: dry matter (DM), 88.54%; neutral detergent fiber, 24.09% of DM; acid detergent fiber, 9.99% of DM; crude protein, 17.39% of DM; in vivo DM digestibility, 93.92%. Using this approach, the bodyweight of MO ewes increases by ~30% from diet initiation to conception, with an additional 20–30% increase in weight from conception to term, while CON ewes exhibit a normal gestational weight gain of only 10–15% Citation[55]. These studies demonstrated that fetuses gestated by MO ewes are approximately 30% heavier than CON fetuses by mid-gestation and exhibited markedly increased pancreatic weights (236% increase) and β-cell numbers compared with fetuses from CON ewes Citation[73], while by late-gestation, pancreatic weights, β-cell numbers and plasma insulin concentrations were markedly lower in fetuses gestated by MO than CON ewes Citation[74]. The authors previously reported that the mitotic rate of β-cells in the pancreas of MO fetuses dramatically increased by mid-gestation Citation[73]; while increased β-cell apoptosis resulted in a reduced number of β-cells in the pancreas of MO fetuses compared with control fetuses at late-gestation Citation[74]. Furthermore, fetuses from MO ewes exhibited increased blood concentrations of IGF-1, cortisol, glucose, cholesterol and fatty acids as well as markedly increased perirenal fat depots compared with fetuses from CON ewes Citation[50,73]. At birth, lambs born to MO ewes exhibited markedly greater adiposity as determined by dual x-ray absorptiometry than lambs from CON ewes Citation[61]. Furthermore, both male and female offspring born to MO mothers exhibit markedly increased appetites, growth rates, adiposity and insulin resistance when subjected to ad libitum feeding at maturity compared with similarly aged offspring from CON ewes Citation[55]. Interestingly, if MO and CON offspring are maintained together from weaning to maturity and only fed to NRC requirements they exhibit no perceptible differences in growth rate or adiposity. These data suggest that our previously proposed requirement for a ‘two hit’ exposure for developmental programming of offspring obesity Citation[75] may be required to illicit clear differences in postnatal metabolism and body composition, the first being exposure to MO, and the second occurring as a result of ad libitum feeding in postnatal life.
Currently proposed intervention strategies
Based on previously presented animal studies, it might be assumed that interventions that improve maternal glucose homeostasis or insulin resistance in obese/or diabetic pregnancies would therefore be expected to reduce the risk of obesity in offspring, but few relevant studies are reported. In a retrospective study, which supports the ‘developmental origins’ hypothesis, treatment (with diet or insulin) of mothers with gestational diabetes eliminated the strong positive association between gestational diabetes and offspring obesity at 5–7 years of age Citation[76]. Several interventional approaches currently under evaluation to improve pregnancy outcome in obese women may theoretically reduce the risk of obesity and diabetes in the offspring. These include lifestyle, for example, diet and physical activity strategies as in the ongoing UPBEAT study in obese women Citation[26], as well as the use of pharmacological agents to improve glucose tolerance Citation[77,78]. The use of pharmacological agents like metformin and insulin in mothers with gestational diabetes has reduced gestational weight gain to a normal range Citation[78]. Furthermore, the children from mothers who were treated with insulin and metformin had a normal bodyfat percentage at 2 years of age Citation[79]. Metformin is also used in treatment of polycystic ovarian syndrome, and a study by Carlsen et al. showed that women who received metformin treatment gained less weight during pregnancy, however, the children born to metformin treated mothers weighed more at 1 year of age compared with children of mothers who only received placebo Citation[80]. Recently, several maternal obese intervention (MOI) studies showed that lifestyle interventions including dietary interventions during pregnancy in obese women resulted in similar gestational weight gain to control women during pregnancy, but did not reduce the risk of large-for-gestational-age newborns Citation[81–84]. A meta-analysis on human MOI studies by Thangaratinam et al. reported that the dietary intervention studies resulted in better pregnancy outcomes among different intervention methods Citation[85]. The authors have recently undertaken studies to determine if an early pregnancy reduction in maternal nutrition to requirements in overnourished/obese ewes will reverse or attenuate the observed negative impacts of MO on fetal growth, adiposity and organ development, and therefore potentially reduce the incidence of obesity, insulin resistance and other components of the metabolic syndrome observed in the resulting offspring. It is felt that the effects of a simple reduction in dietary intake should be evaluated first, before pharmacologic agents are considered for administration to obese pregnant women to increase insulin sensitivity and reduce blood glucose.
Previous studies in our laboratory demonstrated that maternal undernutrition (50% global undernutrition) starting at day 28 of gestation resulted in fetal intrauterine growth restriction (IUGR) by mid-gestation Citation[86] and the resultant offspring exhibited significant metabolic and cardiovascular disturbances as adults (i.e., they exhibited hyperphagia, increased insulin resistance and adiposity and were hypertensive) Citation[87]. It was thus predicted that a dietary intervention where obesogenic diets were reduced from 150 to 100% of NRC requirements (MOI) beginning on day 28 of gestation would be early enough to correct the negative impacts of maternal overnutrition/obesity on the fetus and resulting offspring. Day 28 of pregnancy in the sheep is equivalent to approximately day 50 in human pregnancy, which is approximately the time when women confirm they are pregnant and early enough for a doctor to provide overweight/obese women with a corrective dietary regimen.
Using this MOI model, the authors have investigated the outcomes of early maternal dietary intervention on growth, organ development and hormonal profiles of mid- and late-gestation fetuses and their mothers. The authors found that early maternal dietary intervention reduced the MO-induced increases in baseline cortisol, glucose and insulin in maternal and fetal blood , as well as maternal insulin resistance at both mid- and late-gestation. The observed improvements in endocrine profiles of MOI mothers and fetuses are important in that the offspring born to MOI mothers might be less subjected to programming of metabolic syndrome in postnatal life due to reduced level of these hormones to control level. Perhaps more importantly, the authors were successful in eliminating MO-induced fetal macrosomia during mid-gestation. The reduction in fetal weight at mid-gestation was associated with reduced organ weights in MOI fetuses to CON levels, specifically right and left ventricles of the heart, kidney, pancreas and perirenal fat weights were significantly reduced in MOI fetuses when compared with MO fetuses at mid-gestation . At day 135, while fetal weight was not statistically different between CON, MO and MOI fetuses, MO fetuses continued to exhibited greater left ventricular weights and thicknesses, right ventricular thicknesses, total kidney weight, and total perirenal fat, while pancreatic weights were reduced when compared with CON fetuses . Interestingly, weights and thicknesses of these organs and tissues were returned to CON levels in the MOI fetuses . The data provide the first indication that alterations in fetal organ and tissue growth and development could be prevented by early pregnancy MOI in the face of MO. The authors are currently evaluating the offspring from CON, MO and MOI mothers to determine whether reducing maternal nutrition to recommended levels in early pregnancy of overnourished/obese ewes prevents endocrine and metabolic disturbances in these offspring in adult life.
The hypothalamus, leptin & appetite control
Hypothalamic appetitic centers are programmed during prenatal development to maintain a balance between the hypothalamic orexigenic peptides, neuropeptide-Y and agouti-related protein and the anorexigenic peptides, pro-opiomelanocortin and cocaine- and amphetamine-regulated transcript to sustain proper energy homeostasis Citation[88,89]. During this critical period, neuronal extensions are stimulated from the arcuate nucleus (regarded as the principal site to monitor leptin signaling) towards the paraventricular nucleus, the lateral hypothalamus and the dorsomedial nuclei of the hypothalamus to create a circuitry that will mediate food intake and control energy balance in postnatal life Citation[90]. Development of the hypothalamic neural networks occurs prenatally during late-gestation in larger animals, such as humans and sheep Citation[91], and also in rodents; however, in rodents, continued development and maturation of these pathways is not completed until weaning Citation[92]. The network of interconnected pathways within the hypothalamic circuitry is a system that is subjected to alterations by the internal milieu Citation[93,94]. Permanent alterations in one or more of the relevant pathways, including appetite regulation, altered energy expenditure, tissue metabolism and physical activity during early development could program the development of obesity in postnatal life Citation[95]. If the fetus is exposed to either under- or over-nutrition during this time, the differentiation of the hypothalamic centers responsible for the control of food intake may be altered Citation[96], leading to persistent hyperphagia and associated obesity Citation[97]. Samuelsson et al. showed that mouse offspring born to obese mothers not only exhibited increased adiposity and hyperphagia at birth, but at 3 months of age they were insulin resistant and by 6 months of age, male offspring developed glucose intolerance Citation[98]. As previously, discussed, similar adverse outcomes were seen in adult lambs born to obese mothers. Birthweights of lambs born to obese and control animals were similar, however, dual energy x-ray absorptiometry scans revealed that lambs born to obese ewes exhibited an increased fat-to-lean mass ratio in comparison to controls Citation[75]. When these offspring were placed on an ad libitum feeding trial at 19.5 months of age, they demonstrated hyperphagia, increased adiposity and weight gain, hyperleptinemia, hyperglycemia and insulin resistance in comparison to offspring gestated by control-fed ewes Citation[55]. In addition to rodents and sheep, human offspring of obese mothers show comparable outcomes and may be predisposed to obesity and metabolic disease later in life attributable to greater adiposity and altered glucose and insulin dynamics in fetal and neonatal life Citation[75].
Transgenerational impacts of maternal overnutrition/obesity on offspring
There is now accumulating evidence that maternal diet-induced epigenetic changes can be transmitted across generations. Epigenetics refers to mechanisms that lead to long-term changes in gene expression through chemical modification to or alterations in the packaging of DNA (independent of changes in the DNA sequence) such that the capacity for transcriptional regulation is altered, impacting the fetal phenotype Citation[99]. Typically, epigenetic modifications include DNA methylation, histone modification and RNA interference, mechanisms that are beyond the scope of this review but are further described in a number of excellent review articles Citation[100–104]. Experimentation with animal models, where controlled manipulation of maternal nutrition can be accomplished, has provided solid evidence of dietary manipulation of gene expression Citation[102]. In a unique validation of epigenetic mechanisms, Vonnahme et al. simultaneously studied two flocks of genetically similar Rambouillet/Columbia ewes, derived from a single flock 30 years earlier, which confirmed that environmentally-induced placental phenotypic changes can be passed from generation to generation Citation[103]. One flock was adapted to a nomadic existence with limited nutrition, located near Baggs, Wyoming (Baggs ewes) and the other flock to a sedentary lifestyle with more than adequate nutrition, at the University of Wyoming (UW ewes). Multiparous ewes of similar age, weight and body condition score were assigned to the control (100% NRC recommendations), or a nutrient restriction (50% of the control-fed diet) diet from early to mid-gestation and necropsied. Surprisingly, fetuses gestated by UW ewes, but not those from Baggs ewes showed IUGR, as well as reduced blood glucose Citation[103], and amino acid concentrations Citation[104] compared with fetuses of UW control-fed ewes. These differences resulted from the early conversion of type A placentomes (sites of maternal to fetal nutrient delivery) to more efficient placentomal types (B, C, and D) in Baggs versus UW ewes. By increasing placentomal conversion to more efficient placentomal types, Baggs ewes were better able to provide the nutrition their fetuses required during the bout of maternal nutrient restriction. In a subsequent evaluation of offspring quality, UW and Baggs ewes were again undernourished from early to mid-gestation, as previously described Citation[103], but this time allowed to lamb Citation[105]. When placed on an ad libitum feeding trial as adults, the lambs born to UW ewes but not Baggs ewes exhibited increased appetites and adiposity, hypertension, insulin resistance, decreased nephron numbers, and reduced skeletal muscle mass when compared with their respective controls fed to requirements Citation[106,107]. In addition, recent human epidemiological evidence and appropriately designed dietary interventions in animal models suggest that abnormal nutrition in early life can influence diabetes risk in subsequent generations Citation[108]. In rats, the adult offspring of diabetic mothers maintain glucose homeostasis under basal conditions, but are unable to handle situations stressing their glucose metabolism such as pregnancy, resulting in hyperinsulinemia and hyperglycemia Citation[109]. Fetuses born to these diabetic dams in both the F0 and F1 generations show the same metabolic alterations including macrosomia, islet hyperplasia, β-cell hyperactivity, and hyperinsulinemia as their mothers suggesting transgenerational effects Citation[110]. Studies have also shown that phenotypic alterations can also occur through the paternal lineage in response to overfeeding. Human epidemiological data suggest that increased food availability during the slow prepubertal growth period in grandfathers increases the risk of cardiovascular and diabetes-related deaths in their grandchildren Citation[111–114]. In agreement with these findings, Pentinat et al. reported that 4-month old male mice, overnourished during neonatal development, exhibited obesity, insulin resistance and glucose intolerance Citation[115]. Normally fed male offspring and grand-offspring of these overnourished male mice also exhibited features of the metabolic syndrome with aging Citation[115]. Furthermore, results from this model and other animal models suggest that phenotypes progressively weaken across generations, reiterating the role of epigenetic rather than genetic modifications in mediating phenotypic alterations and metabolic dysfunction Citation[115–118]. Further investigations into epigenetic mechanisms, through human epidemiological data and animal models will undoubtedly provide insight into the molecular mechanisms whereby maternal and paternal malnutrition alters postnatal disease susceptibility and genomic imprinting.
Neonatal leptin & appetite regulation
A neonatal leptin peak that occurs between postnatal day 8 and 21 in rodents and between day 4 and 9 in sheep Citation[89,119,120] is thought to program the appropriate balance of orexigenic and anorexigenic appetitive neuropeptides and influence future leptin sensitivity Citation[89]. The authors have demonstrated that this neonatal leptin peak was eliminated in lambs born to MO mothers Citation[120] as well lambs whose grandmothers were overnourished/obese [Ford et al., Unpublished Data], suggesting a transgenerational effect. In these studies, the daughters of MO mothers were fed only to requirements from conception to maturity and throughout gestation as were the daughters of control-fed mothers. When subjected to an intravenous glucose tolerance test at mid- and late-gestation, however, the daughters of MO ewes exhibited elevated baseline glucose and insulin concentrations and markedly greater insulin resistance than daughters of control-fed mothers Citation[121]. These elevated blood glucose and insulin levels may be the result of metabolic disturbances that were developmentally programmed into the daughters of MO mothers and expressed during pregnancy, a period marked by significant adaptations in maternal pancreatic islet size, secretion capacity and level of insulin at peripheral tissues to meet fetal demands Citation[110]. Bouret et al. has recently reported that leptin is essential for normal development of axonal projections from the arcuate nucleus to surrounding hypothalamic nuclei, thus programming the hypothalamic circuitry responsible for regulating appetite during postnatal life Citation[122]. Impairment in leptin receptor signaling may adversely affect axonal projections from the arcuate nucleus to their targets, affecting energy balance. In postnatal lambs, Muhlhausler et al. reported that mRNA expression of the leptin receptor in the arcuate nucleus of the hypothalamus was inversely related to fat mass Citation[123]. Elimination of this leptin peak may predispose rodent offspring of obese overnourished mothers to increased adiposity and decreased sensitivity to leptin in adulthood Citation[89]. Likewise, lambs born to obese ewes exhibited markedly increased appetites, as well as glucose and insulin dysregulation, increased adiposity, and were hyperleptinemic when compared with offspring from control-fed ewes Citation[122]. OBF1 lambs and subsequently their offspring, OBF2 lambs, exhibited elevated plasma cortisol levels on the day of birth and the beginning of postnatal life compared with CONF1 and CONF2 lambs. Cortisol has an important role in prenatal regulation of cell proliferation and differentiation to mature fetal tissues in preparation for extra-uterine life Citation[124]. Cortisol may cause premature differentiation of adipocytes, possibly altering the timing of the neonatal leptin peak and/or the quantity of leptin secreted. This hypothesis is supported by Long et al. who reported that daughters and granddaughters of ewes administered exogenous glucocorticoids at 0.7 gestation exhibited a similar elimination of the neonatal leptin peak as seen in offspring of overnourished/obese ewes Citation[120,121,125]. Additional research will be required to elucidate the specific mechanism(s) whereby elevated cortisol in the fetal circulation suppresses leptin production in early postnatal life, thereby preventing normal development of the hypothalamic appetitic centers in postnatal life.
Placental function & inflammation
The role of placenta naturally becomes important in the programming events in MO, as any of the perturbing factors must be transmitted across the placenta in order to affect the fetus. Furthermore, placental dysfunction even in the presence of optimal maternal nutrition may result in unfavorable outcomes to fetal growth and development Citation[126]. Dubé et al. recently reported that MO is associated with increased expression of placental fatty acid transporters without altering fetal birthweights in human pregnancies at term Citation[127]. In mice, high fat diets before and during pregnancy caused marked upregulation of glucose transporter 1 and sodium-coupled neutral amino acid transporter 2 protein expression Citation[128]. In sheep, our laboratory and several other laboratories have shown that placental vascularity is reduced in overnourished fetuses than control fetuses in mid- and late-gestation Citation[129–131]. However, fatty acid and glucose transporters are upregulated by MO at mid-gestation and fetuses from obese ewes have significantly increased circulating glucose and triglyceride levels when compared with fetuses of control ewes Citation[50,132].
In human placenta, excessive inflammation can lead to adverse pregnancy outcomes such as spontaneous abortion, preterm labor and IUGR Citation[133]. During an inflammatory response, levels of activated cytotoxic/Th1 lymphocytes and macrophages are elevated at the inflammatory site Citation[134]. Proinflammatory cytokines, including TNF-α, IL-1, IL-2, IL-6, IL-8, IL-12 and IL-18 are Th1 associated cytokines that can promote the differentiation of precursor T cells into Th1 cells Citation[134]. Macrophages express specific membrane markers CD68 or CD11b/CD14 Citation[135], and membrane microbial pattern-recognition receptors such as toll-like receptor (TLR)2 and TLR4 that have been shown to be activated by free fatty acids Citation[136,137]. TNF-α, TLR2 and TLR4 regulate the inflammatory response through NF-κB p65 and c-JNK signaling pathways, both of which promote further production of proinflammatory cytokines Citation[134,138–140]. It has been reported that MO is associated with increased inflammation of the human placenta, in association with elevated macrophage populations (CD68+ and CD11b+ /CD14+ cells) and increased expression of TNF-α, IL-1 and IL-6 Citation[135]. Similarly, the authors reported Citation[140] a greater expression of inflammatory markers in the placental tissue of obese overfed ewes at mid-gestation. Interestingly, these obese overfed ewes exhibited a shortened gestation length (~5 days shorter) than controls Citation[73], suggesting inflammation associated preterm delivery.
Clinical therapeutic implications
In conclusion, MO is a serious and increasing clinical concern and methods must be developed to reduce the proven transgenerational impacts of this condition on offspring obesity and health. Controlled animal studies have provided and continue to provide significant information about obesity-induced developmental and hormonal changes in the fetus during in utero development. As indicated, these authors feel that developing dietary interventions for overnourished/obese pregnant women are a better approach than the utilization of pharmaceuticals which can have unintended consequences on mother and baby.
Expert commentary
While there has been necessary skepticism about in utero programming of human offspring, numerous human epidemiological studies as well as controlled animal studies have been conducted and confirmed that developmental programming does occur in response to a variety of maternal stimuli, including overnutrition/obesity (see for details). Currently, approximately 60% of women of reproductive age are overweight or obese. Furthermore, clinical results have reported that women who are overweight/obese at conception are the most at risk of giving birth to babies who have an increased percentage of bodyfat and go on to become insulin resistant and overweight in later life. Extensive animal research shows that the changes in the levels of the adipose tissue hormone leptin in the first weeks of life can program the setting of appetitic drive. If wrongly set, this predisposes to overeating, abnormal fat deposition, metabolic disease and shortening of life span. It is crucial to identify the signals and mechanisms involved in the transmission of this negative phenotype to offspring at the earliest time they emerge, and to develop diagnostic, preventative and therapeutic strategies to prevent it.
Five-year view
The obesity epidemic worldwide is significant and is accelerating at an astonishing rate, making this problem one of our most serious public health concerns. Thus, it is imperative that we develop increased understandings of the pathways of transmission of this obese phenotype from mother to offspring. While studies have confirmed and begun to elucidate the impacts of MO on fetal organ and tissue development in utero using relevant animal models, we now need to develop more in depth understandings of the molecular and cellular mechanisms involved. Furthermore, we need a greater understanding of the obesity-induced epigenetic mechanisms set in place to alter gene expression in affected offspring. Specifically, a greater focus on fetal hypothalamic, pancreatic and cardiovascular changes are needed to prevent altered appetite control, and the development of insulin resistance/obesity, hypertension and other components of the metabolic syndrome in postnatal life. In addition to pharmacological interventions, which may cause unintended consequences, recent data generated in humans trials and controlled animal studies have suggested that alterations in maternal diet can have a significant impact on fetal and offspring phenotype. Additional maternal nutritional interventions will be accomplished in obese/overweight women in clinical practice, as well as with animal models, providing vital information on optimal timing of dietary interventions, as well as the relative contributions of different dietary components in preventing offspring obesity and associated metabolic disease in postnatal life.
Table 1. Comparison of glucose and selected hormones in the blood of fetuses from maternal obese and maternal obese intervention mothers at mid- (day 75) and late-gestation (day 135) relative to fetuses from control mothers.
Table 2. Comparison of selected fetal characteristics of fetuses from maternal obese and maternal obese intervention mothers at mid- (day 75) and late-gestation (day 135) relative to fetuses from control mothers.
Key issues
• Maternal obesity is a significant public health concern and is increasing at an alarming rate worldwide.
• Not only does obesity during pregnancy lead to increased maternal health risks, it is also linked to increased adiposity and components of the metabolic syndrome in the children and grandchildren of obese women.
• Diet-induced maternal obesity from conception results in fetal macrosomia as well as altered growth and development of the hypothalamus, pancreas and heart.
• Fetuses gestated by obese mothers have increased adiposity, are hyperglycemic and hyperinsulinemic, and have elevated blood cortisol levels, a hormone known to alter tissue growth and differentiation.
• Offspring born to obese mothers fail to exhibit a normal leptin surge in the early postnatal period, which functions to set the hypothalamic appetite centers regulating appetite in later life.
• Adult offspring of obese mothers exhibit increased appetites, are insulin resistant and have increased adiposity, when compared with adult offspring born to lean mothers.
• Reduction in food intake to required levels in early pregnancy in overfed/obese mothers eliminates macrosomia and returns fetal tissue and organ development and endocrine patterns to control levels.
• Diet manipulation of obese women during pregnancy may be effective in the production of normal healthy offspring.
References
- Burke JP, Williams K, Gaskill SP, Hazuda HP, Haffner SM, Stern MP. Rapid rise in the incidence of Type 2 diabetes from 1987 to 1996: results from the San Antonio Heart Study. Arch. Intern. Med. 159(13), 1450–1456 (1999).
- Caballero E. Obesity, diabetes, and the metabolic syndrome: new challenges in antipsychotic drug therapy. CNS Spectr. 8(11 Suppl. 2), 19–22 (2003).
- Friedrich MJ. Epidemic of obesity expands its spread to developing countries. JAMA 287(11), 1382–1386 (2002).
- King H, Aubert RE, Herman WH. Global burden of diabetes, 1995–2025: prevalence, numerical estimates, and projections. Diabetes Care 21(9), 1414–1431 (1998).
- Littorin B, Nyström L, Gullberg B et al. Increasing body mass index at diagnosis of diabetes in young adult people during 1983–1999 in the Diabetes Incidence Study in Sweden (DISS). J. Intern. Med. 254(3), 251–256 (2003).
- Must A, Spadano J, Coakley EH, Field AE, Colditz G, Dietz WH. The disease burden associated with overweight and obesity. JAMA 282(16), 1523–1529 (1999).
- Narayan KM, Boyle JP, Geiss LS, Saaddine JB, Thompson TJ. Impact of recent increase in incidence on future diabetes burden: USA, 2005–2050. Diabetes Care 29(9), 2114–2116 (2006).
- Sorensen TIA. The changing lifestyle in the world. Diabetes Care 23(Suppl. 2), B1–B4 (2000).
- Zimmet P. The burden of Type 2 diabetes: are we doing enough? Diabetes Metab. 29(4 Pt 2), 6S9–6S18 (2003).
- ACOG Committee Opinion Number 315. Obesity in pregnancy. Obstet. Gynecol. 106(3), 671–675 (2005).
- Chopra M, Galbraith S, Darnton-Hill I. A global response to a global problem: the epidemic of overnutrition. Bull. World Health Organ. 80(12), 952–958 (2002).
- Reilly JJ, Methven E, McDowell ZC et al. Health consequences of obesity. Arch. Dis. Child. 88(9), 748–752 (2003).
- Wardle J, Brodersen NH, Cole TJ, Jarvis MJ, Boniface DR. Development of adiposity in adolescence: five year longitudinal study of an ethnically and socioeconomically diverse sample of young people in Britain. BMJ 332(7550), 1130–1135 (2006).
- Whitaker RC, Wright JA, Pepe MS, Seidel KD, Dietz WH. Predicting obesity in young adulthood from childhood and parental obesity. N. Engl. J. Med. 337(13), 869–873 (1997).
- Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birthweight, maternal obesity, and gestational diabetes mellitus. Pediatrics 115(3), e290–e296 (2005).
- Gale CR, Javaid MK, Robinson SM, Law CM, Godfrey KM, Cooper C. Maternal size in pregnancy and body composition in children. J. Clin. Endocrinol. Metab. 92(10), 3904–3911 (2007).
- Koupil I, Toivanen P. Social and early-life determinants of overweight and obesity in 18-year-old Swedish men. Int. J. Obes. (Lond.) 32(1), 73–81 (2008).
- Mingrone G, Manco M, Mora ME et al. Influence of maternal obesity on insulin sensitivity and secretion in offspring. Diabetes Care 31(9), 1872–1876 (2008).
- Dabelea D. The predisposition to obesity and diabetes in offspring of diabetic mothers. Diabetes Care 30(Suppl. 2), S169–S174 (2007).
- Vohr BR, Boney CM. Gestational diabetes: the forerunner for the development of maternal and childhood obesity and metabolic syndrome? J. Matern. Fetal. Neonatal. Med. 21(3), 149–157 (2008).
- Crozier SR, Inskip HM, Godfrey KM et al.; Southampton Women’s Survey Study Group. Weight gain in pregnancy and childhood body composition: findings from the Southampton Women’s Survey. Am. J. Clin. Nutr. 91(6), 1745–1751 (2010).
- Lawler DA, Frazer A, Lindsay RS et al. Association of existing diabetes, gestational diabetes and glycosuria in pregnancy withmacrosomia and offspring body mass index, waist and fat mass in later childhood: findings from a prospective pregnancy cohort. Diabetologia 53(1), 89–97 (2010).
- Griffiths LJ, Dezateux C, Cole TJ. Differential parental weight and height contributions to offspring birthweight and weight gain in infancy. Int. J. Epidemiol. 36(1), 104–107 (2007).
- Lawlor DA, Timpson NJ, Harbord RM et al. Exploring the developmental overnutrition hypothesis using parental-offspring associations and FTO as an instrumental variable. PLoS Med. 5(3), e33 (2008).
- Catalano PM, Presley L, Minium J, Hauguel-de Mouzon S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care 32(6), 1076–1080 (2009).
- Nelson SM, Matthews P, Poston L. Maternal metabolism and obesity: modifiable determinants of pregnancy outcome. Hum. Reprod. Update 16(3), 255–275 (2010).
- Lawlor DA, Smith GD, O’Callaghan M et al. Epidemiologic evidence for the fetal overnutrition hypothesis: findings from the mater-university study of pregnancy and its outcomes. Am. J. Epidemiol. 165(4), 418–424 (2007).
- Harvey NC, Poole JR, Javaid MK et al.; SWS Study Group. Parental determinants of neonatal body composition. J. Clin. Endocrinol. Metab. 92(2), 523–526 (2007).
- Parsons TJ, Power C, Manor O. Fetal and early life growth and body mass index from birth to early adulthood in 1958 British cohort: longitudinal study. BMJ 323(7325), 1331–1335 (2001).
- Quek CM, Koh K, Lee J. Parental body mass index: a predictor of childhood obesity? Ann. Acad. Med. Singap. 22(3), 342–347 (1993).
- Shields BM, Knight BA, Powell RJ, Hattersley AT, Wright DE. Assessing newborn body composition using principal components analysis: differences in the determinants of fat and skeletal size. BMC Pediatr. 6, 24 (2006).
- Poston L. Developmental programming and diabetes – The human experience and insight from animal models. Best Pract. Res. Clin. Endocrinol. Metab. 24(4), 541–552 (2010).
- Fraser A, Tilling K, Macdonald-Wallis C et al. Association of maternal weight gain in pregnancy with offspring obesity and metabolic and vascular traits in childhood. Circulation 121(23), 2557–2564 (2010).
- Di Cianni G, Miccoli R, Volpe L et al. Maternal triglyceride levels and newborn weight in pregnant women with normal glucose tolerance. Diabet. Med. 22(1), 21–25 (2005).
- Sewell MF, Huston-Presley L, Super DM, Catalano P. Increased neonatal fat mass, not lean body mass, is associated with maternal obesity. Am. J. Obstet. Gynecol. 195(4), 1100–1103 (2006).
- Pederson J. Weight and length at birth of infants of diabetic mothers. Acta Endocrinol. 6(4), 330–342 (1954).
- Catalano PM, Tyzbir ED, Allen SR, McBean JH, McAuliffe TL. Evaluation of fetal growth by estimation of neonatal body composition. Obstet. Gynecol. 79(1), 46–50 (1992).
- Hammami M, Walters JC, Hockman EM, Koo WW. Disproportionate alterations in body composition of large for gestational age neonates. J. Pediatr. 138(6), 817–821 (2001).
- Silverman BL, Rizzo TA, Cho NH, Metzger BE. Long-term effects of the intrauterine environment. The Northwestern University Diabetes in Pregnancy Center. Diabetes Care 21(Suppl. 2), B142–B149 (1998).
- Dabelea D, Hanson RL, Lindsay RS et al. Intrauterine exposure to diabetes conveys risks for Type 2 diabetes and obesity: a study of discordant sibships. Diabetes 49(12), 2208–2211 (2000).
- Whitaker RC, Dietz WH. Role of the prenatal environment in the development of obesity. J. Pediatr. 132(5), 768–776 (1998).
- Gillman MW, Rifas-Shiman S, Berkey CS, Field AE, Colditz GA. Maternal gestational diabetes, birthweight, and adolescent obesity. Pediatrics 111(3), e221–e226 (2003).
- Bayol SA, Simbi BH, Bertrand JA, Stickland NC. Offspring from mothers fed a ‘junk food’ diet in pregnancy and lactation exhibit exacerbated adiposity that is more pronounced in females. J. Physiol. (Lond.) 586(13), 3219–3230 (2008).
- Sullivan EL, Grove KL. Metabolic imprinting in obesity. Forum Nutr. 63, 186–194 (2010).
- Pedersen J. Weight and length at birth of infants of diabetic mothers. Acta Endocrinol. 16(4), 330–342 (1954).
- Symonds ME, Sebert SP, Budge H. The impact of diet during early life and its contribution to later disease: critical checkpoints in development and their long-term consequences for metabolic health. Proc. Nutr. Soc. 68(4), 416–421 (2009).
- Herrera E, Amusquivar E. Lipid metabolism in the fetus and the newborn. Diabetes Metab. Res. Rev. 16(3), 202–210 (2000).
- Catalano PM, Farrell K, Thomas A et al. Perinatal risk factors for childhood obesity and metabolic dysregulation. Am. J. Clin. Nutr. 90(5), 1303–1313 (2009).
- Hamilton JK, Odrobina E, Yin J, Hanley AJ, Zinman B, Retnakaran R. Maternal insulin sensitivity during pregnancy predicts infant weight gain and adiposity at 1 year of age. Obesity (Silver Spring) 18(2), 340–346 (2010).
- Zhu MJ, Ma Y, Long NM, Du M, Ford SP. Maternal obesity markedly increases placental fatty acid transporter expression and fetal blood triglycerides at midgestation in the ewe. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299(5), R1224–R1231 (2010).
- Grayson BE, Levasseur PR, Williams SM, Smith MS, Marks DL, Grove KL. Changes in melanocortin expression and inflammatory pathways in fetal offspring of nonhuman primates fed a high-fat diet. Endocrinology 151(4), 1622–1632 (2010).
- Aerts L, Sodoyez-Goffaux F, Sodoyez JC, Malaisse WJ, Van Assche FA. The diabetic intrauterine milieu has a long-lasting effect on insulin secretion by B cells and on insulin uptake by target tissues. Am. J. Obstet. Gynecol. 159(5), 1287–1292 (1988).
- Plagemann A, Harder T, Kohlhoff R, Rohde W, Dörner G. Overweight and obesity in infants of mothers with long-term insulin-dependent diabetes or gestational diabetes. Int. J. Obes. Relat. Metab. Discord 21(6), 451–456 (1997).
- Sullivan EL, Smith MS, Grove KL. Perinatal exposure to high-fat diet programs energy balance, metabolism and behavior in adulthood. Neuroendocrinology 93(1), 1–8 (2011).
- Long NM, George LA, Uthlaut AB et al. Maternal obesity and increased nutrient intake before and during gestation in the ewe results in altered growth, adiposity, and glucose tolerance in adult offspring. J. Anim. Sci. 88(11), 3546–3553 (2010).
- Francis CC, Bope AA, MaWhinney S, Czajka-Narins D, Alford BB. Body composition, dietary intake, and energy expenditure in nonobese, prepubertal children of obese and nonobese biological mothers. J. Am. Diet. Assoc. 99(1), 58–65 (1999).
- Gluck ME, Venti CA, Lindsay RS, Knowler WC, Salbe AD, Krakoff J. Maternal influence, not diabetic intrauterine environment, predicts children’s energy intake. Obesity (Silver Spring). 17(4), 772–777 (2009).
- Kral TV, Rauh EM. Eating behaviors of children in the context of their family environment. Physiol. Behav. 100(5), 567–573 (2010).
- Li M, Sloboda DM, Vickers MH. Maternal obesity and developmental programming of metabolic disorders in offspring: evidence from animal models. Exp. Diabetes Res. 2011, 592408 (2011).
- Samper BP, Vanhoose, Winfield HM et al. Cafeteria diet is a robust model of human metabolic syndrome with liver and adipose inflammation: comparison to high fat diet. Obesity 19(6), 1109–1117 (2011).
- Anderson MS, Thamotharan M, Kao D et al. Effects of acute hyperinsulinemia on insulin signal transduction and glucose transporters in ovine fetal skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288(2), R473–R481 (2005).
- Anderson MS, He J, Flowers-Ziegler J, Devaskar SU, Hay WW Jr. Effects of selective hyperglycemia and hyperinsulinemia on glucose transporters in fetal ovine skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 281(4), R1256–R1263 (2001).
- Anderson MS, Flowers-Ziegler J, Das UG, Hay WW Jr, Devaskar SU. Glucose transporter protein responses to selective hyperglycemia or hyperinsulinemia in fetal sheep. Am. J. Physiol. Regul. Integr. Comp. Physiol. 281(5), R1545–R1552 (2001).
- Battaglia FC, Meschia GE. An introduction to fetal physiology. Orlando Academic Press, Orlando, FL, USA, 1–257 (1986).
- DiGiacomo JE, Hay WW Jr. Effect of hypoinsulinemia and hyperglycemia on fetal glucose utilization. Am. J. Physiol. 259(4 Pt 1), E506–E512 (1990).
- Hay WW Jr, DiGiacomo JE, Meznarich HK, Hirst K, Zerbe G. Effects of glucose and insulin on fetal glucose oxidation and oxygen consumption. Am. J. Physiol. 256(6 Pt 1), E704–E713 (1989).
- Hay WW Jr. Regulation of placental metabolism by glucose supply. Reprod. Fert. Dev. 7(3), 365–375 (1995).
- Limesand SW, Rozance PJ, Smith D, Hay WW Jr. Increased insulin sensitivity and maintenance of glucose utilization rates in fetal sheep with placental insufficiency and intrauterine growth restriction. Am. J. Physiol. Endocrinol. Metab. 293(6), E1716–E1725 (2007).
- Wallace JM, Milne JS, Aitken RP, Hay WW Jr. Sensitivity to metabolic signals in late-gestation growth-restricted fetuses from rapidly growing adolescent sheep. Am. J. Physiol. Endocrinol. Metab. 293(5), E1233–E1241 (2007).
- Anthony RV, Scheaffer AN, Wright CD, Regnault TR. Ruminant models of prenatal growth restriction. Reprod. Suppl. 61, 183–194 (2003).
- NRC. Nutrient requirements of sheep (6th Edition). National Academy Press, Washington, DC, USA (1985).
- Zhu MJ, Han B, Tong J et al. AMP-activated protein kinase signalling pathways are down regulated and skeletal muscle development impaired in fetuses of obese, over-nourished sheep. J. Physiol. (Lond.) 586(10), 2651–2664 (2008).
- Ford SP, Zhang L, Zhu M et al. Maternal obesity accelerates fetal pancreatic beta-cell but not α-cell development in sheep: prenatal consequences. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297(3), R835–R843 (2009).
- Zhang L, Long NM, Hein SM, Ma Y, Nasthanielsz PW, Ford SP. Maternal obesity in ewesresults in reduced fetal pancreatic β-cell number in late gestation and decreased circulating insulinconcentration at term. Dom. Anim. Endocrinol. 40(1), 30–39 (2011).
- George LA, Uthlaut AB, Long NM et al. Different levels of overnutrition and weight gain during pregnancy have differential effects on fetal growth and organ development. Reprod. Biol. Endocrinol. 8, 75 (2010).
- Hillier TA, Pedula KL, Schmidt MM, Mullen JA, Charles MA, Pettitt DJ. Childhood obesity and metabolic imprinting: the ongoing effects of maternal hyperglycemia. Diabetes Care 30(9), 2287–2292 (2007).
- Feig DS, Briggs GG, Koren G. Oral antidiabetic agents in pregnancy and lactation: a paradigm shift? Ann. Pharmacother. 41(7), 1174–1180 (2007).
- Rowan JA, Hague WM, Gao W, Battin MR, Moore MP; MiG Trial Investigators. Metformin versus insulin for the treatment of gestational diabetes. N. Engl. J. Med. 358(19), 2003–2015 (2008).
- Rowan JA, Rush EC, Obolonkin V, Battin M, Wouldes T, Hague WM. Metformin in gestational diabetes: the offspring follow-up (MiG TOFU): body composition at 2 years of age. Diabetes Care 34(10), 2279–2284 (2011).
- Carlsen SM, Martinussen MP, Vanky E. Metformin’s effect on first-year weight gain: a follow-up study. Pediatrics 130(5), e1222–e1226 (2012).
- Quinlivan JA, Julania S, Lam L. Antenatal dietary interventions in obese pregnant women to restrict gestational weight gain to Institute of Medicine recommendations: a meta-analysis. Obstet. Gynecol. 118(6), 1395–1401 (2011).
- Vinter CA, Jensen DM, Ovesen P, Beck-Nielsen H, Jørgensen JS. The LiP (Lifestyle in Pregnancy) study: a randomized controlled trial of lifestyle intervention in 360 obese pregnant women. Diabetes Care 34(12), 2502–2507 (2011).
- Dodd JM, Grivell RM, Crowther CA, Robinson JS. Antenatal interventions for overweight or obese pregnant women: a systematic review of randomised trials. BJOG 117(11), 1316–1326 (2010).
- Tanentsapf I, Heitmann BL, Adegboye AR. Systematic review of clinical trials on dietary interventions to prevent excessive weight gain during pregnancy among normal weight, overweight and obese women. BMC Pregnancy Childbirth 11, 81 (2011).
- Thangaratinam S, Rogozinska E, Jolly K et al. Effects of interventions in pregnancy on maternal weight and obstetric outcomes: meta-analysis of randomised evidence. BMJ 344, e2088 (2012).
- Vonnahme KA, Hess BW, Hansen TR et al. Maternal undernutrition from early- to mid-gestation leads to growth retardation, cardiac ventricular hypertrophy, and increased liver weight in the fetal sheep. Biol. Reprod. 69(1), 133–140 (2003).
- Ford SP, Hess BW, Schwope MM et al. Maternal undernutrition during early gestation in the eweresults in altered growth, adiposity and glucose intolerance in male offspring. J. Anim. Sci. 85(Suppl. 2), 1285–1294 (2006).
- Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature 404(6778), 661–671 (2000).
- Yura S, Itoh H, Sagawa N et al. Role of premature leptin surge in obesity resulting from intrauterine undernutrition. Cell Metab. 1(6), 371–378 (2005).
- Bouret SG, Draper SJ, Simerly RB. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science 304(5667), 108–110 (2004).
- Gardner DS, Rhodes P. Developmental origins of obesity: programming of food intake or physical activity? Adv. Exp. Med. Biol. 646, 83–93 (2009).
- Grove KL, Grayson BE, Glavas MM, Xiao XQ, Smith MS. Development of metabolic systems. Physiol. Behav. 86(5), 646–660 (2005).
- Kalra SP. Appetite and bodyweight regulation: is it all in the brain? Neuron 19(2), 227–230 (1997).
- Berthoud HR, Morrison C. The brain, appetite, and obesity. Annu. Rev. Psychol. 59, 55–92 (2008).
- Taylor PD, Poston L. Developmental programming of obesity in mammals. Exp. Physiol. 92(2), 287–298 (2007).
- Ravelli GP, Stein ZA, Susser MW. Obesity in young men after famine exposure in utero and early infancy. N. Engl. J. Med. 295(7), 349–353 (1976).
- Plagemann A, Harder T, Rake A et al. Observations on the orexigenic hypothalamic neuropeptide Y-system in neonatally overfed weanling rats. J. Neuroendocrinol. 11(7), 541–546 (1999).
- Samuelsson AM, Matthews PA, Argenton M et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension 51(2), 383–392 (2008).
- Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell 128(4), 635–638 (2007).
- Bernstein E, Allis CD. RNA meets chromatin. Genes Dev. 19(14), 1635–1655 (2005).
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33(Suppl.), 245–254 (2003).
- Heerwagen MJ, Miller MR, Barbour LA, Friedman JE. Maternal obesity and fetal metabolic programming: a fertile epigenetic soil. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299(3), R711–R722 (2010).
- Vonnahme KA, Hess BW, Nijland MJ, Nathanielsz PW, Ford SP. Placentomal differentiation may compensate for maternal nutrient restriction in ewes adapted to harsh range conditions. J. Anim. Sci. 84(12), 3451–3459 (2006).
- Jobgen WS, Ford SP, Jobgen SC et al. Baggs ewes adapt to maternal undernutrition and maintain conceptus growth by maintaining fetal plasma concentrations of amino acids. J. Anim. Sci. 86(4), 820–826 (2008).
- Burt BE, Hess BW, Nathanielsz PW, Ford SP. Flock differences in the impact of maternal dietary restriction on offspring growth and glucose tolerance in female offspring. Soc. Reprod. Fertil. Suppl. 64, 411–424 (2007).
- Ford SP, Hess BW, Schwope MM et al. Maternal undernutrition during early to mid-gestation in the ewe results in altered growth, adiposity, and glucose tolerance in male offspring. J. Anim. Sci. 85(5), 1285–1294 (2007).
- Gilbert JS, Lang AL, Grant AR, Nijland MJ. Maternal nutrient restriction in sheep: hypertension and decreased nephron number in offspring at 9 months of age. J. Physiol. 565(Pt. 1), 137–147 (2005).
- Gallou-Kabani C, Junien C. Nutritional epigenomics of metabolic syndrome: new perspective against the epidemic. Diabetes 54(7), 1899–1906 (2005).
- Aerts L, Van Assche FA. Ultrastructural evaluation of B-cell recruitment in virgin and pregnant offspring of diabetic mothers. Diabetes Res. Clin. Pract. 41(1), 9–14 (1998).
- Aerts L, Van Assche FA. Animal evidence for the transgenerational development of diabetes mellitus. Int. J. Biochem. Cell Biol. 38(5–6), 894–903 (2006).
- Kaati G, Bygren LO, Pembrey M, Sjöström M. Transgenerational response to nutrition, early life circumstances and longevity. Eur. J. Hum. Genet. 15(7), 784–790 (2007).
- Pembrey ME, Bygren LO, Kaati G et al.; ALSPAC Study Team. Sex-specific, male-line transgenerational responses in humans. Eur. J. Hum. Genet. 14(2), 159–166 (2006).
- Kaati G, Bygren LO, Edvinsson S. Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur. J. Hum. Genet. 10(11), 682–688 (2002).
- Bygren LO, Kaati G, Edvinsson S. Longevity determined by paternal ancestors’ nutrition during their slow growth period. Acta Biotheor. 49(1), 53–59 (2001).
- Pentinat T, Ramon-Krauel M, Cebria J, Diaz R, Jimenez-Chillaron JC. Transgenerational inheritance of glucose intolerance in a mouse model of neonatal overnutrition. Endocrinology 151(12), 5617–5623 (2010).
- Zambrano E, Martínez-Samayoa PM, Bautista CJ et al. Sex differences in transgenerational alterations of growth and metabolism in progeny (F2) of female offspring (F1) of rats fed a low protein diet during pregnancy and lactation. J. Physiol. (Lond.) 566(Pt 1), 225–236 (2005).
- Benyshek DC, Johnston CS, Martin JF. Glucose metabolism is altered in the adequately-nourished grand-offspring (F3 generation) of rats malnourished during gestation and perinatal life. Diabetologia 49(5), 1117–1119 (2006).
- Jimenez-Chillaron JC, Isganaitis E, Charalambous M et al. Intergenerational transmission of glucose intolerance and obesity by in utero undernutrition in mice. Diabetes 58(2), 460–468 (2009).
- Kirk SL, Samuelsson AM, Argenton M et al. Maternal obesity induced by diet in rats permanently influences central processes regulating food intake in offspring. PLoS ONE 4(6), e5870 (2009).
- Long NM, Ford SP, Nathanielsz PW. Maternal obesity eliminates the neonatal lamb plasma leptin peak. J. Physiol. (Lond.) 589(Pt 6), 1455–1462 (2011).
- Shasa DR, Long NM, Nathanielsz PW, Ford SP. Maternal obesity and overnutritionis associated with reduced systemic progesterone during the estrous cycle of adult female offspring and a marked increase in insulin resistance at midpregnancy. Presented at: 44th Annual Meeting of Society for the Study of Reproduction. Portland, OR, USA, 31 July-4 August 2011.
- Bouret SG, Bates SH, Chen S, Myers MG Jr, Simerly RB. Distinct roles for specific leptin receptor signals in the development of hypothalamic feeding circuits. J. Neurosci. 32(4), 1244–1252 (2012).
- Muhlhausler BS, Adam CL, Findlay PA, Duffield JA, McMillen IC. Increased maternal nutrition alters development of the appetite-regulating network in the brain. FASEB J. 20(8), 1257–1259 (2006).
- Forhead AJ, Gillespie CE, Fowden AL. Role of cortisol in the ontogenic control of pulmonary and renal angiotensin-converting enzyme in fetal sheep near term. J. Physiol. (Lond.) 526 Pt 2, 409–416 (2000).
- Long NM, Ford SP, Nathanielsz PW. Multigenerational effects of fetal dexamethasone exposure on the hypothalamic-pituitary-adrenal axis of first- and second-generation female offspring. Am. J. Obstet. Gynecol. 208(3), 217.e1–217.e8 (2013).
- Burdge GC, Lillycrop KA. Nutrition, epigenetics, and developmental plasticity: implications for understanding human disease. Annu. Rev. Nutr. 30, 315–339 (2010).
- Dubé E, Gravel A, Martin C et al. Modulation of fatty acid transport and metabolism by maternal obesity in the human full-term placenta. Biol. Reprod. 87(1), 14 (2012).
- Jones HN, Woollett LA, Barbour N, Prasad PD, Powell TL, Jansson T. High-fat diet before and during pregnancy causes marked up-regulation of placental nutrient transport and fetal overgrowth in C57/BL6 mice. FASEB J. 23(1), 271–278 (2009).
- Ma Y, Zhu MJ, Zhang L, Hein SM, Nathanielsz PW, Ford SP. Maternal obesity and overnutrition alter fetal growth rate and cotyledonary vascularity and angiogenic factor expression in the ewe. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299(1), R249–R258 (2010).
- Redmer DA, Wallace JM, Reynolds LP. Effect of nutrient intake during pregnancy on fetal and placental growth and vascular development. Domest. Anim. Endocrinol. 27(3), 199–217 (2004).
- Reynolds LP, Borowicz PP, Vonnahme KA et al. Animal models of placental angiogenesis. Placenta 26(10), 689–708 (2005).
- Long NM, Rule DC, Zhu MJ, Nathanielsz PW, Ford SP. Maternal obesity upregulates fatty acid and glucose transporters and increases expression of enzymes mediating fatty acid biosynthesis in fetal adipose tissue depots. J. Anim. Sci. 90(7), 2201–2210 (2012).
- Orsi NM, Tribe RM. Cytokine networks and the regulation of uterine function in pregnancy and parturition. J. Neuroendocrinol. 20(4), 462–469 (2008).
- Hayden MS, Ghosh S. Shared principles in NF-κB signaling. Cell 132(3), 344–362 (2008).
- Challier JC, Basu S, Bintein T et al. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 29(3), 274–281 (2008).
- Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2(8), 675–680 (2001).
- Nguyen MT, Favelyukis S, Nguyen AK et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via toll-like receptors 2 and 4 and JNK-dependent pathways. J. Biol. Chem. 282(48), 35279–35292 (2007).
- Arima N, Kuziel WA, Grdina TA, Greene WC. IL-2-induced signal transduction involves the activation of nuclear NF-κB expression. J. Immunol. 149(1), 83–91 (1992).
- Takeda K, Noguchi T, Naguro I, Ichijo H. Apoptosis signal-regulating kinase 1 in stress and immune response. Annu. Rev. Pharmacol. Toxicol. 48, 199–225 (2008).
- Zhu MJ, Du M, Nathanielsz PW, Ford SP. Maternal obesity up-regulates inflammatory signaling pathways and enhances cytokine expression in the mid-gestation sheep placenta. Placenta 31(5), 387–391 (2010).
Website
- WHO. Global Strategy on Diet, Physical Activity and Health. www.who.int/dietphysicalactivity/childhood/en
Maternal obesity: how big an impact does it have on offspring prenatally and during postnatal life?
To obtain credit, you should first read the journal article. After reading the article, you should be able to answer the following, related, multiple-choice questions. To complete the questions (with a minimum 70% passing score) and earn continuing medical education (CME) credit, please go to www.medscape.org/journal/expertendo. Credit cannot be obtained for tests completed on paper, although you may use the worksheet below to keep a record of your answers. You must be a registered user on Medscape.org. If you are not registered on Medscape.org, please click on the New Users: Free Registration link on the left hand side of the website to register. Only one answer is correct for each question. Once you successfully answer all post-test questions you will be able to view and/or print your certificate. For questions regarding the content of this activity, contact the accredited provider, [email protected]. For technical assistance, contact [email protected]. American Medical Association's Physician's Recognition Award (AMA PRA) credits are accepted in the US as evidence of participation in CME activities. For further information on this award, please refer to http://www.ama-assn.org/ama/pub/category/2922.html. The AMA has determined that physicians not licensed in the US who participate in this CME activity are eligible for AMA PRA Category 1 Credits™. Through agreements that the AMA has made with agencies in some countries, AMA PRA credit may be acceptable as evidence of participation in CME activities. If you are not licensed in the US, please complete the questions online, print the AMA PRA CME credit certificate and present it to your national medical association for review.
Activity Evaluation: Where 1 is strongly disagree and 5 is strongly agree
1. You are seeing a 24-year-old primiparous woman for her initial prenatal examination. Her pre-pregnancy body mass index (BMI) was 34 kg/m2, and she has already gained 5 kg in the first 6 weeks of her pregnancy. What should you consider regarding the epidemiology of obesity in pregnancy and its potential complications for her offspring?
□ A The rate of obesity among pregnant women in the United States has fallen under 10%
□ B There is no established link between obesity during childhood and adulthood
□ C The effect of maternal obesity on offspring appears to be rendered insignificant by age 5 years
□ D Higher birthweight is associated with higher BMI later in life
2. What should you consider regarding placental factors and the development of fetal organs in this obese patient?
□ A Maternal triglycerides cross the placenta directly
□ B Maternal obesity is associated with increased placental vascularity
□ C Maternal obesity is associated with increased weight of the fetal heart
□ D Maternal obesity is associated with increased weight of the fetal pancreas
3. According to the current review, what is the preferred method of treatment for this patient’s obesity?
□ A Diet only
□ B Diet plus a thiazolidinedione
□ C Diet plus metformin
□ D Diet plus insulin
4. The patient eventually delivers a large-for-gestational-age infant at term. What should you consider regarding the metabolic effects of maternal obesity?
□ A The normal peak in neonatal leptin levels may be blunted
□ B The child can be expected to be highly sensitive to leptin for years
□ C Cortisol levels should be low in the neonatal period
□ D Maternal obesity generally has no effect on neonatal insulin levels