RFLP: Restriction fragment length polymorphism; SNP: Single-nucleotide polymorphism.
Hemophilia A is a common inherited X-linked bleeding disorder resulting from a wide variety of mutations in the Factor VIII (FVIII) gene located on long arm of X-chromosome at the Xq28 locus. It is characterized by deficiency in FVIII clotting activity resulting in prolonged bleeding after minor injury and spontaneous deep hematomas frequently found in the joints, muscle or CNS. The phenotypic expression of the disease is constant in families. It varies in severity and may be categorized on the basis of FVIII bioactivity into mild, moderate and severe hemophilia. Severe cases have under 1% clotting activity and present with spontaneous bleeding in the first year of life. Moderate severity is evident as bleeding after minor injuries and cases have FVIII activity between 1 and 5%. Mild cases with hemophilia present later in life, with clotting activity between 5 and 35% and have abnormal bleeding after major injuries, surgery or tooth extractions Citation[101].
The disease affects approximately one in 5000 males worldwide Citation[1]. Diagnosis of a case with hemophilia can be conveniently established with a FVIII assay in cases with a clinical and laboratory picture of a coagulation disorder. Genetic analysis is required to determine the carrier status of females and to determine the causative mutation in the male proband. In view of the high incidence as well as morbidity and mortality associated with hemophilia A, it is one of the target diseases for which screening facilities are mandatory, even in developing nations. Where resources are limited, cost-effective, low-infrastructure and low-expertise methods of genetic analysis are required to establish effective facilities. Although ideally a direct mutation detection approach is most accurate and reliable in expert hands, we feel that a PCR-restriction fragment length polymorphism (RFLP)-based approach combined with screening for FVIII inversion mutation forms an acceptable and cost-effective modality for the genetic diagnosis of carriers with hemophilia A.
Modalities of carrier detection
Indirect approach: PCR-RFLP-based linkage analysis
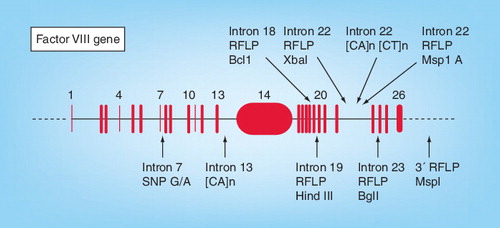
Until the early 1990s, linkage analysis was used to determine female carrier status in families with hemophilia A. The polymorphic loci used in linkage analysis are RFLP, variable number tandem repeats and short tandem repeats. depicts intragenic loci used for carrier screening. These polymorphisms are simple to detect and we use a PCR-RFLP method with gel electrophoresis and ethidium bromide staining or silver staining. Dimorphisms in the FVIII gene that can be analyzed by PCR include those detected by digestion with BclI in intron 18 Citation[3], HindIII in intron 19 Citation[4] and G/A in intron 7, which can be detected using an introduced AlwNI site Citation[5].
Using three common markers (HindIII, Bcl1 and Xba1), we have been able to determine carrier status in 77 out of 100 families registered with us. HindIII is in strong linkage disequilibrium with BclI and we found 35 families of the 53 that were informative for Bcl1 were also informative for HindIII. The fragment amplified for HindIII dimorphism analysis contains a constant HindIII restriction enzyme site and one variable site Citation[2].
Intragenic markers used included dinucleotide repeats and several dimorphisms within the FVIII gene. Dinucleotide repeats in introns 13 and 22 have been used in some studies as the first choice of markers as they have the high rates of heterozygosity Citation[6].
A dimorphism within intron 22, MspA1I, has been described which is in close proximity to XbaI dimorphism but is not in complete linkage disequilibrium and may be useful in some family studies Citation[7].
Even in the ideal set up where high-throughput assays are available for direct mutation detection, complete gene sequencing protocols are being followed and intragenic linked markers are still useful. They may be of particular value when a family has been investigated previously by linked markers and the mutation has not been identified, verified or found and in families with a large deletion, where a mutation-specific PCR product cannot readily be amplified. It is also useful for tracking the origin of de novo mutations Citation[101].
Linkage analysis limitations
The technique requires families and cannot be done on isolated individuals. It presumes that relationships as stated are correct. The frequency of recombination is a major concern and may vary from 0 to 5%. Recombination events between FVIII and the extragenic site occur in up to 5% of meioses but have not been observed between hemophilic mutations and intragenic sites. Extragenic sites should therefore be avoided for linkage analysis. Most clinically useful markers do not have heterozygosity rates of more than 50–80%. Population variations are known and different ethnic groups have variable heterozygosity rates for different markers. summarizes some of these differences for the Bcl1, HindIII and Xba1 markers. Interpretation of results is also tricky in families with sporadic cases and female relatives can only be excluded from being carriers where they do not share an allele with the affected male. Females who are linked to the affected allele may not carry the mutation when the affected male may have a de novo disease-causing mutation. Somatic mosaicism may occur in as many as 15% of probands with a point mutation and no known family history of hemophilia A Citation[8]. Chances that the mother is a carrier of a de novo disease-causing mutation are high with possibilities of a germline mutation at the time of her conception or a somatic mutation very early in embryogenesis, resulting in somatic mosaicism.
Direct mutation analysis
The FVII gene spans 186 kb with 26 exonic regions varying in size from 69 bp (exon 5) to 3.1 kb (exon 14). The mRNA is 9 kb. The most frequent mutation observed in 45% of severe cases is the inversion mutation at intron 22 Citation[9]. The intron 1 mutation is an inversion between a 1-kb sequence in intron 1 and an inverted repeat 5´ to the FVIII gene Citation[10] is also associated with a severe phenotype. The inversions can be detected by a long PCR-based protocol Citation[11,12]. Initially Southern blot and, recently, inverse PCR have also been used to detect the intron 22 inversion. We have found the inverse PCR easier to standardize than the long PCR, which is not very consistent with long-term stored or fragmented DNA.
A listing of mutation types can be found at The Hemophilia A Mutation, Structure, Test and Resource Site (HAMSTeRS) online resource Citation[102]. A total of 797 unique single-base (point) mutations have been described in the Hemophilia Mutation Database updated to 2007 Citation[101]. Total unique mutations of all types amount to 943 Citation[102]. Point mutations leading to new stop codons are associated with a severe phenotype, as are most frameshift mutations. Splice site mutations are often severe but may be mild, depending on the specific change and location. Missense mutations occur in fewer than 20% of individuals with severe hemophilia A but nearly all of those with mild or moderately severe bleeding tendencies Citation[9]. Diagnosis of these mutations requires PCR amplification of 26 exonic regions with mutation prescreening using single-strand conformation polymorphism (SSCP) or conformation-sensitive gel electrophoresis (CSGE) followed by DNA sequencing. Direct sequencing is the gold standard. Streamlined methods, appropriate infrastructure and rapid timescale of a diagnostic setting with expertise for full sequence analysis are required.
Mutation validation is mandatory to confirm it as a causative mutation. The possibility of a causative mutation lying outside the regions of the FVIII exons amplified may reduce sensitivity of this test. When a novel nucleotide change is found, caution should be exercised before deciding that it is the one responsible for disease. The entire FVIII gene should be analyzed for sequence alterations. Citation[13]. Mutation scanning and sequence analysis cannot detect gene deletions and rearrangements in females, except by quantitative methods available in limited research settings. Whereas termination, deletion and insertion mutations are frequently causative, missense and other changes may not be. A microarray approach identified 96% of known point mutations in a selected portion of the FVIII coding sequence Citation[14]. However, because of the large number of distinct hemophilic mutations, and new ones being reported worldwide, it remains unclear whether such an approach would be viable at present.
Apart from a definitive diagnosis, the advantages of the direct mutation detection in sporadic cases and incomplete, as well as noninformative, families is that once the tedious process of defining mutation in a family is accomplished, sequencing analysis for the single exon would diagnose other cases and carriers within the family. Further structural changes occurring in the FVIII protein due to mutation can be predicted and correlated with the possibility of FVIII inhibitor development in some cases Citation[15,16].
Combined approach to carrier detection
We have found that the following approach is cost effective and can be used in peripheral diagnostic laboratories in developing countries:
• Clinical evaluation, bleeding profile and FVIII assay in cases with hemophilia for diagnostic confirmation. Severity of hemophilia in the family is categorized as mild, moderate or severe;
• Pedigree analysis: An accurate family history should be obtained and summarized as a pedigree. Minimum information includes three generations: all of the proband’s first-degree relatives (parents, siblings and offspring) and second-degree relatives (aunts, uncles and grandparents) and their state of health. Complicated family histories with consanguineous matings require extended pedigrees. Ethnic background is recorded. Suspected carriers and cases with hemophilia are defined;
• FVII assay in suspected carriers and cases with hemophilia: the normal range for FVIII clotting activity is 50–150%. hemophilia A carrier females may have a FVIII clotting activity between 35 and 50%, and in 10%, lower than 35% Citation[16].
• Intron 22 inversion mutation is detected by a long PCR or inverse PCR approach in cases with moderate-to-severe hemophilia. In cases with mild hemophilia, an indirect linkage analysis is done as a first-line approach;
• Linkage analysis using PCR-RFLP for Bcl1, HindIII and Xba1 and intron 13 CA repeat analysis (in the Indian context). These may vary depending on the informativity of the marker in the population;
• Direct mutation analysis in carriers of noninformative families: We have used an SSCP screening of amplified exons with DNA sequencing of exonic segments with altered mobility. Full direct sequencing of all exonic regions is ideal and can be standardized and implemented in reference laboratories with adequate infrastructure and technical expertise. The prescribed protocol for defining causative mutations has to be followed Citation[13];
• Prenatal diagnosis: The carrier status of a woman at risk should be established prior to pregnancy or as early in a pregnancy as possible. Chorionic villus sampling at approximately 10–12 weeks of gestation or by amniocentesis usually performed at approximately 15–18 weeks’ gestation. If the karyotype is 46, XY, DNA extracted from fetal cells can be analyzed for the known mutation in families or linkage analysis for informative intragenic markers.
Genetic counseling
Genotype and phenotype testing should be discussed with parents along with accuracy and limitations of the test. Women with positive carrier results should be advised of all reproductive choices including abortion, adoption and in vitro fertilization. Risk for each testing procedure should be discussed taking into consideration the testing facilities available to the patients.
Implementation of DNA based carrier testing for prevention of genetic diseases in population involves technology development, personnel training and technique perfection. Major financial expenditure is involved. However, the impact of these measures is dependant to a large extent on the acceptability of the carrier testing Citation[17]. Facilities for testing may be available but social, economic and knowledge constraints may limit their implementation. In the course of our genetic studies and counseling we subjected cases and their families to a questionnaire assessing their socio-economic and educational status as well as their responses to counseling and testing procedure. Most of the 104 families studied were in the low socioeconomic group yet it was encouraging to note that the acceptability of the genetic carrier diagnosis as well as communication within the families was good.
The effective prevention of hemophilia, especially in developing countries, can be achieved through accurate carrier detection and prenatal diagnosis. It is encouraging to see the work of various hemophilia organizations, in our case, the Hemophilia Federation of India, which provides genetic counseling and treatment support with efforts to improve the quality of life of patients with hemophilia and their families.
Table 1. Regional and ethnic variation is allele frequency and heterozygosity rates.
Financial & competing interests disclosure
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
- Bolton-Maggs PHB, Pasi JK. Haemophilias A and B. Lancet361, 1801–1809 (2003).
- Raza ST, Husain N, Kumar A. Screening for hemophilia carriers: utility of PCR-RFLP-based polymorphism analysis. Clin. Appl. Thromb. Hemost.15, 78–83 (2009).
- Kogan SC, Doherty M, Gitschier J. An improved method for prenatal diagnosis of genetic diseases by analysis of amplified DNA sequences. Application to hemophilia A. N. Engl. J. Med.317, 985–990 (1987).
- Graham JB, Kunkel GR, Fowlkes DM, Lord ST. The utility of a HindIII polymorphism of Factor VIII examined by rapid DNA analysis. Br. J. Haematol.76, 75–79 (1990).
- Kogan S, Gitschier J. Mutations and a polymorphism in the FVIII gene discovered by denaturing gradient gel electrophoresis. Proc. Natl Acad. Sci. USA87, 2092–2096 (1990).
- Lalloz MR, Schwaab R, McVey JH, Michaelides K, Tuddenham EG. Haemophilia A diagnosis by simultaneous analysis of two variable dinucleotide tandem repeats within the Factor VIII gene. Br. J. Haematol.86, 804–809 (1994).
- Bowen DJ, De Brasi CD, Larripa IB, Collins PW. A new polymorphism in the human factor VIII gene: implications for linkage analysis in haemophilia A and for the evolution of int22h sequences. Br. J. Haematol.111, 544–548 (2000).
- Leuer M, Oldenburg J, Lavergne JM et al. Somatic mosaicism in hemophilia A: a fairly common event. Am. J. Hum. Genet.69, 75–87 (2001).
- Kaufman RJ, Antonarakis SE, Fay PJ. Factor VIII and hemophilia A. In: Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 5th edition. Colman RW, Marder VJ, Clowes AW, George JN, Goldhaber SZ (Eds). Lippincott–Raven, PA, USA, 151–175 (2006)
- Bagnall RD, Waseem N, Green PM, Giannelli F. Recurrent inversion breaking intron 1 of the Factor VIII gene is a frequent cause of severe hemophilia A. Blood99, 168–174 (2002).
- Liu Q, Nozari G, Sommer SS. Single-tube polymerase chain reaction for rapid diagnosis of the inversion hotspot of mutation in hemophilia A. Blood92, 1458–1459 (1998). Blood93, 2141 (1998) (Erratum).
- Liu Q, Sommer SS. Subcycling-PCR for multiplex long distance amplification of regions with high and low GC content: application to the inversion hotspot in the FVIII gene. Biotechniques25, 1022–1028 (1998).
- Keeney S, Mitchell M, Goodeve A. The molecular analysis of haemophilia A: a guideline from the UK haemophilia centre doctor’s organization haemophilia genetics laboratory network. Haemophilia11, 387–397(2005).
- Berber E, Leggo J, Brown C et al. DNA microarray analysis for the detection of mutations in hemophilia A. J. Thromb. Haemost.4, 1756–1762 (2006).
- Hay CR, Brown S, Collins PW, Keeling DM, Liesner R. The diagnosis and management of Factor VIII and IX inhibitors: a guideline from the United Kingdom Haemophilia Centre Doctors Organisation. Br. J. Haematol.133, 591–605 (2006).
- Salviato R, Belvini D, Radossi P et al.F8 gene mutation profile and ITT response in a cohort of Italian haemophilia A patients with inhibitors. Haemophilia13, 361–372 (2007).
- Vare Kamp I, Suurmeiher TP, Brocker-Vriends AH et al. Carrier testing and prenatal diagnosis for hemophilia: experiences and attitude of 549 potential obligate carriers. Am. J. Med. Genet.37(1), 147–154 (1990).
- Peake IR, Lillicrap DP, Boulyjenkov V et al. Haemophilia: strategies for carrier detection and prenatal diagnosis. Bull. World Health Organ71(3–4), 429–458 (1993).
- Shetty S, Ghosh K, Pathare A, Colah R, Badakare S, Mohanty D. Factor VIII and IX gene polymorphisms and carrier analysis in Indian population. Am. J. Hematol.54, 271–275 (1997).
- Chowdhury MR, Herrmann FH, Schroder W et al. Factor VIII gene polymorphisms in the Asian Indian population. Haemophilia6, 625–630 (2000).
- Srinivasan A, Mukhopadhyay S, Karim Z et al. Factor VIII gene polymorphisms in north Indian population a consensus algorithm for carrier analysis of hemophilia A. Clin. Chim. Acta325, 177–181(2002).
- Pandey GS, Phadke SR, Mittal B. Carrier analysis and prenatal diagnosis of Hemophilia A in North India. Int. J. Mol. Med.10, 661–664 (2002).
- Jayandharan G, Shaji RV, George B, Chandy M, Srivastava A. Informativeness of linkage analysis for genetic diagnosis of Hemophilia A in India. Haemophilia10, 553–559 (2004).
- Peake IR. Registry of DNA polymorphisms within or close to the human Factor VIII and Factor IX genes. for the Factor VIII/IX subcommittee of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb. Haemost.30(67), 227–280 (1992).
- Van-de-Water NS, Ridgway D, Ockelford PA. Restriction fragment length polymorphisms associated with the Factor VIII and Factor IX genes in Polynesians. J. Med. Genet.28, 171–176 (1991).
- Tuddenham GD, Goldman E, McGraw A, Kernoff PBA. Haemophilia A: carrier detection and prenatal diagnosis by linkage analysis using DNA polymorphism. J. Clin. Pathol.40, 971–977 (1987).
- Antonarakis SE, Waber PG, Kittur SD et al. Hemophilia A. Detection of molecular defects and of carriers by DNA analysis. N. Engl. J. Med.313, 842–848 (1985).
- Tagariello G, Belvini D, Salviato R et al. Experience of a single Italian center in genetic counseling for hemophilia: from linkage analysis to molecular diagnosis. Haematologica85, 525–529 (2000).
- Aseev M, Surin V, Baboev K et al. Allele frequencies and molecular diagnosis in Haemophilia A and B patients from Russia and from some Asian Republics of the former U.S.S.R. Prenat. Diagn.14, 513–522 (1994).
- Chan V, Chan TK, Liu VWS, Wong ACK. Restriction fragment length polymorphisms associated with Factor VIII: C gene in Chinese. Hum. genet.79, 128–131 (1988).
- Soares RP, Chamone DA, Bydlowski SP. Factor VIII gene inversions and polymorphisms in Brazilian patients with haemophilia A: carrier detection and prenatal diagnosis. Haemophilia7(3), 299–305 (2001).
- Nafa K, Meriane F, Reghis A et al. Investigation of factor VIII:C gene restriction fragment length polymorphisms and search for deletions in hemophiliac subjects in Algeria. Hum. Genet.84(5), 401–405 (1990).
- Zadeh-Vakilli A, Eshghi P, Rastegar G. Efficiency of Bcl1 RFLP for detection of Hemophilia A carriers in Sistan and Baluchistan Province, southeast of Iran. Iran J. Med. Sci.33(1), 33 (2008).
Websites
- Brower CL & Thompson AR. Hemophilia A www.geneclinics.org/profiles/hemo-a/details.html
- The Haemophilia A Mutation, Structure, Test and Resource Site http://europium.csc.mrc.ac.uk/WebPages/Main/main.htm