Abstract
Technologies for the correct and timely diagnosis of bloodstream infections are urgently needed. Molecular diagnostic methods have yet to have a major impact on the diagnosis of bloodstream infections; however, new methods are being developed that are beginning to address key issues. In this article, we discuss the key needs and objectives of molecular diagnostics for bloodstream infections and review some of the currently available methods and how these techniques meet key needs. We then focus on a new method that combines nucleic acid amplification with mass spectrometry in a novel approach to molecular diagnosis of bloodstream infections.
ESI: Electrospray ionization.
The primer coverage of the rDNA is represented by the gray background. Exceptions are indicated by red boxes.
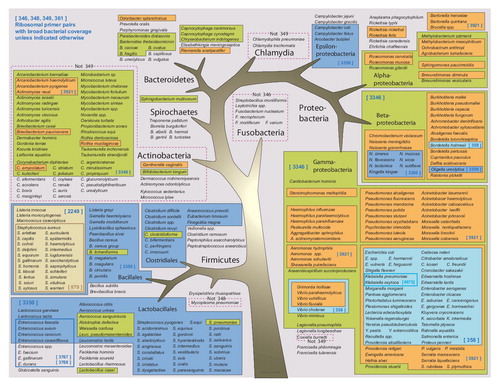
Figure 3. (A) Base composition signatures of Candida spp. Columns correspond to the three chromosomal targets and the mitochondrial rDNA target. AGCT counts for each target are shown in the boxes. (B) Color coding. To create this visualization, base compositions from each primer pair (A, G, C, T) were first mapped into a 3D space according to their relative proportions of G+C, A+C and A+G. This enabled the comparison of base compositions representing amplicons of different lengths, while reducing to three the number of independent axes. For each axis, one end is in turn associated with a primary color; thus, pure hues of cyan, magenta and yellow are associated with the maximum values of G+C, A+C and A+G, respectively. Conversely, the complementary colors are found on the other end of these axes, so pure hues of red, green, and blue are associated with the minimum values of G+C, A+C and A+G, respectively (or equivalently, with the maximum values of A+T, G+T and C+T, respectively). In between, the median values are associated with white on each one of the three axes. Individual color components can thus be extrapolated for each cell, blending the amounts of cyan/white/red, magenta/white/green and yellow/white/blue that correspond to the cell’s G+C, A+C and A+G metrics, respectively. Black ink was added for base compositions representing amplicons whose lengths differ from the consensus length at that particular locus. The process is repeated for each one of the nine loci independently and colors cannot be compared from column to column.
Colored cells represent individual base-composition signatures found for the 332 organisms tested (rows) in the nine loci (columns) that were used for broad bacterial identification. Row height occasionally varies as two or three distinct base counts may be found within the same locus for some organisms. Cells are left blank when no signature was found for the corresponding organism and locus.

A blood specimen from a patient co-infected with Staphylococcus aureus containing a mecA gene and Pseudomonas aeruginosa as analyzed by PCR/electrospray ionization/mass spectrometry. Individual spectra from six of the nine bacteria-targeted amplifications are shown. M/Z was converted to the M domain and the scales were selected to visualize all detected peaks.
CoNS: Coagulase-negative staphylococci.
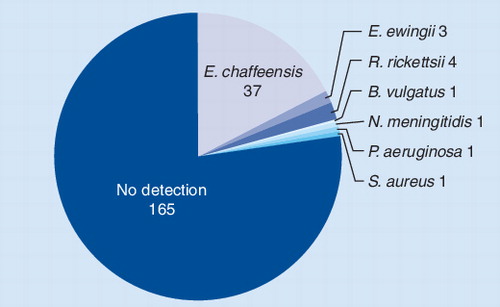
Pie chart showing the distribution of pathogens detected by PCR/ESI-MS from patients with suspected tick-borne infections. Of the 213 specimens tested, 40 (18.8%) tested positive for Ehrlichia chaffeensis or Ehrlichia ewingii.
Need for improving current diagnostic tools for bloodstream infections
There is a major unmet need to shorten and improve current laboratory procedures for the detection and identification of microorganisms responsible for bloodstream infections. An ideal diagnostic technology would identify the infecting organism(s) and also the determinants of antibiotic resistance in a timely manner so that appropriate therapy could begin as soon as possible Citation[1]. In the case of the most serious bloodstream infections associated with septic shock, speed is of the essence. It has been shown that the risk of mortality in septic shock increases substantially by the hour if appropriate antimicrobial therapy is delayed. For patients with septic shock, Kumar et al. have reported that each hour of delay in effective therapy is associated with a 7.6% decrease in survival Citation[2]. The survival rate of severe sepsis goes from approximately 80% if effective therapy is given within the first hour of the onset of symptoms to less than 10% if effective therapy is not given by 24 h. The International Committee on Surviving Sepsis has proposed a ‘strong recommendation’ that patients who show symptoms of sepsis be treated empirically within the first hour with broad-spectrum antibiotics, even though a more targeted therapy would be preferred Citation[3]. The committee also strongly recommended that blood be drawn before administration of antimicrobial therapy so that therapy can be appropriately targeted when culture results are available.
The current gold standard of bloodstream microbial detection and identification is automatic, continuous monitoring liquid culture, followed by Gram stain, subculturing and use of phenotypic methods to identify the organism and its susceptibilities. A major limitation to culture is the time required to complete the process, which ranges from 1 to 5 days or more. This timeline is inconsistent with the need to obtain rapid answers to guide therapy. In addition, culture methods miss fastidious organisms that are difficult or impossible to culture. Culture can also be confounded if antibiotics are administered before the blood is sampled. Blood cultures are reported to be negative in more than 50% of the cases where true bacterial or fungal sepsis is believed to exist Citation[3,4]. This has driven the recommendation by the International Sepsis Committee that decisions on antibiotic administration, changes in therapy or its discontinuation be based on clinical judgment rather than culture results. Thus, blood culture is not an ideal gold standard. The results come too late, are incomplete and are potentially misleading, to the point where the recommendations are to ignore them in many cases.
Potential for molecular methods
It is remarkable that in this modern era of molecular technology, no molecular method has replaced or is even used broadly as an add-on to blood culture in clinical laboratories for the diagnosis of disease caused by infectious organisms Citation[5,6]. Molecular methods have the potential to overcome many limitations of culture . The ideal molecular method would analyze a patient’s blood and provide all the information needed to immediately direct the optimal antimicrobial therapy for bacterial or fungal infections, and could subsequently provide data to assess the therapy by measuring the clearance of microbial nucleic acids from the blood over time. Although no currently available molecular method is sufficiently rapid, accurate or informative to achieve these goals, these should be the long-term objectives for diagnosis of bloodstream infections.
An achievable goal in the near term is to analyze blood in parallel with culture methods and identify the pathogens, including unculturable organisms, responsible for infection and some of the key determinants of drug resistance well before the culture results are available. All of the molecular determinants of drug resistance are not yet known, yet genes have been identified in some species, including genes encoding methicillin resistance, vancomycin resistance and carbapenem resistance, that can be measured using molecular methods.
If available, quantitative measurements would be broadly used in microbiology. Many years of research studies using quantitative microbiology on solid media have demonstrated that quantitative measurements provide clinically valuable information Citation[7]. For example, bacterial load is predictive of the occurrence of complications and death Citation[7]. However, the inability to measure bacterial load routinely has precluded a complete understanding of the value of this metric. Quantification of bacteria in blood is difficult to achieve by culture methods and is rarely practiced in clinical laboratories because it requires subsequent plating on solid media rather than incubation of blood in liquid culture. The time required for liquid culture bottles to become positive provides some suggestion of bacterial load, but is a weak quantitative metric and varies with the microbe or microbes present.
Molecular tests have the potential to be quantitative. Quantitative molecular measurements have been standard in managing chronic viral infections, such as HIV and hepatitis C (HCV), for many years. Several factors preclude quantitative measurements in bloodstream infections. First, the range of bacterial concentrations in infected patients is broad and the low-end challenges the lower limit of detection of current molecular methods. Many published studies have shown that the number of recoverable colony-forming units (CFU) of bacteria in the blood of adult patients with clinically significant bacteremia is low, typically in the range of 1–30 CFU/ml Citation[8–10]; in children, the levels of bacteria are substantially higher, in excess of 100 CFU/ml Citation[7]. However, the CFU measured by culture microbiology represents only the viable organisms that survive the plating process and does not count dead cells, cells that cannot form colonies or free microbial DNA that may have been liberated from lysed cells in the blood compartment. Second, sample preparation methods to extract pathogen DNA from whole blood in sufficient purity and quantity for molecular methods are inadequate. Third, infections can by caused by many dozens of different organisms and quantitative molecular methods that encompass all the potential organisms are lacking. Thus, the true concentration of pathogen DNA available for molecular analysis in patients with bloodstream infections is seldom known. However, recent data suggest that quantitative data on bacterial DNA load may have value. In patients with pneumococcal pneumonia, a high bacterial DNA load, measured by quantitative PCR for Streptococcus pneumoniae DNA, was a major determining factor for shock and organ failure Citation[11]. In patients with pneumococcal pneumonia, bacterial DNA load in the blood is associated with the likelihood of death, the risk of septic shock and the likely need for mechanical ventilation Citation[11].
Species identification & antibiotic resistance determination from culture samples
A stepping-stone to the direct analysis of blood specimens is the use of molecular methods to identify bacterial and fungal species after they have been grown in culture bottles. The acute disadvantages of analyzing specimens after they have grown in culture are the time delay and the bias generated by a culture step. Of course, unculturable organisms cannot be identified this way at all. Nevertheless, the use of molecular methods to identify organisms after culture has the potential to be faster than the standard phenotypic definitive identification and antimicrobial susceptibility testing, which requires additional 24–72 h after the culture becomes positive. To take advantage of the rapid enrichment of automated blood culture instruments, several studies have reported that use of peptide and/or nucleic acid probes, PCR and other nucleic acid amplification techniques can rapidly identify organisms from flagged blood cultures Citation[12–25].
A molecular method employing peptide nucleic acid (PNA) FISH is commercially available from AdvanDx (MA, USA). In their first product (Staphylococcus aureus PNA FISH), the PNA FISH uses PNA probes targeting S. aureus 16S rRNA to directly identify S. aureus from positive blood culture bottles Citation[13,18,24]. Available as a kit containing all the necessary reagents and approved by the US FDA, this assay distinguishes between S. aureus or non-S. aureus within hours. Briefly, one drop of the blood culture broth is added to a slide containing a drop of fixation solution. After 5 min, one drop of S. aureus PNA probe and a coverslip are added. The probe and specimen are incubated for 90 min. Coverslips are removed and slides are immersed in a preheated wash solution for 30 min. After air drying the slides, one drop of mounting medium and a coverslip are added and slides are examined under a fluorescence microscope and a 100× oil objective Citation[26]. This assay has been extended to identify other bacterial and fungal pathogens Citation[27,28].
The StaphPlex system (Qiagen, CA, USA) was designed for simultaneous species-level identification and detection of Panton–Valentine leukocidin (PVL) and several antimicrobial-resistance determinants of staphylococci directly from blood culture medium in which Gram-positive cocci in clusters were seen by using Gram staining. The StaphPlex system uses a unique target-enriched multiplex PCR method Citation[29] to amplify and detect 18 Staphylococcus-specific genes simultaneously in one reaction. The tuf gene target provides identification and differentiation of coagulase-negative staphylococci, and the nuc gene target is specific for S. aureus. The mecA gene confers resistance to methicillin. The aacA gene is responsible for resistance to aminoglycosides. The ermA and ermC genes contribute to resistance to macrolides, lincosamides and streptogramins. The tetM and tetK genes are responsible for resistance to tetracycline. The amplified products are further characterized by using a Luminex suspension array. The whole process, from the blood culture medium processing to results reporting, can be performed within 5 h, which greatly shortens the time usually needed for phenotypic identification and antimicrobial susceptibility testing Citation[30].
A broad new approach to postculture identification of bacterial species has been recently reported using MALDI-time of flight (TOF) mass spectrometry (MS; Bruker Daltonics, Bremen, Germany) Citation[31]. Colonies from an agar plate or liquid culture are mixed with a MALDI matrix and rapidly analyzed by MS Citation[31,32]. The MALDI-TOF system works by comparison of the mass spectral signals obtained from postculture specimens with a database of spectra from reference standard spectra. A significant advantage of this methodology is that an answer is provided rapidly with a minimal amount of labor compared with conventional methods. Seng and colleagues reported 95.4% success in postculture bacterial identification by MALDI-TOF MS; 84.1% of pathogens were identified at the species level and 11.3% were identified at the genus level, making this method a candidate to replace Gram staining, which is the most likely niche for this technology in the laboratory Citation[32]. It is still necessary to culture bacteria before analysis by MALDI-TOF, and drug resistance must still be determined by conventional methods. Identification of mixed populations of bacteria will probably be difficult owing to dynamic range issues in the mass spectrometer; La Scola and Raoult reported that, of 22 culture samples containing two or more organisms, only one species was identified in 18 of the samples Citation[31]. However, due to ease of use and ability to speciate rapidly, this method has the potential to replace the cumbersome and labor-intensive efforts required to identify the species following culture. It is likely that the accuracy and dynamic range of this method will improve over time as the quality of the databases and general methodology improves.
Direct amplification & detection methods from blood samples
The holy grail of molecular methods is the direct analysis of blood or other patient specimens to identify the organisms present and, to the extent possible, identify the presence of genes associated with drug resistance. In recent years a number of molecular-based products have been developed for the diagnosis of bloodstream infections Citation[33–37]. All follow the paradigm of bacterial and/or fungal lysis, nucleic acid extraction and purification, and amplification by PCR followed by a back-end detection or identification method. A number of products have been CE marked, although none are currently approved by the FDA for use in the USA. A list of the products for molecular analysis of bloodstream infections and the basic properties of each is shown in .
SeptiFast®
The Roche product SeptiFast®Citation[38] has been available longer than other molecular-based tests and has been evaluated in a number of published clinical studies Citation[39–47]. SeptiFast uses real-time PCR in a nonquantitative mode to identify ten bacteria at the species level, several more at the genus level, as well as five Candida species and Aspergillus fumagatus. This assay reportedly identifies the 25 organisms that account for more than 90% of the culturable pathogens associated with sepsis. No unculturable organisms are identified nor are most of the highly fastidious organisms that are difficult to culture. The SeptiFast procedure involves extraction of nucleic acid from 1.5 ml of whole blood using mechanical lysis with ceramic beads and manual spin column-based nucleic acid purification under a contamination-controlled workflow Citation[38]. Human DNA from white cells in blood and pathogen DNA are isolated together. Nucleic acid preparation is followed by placing 50-µl aliquots of the elute into three real-time PCR reactions that amplify Gram-positive and Gram-negative bacteria and fungi. Fluorescent probes are used to identify the pathogens.
Clinical studies using SeptiFast on blood samples from hospital patient populations with suspected sepsis Citation[39–41,43,46,47], emergency department patients with suspected sepsis Citation[44] and febrile neutropenic patients Citation[45,48] have been reported. From these studies, some important themes have emerged. First, using blood culture as a reference ‘gold standard’ to compare molecular methods is problematic. As described earlier, culture fails to identify more than 50% of the cases of sepsis believed to be caused by bacteria or fungi based on clinical and other criteria Citation[3]. Unsurprisingly, SeptiFast consistently identified more positive specimens than blood culture methods. These potential ‘false positives’ were frequently deemed clinically significant based on chart review of clinical data, other analytical evidence of infection or disease severity Citation[39], and were often subsequently confirmed after isolation of the pathogen from relevant clinical samples Citation[49]. SeptiFast-positive/culture-negative results could conceivably come from nonviable organisms in the blood (resulting from ongoing antibiotic treatment), cell-free DNA released from infected or colonized remote infection sites, or antibiotic interference with culture. Thus, evaluation of molecular diagnosis of sepsis requires a reference method based on multiple data types since blood culture does not detect many true sepsis cases Citation[3].
On the other hand, culture consistently identified some organisms that were not identified by SeptiFast, possibly due to the larger volume of blood analyzed by culture and the lower limit of SeptiFast detection of approximately 3–30 CFU/ml Citation[38]. In addition, some organisms that cause sepsis are not detected by the SeptiFast method. Nevertheless, SeptiFast and blood culture results were usually in agreement, suggesting that SeptiFast can add value as an adjunct to blood culture by both identifying pathogens not identified by blood culture and by identifying pathogens more rapidly than blood culture. SeptiFast-negative/culture-negative specimens from patients deemed to be infected based on clinical observations or other molecular markers could result from unculturable organisms, since SeptiFast identifies only culturable organisms.
The time required to conduct a SeptiFast analysis is less than 6 h; however, the time to the final results in real clinical settings may be significantly longer. In a study that modeled a one batch per day model, the median time for a SeptiFast result was 27 h; it was 18 h for the two batch per day model Citation[40]. Thus, the practical considerations of specimen transport, batching specimens for testing and result reporting increase the time from the theoretical minimum turn-around time substantially. Nevertheless, even at 27 h, the SeptiFast results are available more rapidly than blood-culture results, and SeptiFast provides a measurable amount of ‘gainable days’ of adequate antimicrobial therapy Citation[41]. This combination of shorter times to microbe identification and detection of organisms missed by culture proves the value of molecular methods. These pioneering SeptiFast studies were the first significant commercial attempts to use molecular methods to identify the organisms present in patients with bloodstream infections, and these studies have paved the way for future molecular methods and have established the benchmarks by which the value of newer molecular methods can be assessed.
SeptiTest®& LOOXSTER®/VYOO™
An important consideration in molecular analysis of bloodstream infections is the fact that blood contains human DNA located in circulating white blood cells. When whole blood is treated to extract and purify pathogen DNA, human DNA is co-isolated in great excess relative to pathogen DNA. The presence of excess human DNA is inhibitory to PCR and decreases the sensitivity of pathogen detection in bloodstream infections Citation[50–52]. Although PCR sensitivity could theoretically be increased by extracting DNA from larger volumes of blood, the co-extraction of increasingly proportionate amounts of human DNA results in diminishing returns.
The MolZym product known as SeptiTest® incorporates a unique sample-preparation methodology that reduces the burden of human DNA in an extracted blood sample. The method employs gentle lysis of white blood cells followed by DNase treatment to remove human DNA. This is followed by lysis of bacterial and fungal cells under more severe conditions. Thus, pathogen DNA is recovered in the presence of a greatly reduced burden of contaminating human DNA. PCR amplification of 16S ribosomal DNA and sequencing are used to identify the microorganisms present in the sample. In theory, the MolZym sample preparation method can be used with any back-end molecular test. In analytical studies with spiked specimens, the sensitivity of SeptiTest was reportedly increased up to 1000-fold for spiked blood specimens relative to conventional spin column-based technology with a lower limit of detection of 50 CFU/ml of blood Citation[53–56]. A potential disadvantage of this approach is that freely circulating DNA in the blood from pathogens released from remote site infections is digested along with the human white cell DNA. Likewise, intracellular pathogens with easy to lyse membranes might be lost. The relative quantities of these sources of DNA in bloodstream infections are not known.
The SIRS-Lab product LOOXSTER® uses a strategy that exploits the methylation differences between bacterial/fungal DNA and human DNA to enrich the clinical sample in pathogen DNA by affinity chromatography followed by 16S rDNA gene amplification using a product known as VYOO™ Citation[57]. In analytical studies, the approach resulted in substantial alteration of the pathogen–human DNA ratio. Approximately 90% of eukaryotic DNA was removed. This significantly decreased the signal loss in amplification due to human DNA with sensitivity elevated at least tenfold compared samples not subjected to pathogen DNA enrichment. Early studies with small numbers of human clinical specimens are also beginning to show promising results for this approach Citation[41,57].
PCR/electrospray ionization/MS for bloodstream infections
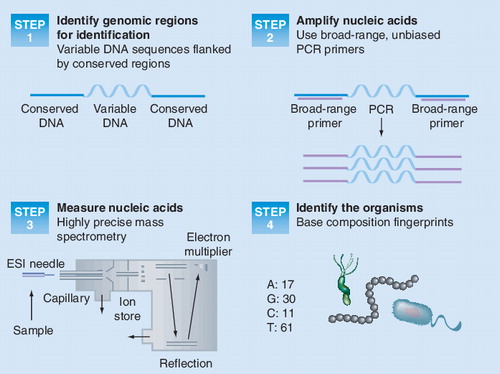
A new strategy for the molecular detection of bloodstream infections couples broad-range PCR amplification to electrospray ionization/MS (PCR/ESI-MS). Previous versions of this method, invented by Ibis Biosciences, now a part of Abbott Molecular, were known commercially as TIGER Citation[58] or the Ibis T5000 Citation[59]. The current commercial hardware platform that conducts the MS analysis is now known as the Abbott PLEX-ID and the assay for direct analysis of bloodstream infections that runs on the PLEX-ID is called the BAC Spectrum Assay. This technique uses primers designed to genomic regions highly conserved regions across the bacterial or fungal domains of life. The method was initially developed for the identification of microbes, including previously unknown or unculturable organisms in samples where multiple microbes may be present, primarily for biodefense applications Citation[58,60,61]. It is now being developed for diagnosis of bloodstream infections.
The steps in the PCR/ESI-MS process are illustrated in and the PCR/ESI-MS assay is compared with other methods in . Briefly, multiple pairs of primers are used to amplify carefully selected regions of bacterial or fungal genomes; the primer target sites are broadly conserved but the amplified region carries information on the microbe’s identity in its nucleotide base composition. Regions like this appear in the DNA that encodes ribosomal RNA and in housekeeping genes that encode essential proteins. Following PCR amplification, a fully automated ESI MS analysis is performed on the PCR/ESI-MS instrument. The mass spectrometer effectively weighs the PCR amplicons, or the mixture of amplicons, with sufficient mass accuracy that the composition of A, G, C and T can be deduced for each amplicon present. The base compositions are compared with a database of calculated base compositions derived from the sequences of known organisms and to signatures from reference standards previously determined via PCR/ESI-MS .
Although not as information rich as the sequence (the linking order of A, G, C and T bases is not determined using ESI-MS), for diagnostic purposes the nucleotide composition of a nucleic acid has the same practical value. For example, in the PLEX-ID BAC Spectrum assay, eight PCR reactions yield sufficient information in the base compositions of the amplicons to identify most bacteria to the species level. A critical advantage of the PCR/ESI-MS technology is that a probe is not required. When designing a probe, the target nucleic acid sequence must be known. The difference between the PCR/ESI-MS approach and probe-based technologies is that the mass spectrometer does not anticipate the amplification product being measured. The amplicon is weighed, the base composition fingerprint that corresponds to the weight is calculated, and the resulting fingerprint is compared with those of all known organisms in a database. In the event that there is no match of the measured base composition with a sequence in the database, the nearest neighbor organism is identified in a manner similar to that used in identification of related organisms using sequence data, such as the Blast search algorithm.
As with broad PCR followed by sequencing, use of the PCR/ESI-MS is equivalent to running many thousands of specific tests, including tests that have not yet been developed, because the identity of the infectious organism does not need to be anticipated. The PCR/ESI-MS platform not only identifies organisms present in a clinical sample but is also capable of providing information about the microbe, such as its strain type, whether or not it contains genes that mediate resistance to certain drugs and whether it carries certain virulence factors Citation[62,63].
In any nucleic acid-based microbial identification schema, whether it be based on amplification followed by probes, sequencing or MS, it is essential to analyze the regions of the microbial genome that contain the information needed for identification of important bacterial and fungal species. 16S ribosomal DNA has been widely used for microbial identification. However, not all regions of the 16S ribosomal DNA are equivalent – not all provide a universal amplification target and not all have useful information content. It is, therefore, advantageous to amplify and analyze multiple target sites within the ribosomal DNA. A major strength of the PCR/ESI-MS method is that the mass spectrometer is capable of analyzing multiple amplification products in a fast and automated fashion. With the PLEX-ID instrument, an amplification reaction can be analyzed in the mass spectrometer in 30 s, enabling analysis of approximately 30 microtiter plates in a 24-h period. This high-throughput capability allows each specimen to be interrogated with multiple PCR reactions to increase both the breadth of coverage and the resolving power, allowing differentiation of closely related species.
The BAC Spectrum assay for bloodstream infections analyzes the bacterial domain of life with eight amplification reactions and nine primer pairs (two primer pairs are multiplexed) and combines the information from all reactions to identify the bacteria present. Of the nine primer pairs, three are targeted to different regions of 16S ribosomal DNA, one is targeted to 23S ribosomal DNA, and the remaining four are targeted to genes encoding universally conserved housekeeping genes tufB, rplB, valS and rpoB (two regions). The coverage of the bacterial domain of life by the PCR/ESI-MS assay is illustrated in . Primers targeting housekeeping genes are important for two reasons. First, it is frequently the case that the information needed to distinguish closely related species is not contained within the ribosomal DNA sequence, so the mass spectral signatures obtained from these housekeeping gene targets supplements the information obtained from the ribosomal targets. The ability to distinguish species of bacteria by integrating information from multiple signatures has been described previously Citation[59]. Second, the dynamic range of amplification of mixed populations of microbes is enhanced by using primer pairs that have nonoverlapping coverage. For example, when the ribosomal primer pairs amplify mixed populations of microbes, the lower abundance representatives are detected at reduced sensitivity owing to competitive amplification of the higher abundance species. When primer pairs that do not overlap in coverage are included in the analysis, like those to the housekeeping genes, this competition is avoided. This is exemplified later in a section describing detection of mixtures.
In addition to bacterial identification, presence of three important drug-resistance markers are simultaneously interrogated by the assay. The first is mecA, which is a molecular determinant of antibiotic resistance to β-lactam antibiotics Citation[64]. The second are the vanA and vanB genes, which encode resistance to vancomycin Citation[65]. Finally, the blaKPC gene, which encodes resistance to the carbapenem class of antibiotics in Klebsiella pneumoniae and other Gram-negative bacilli Citation[66], is interrogated.
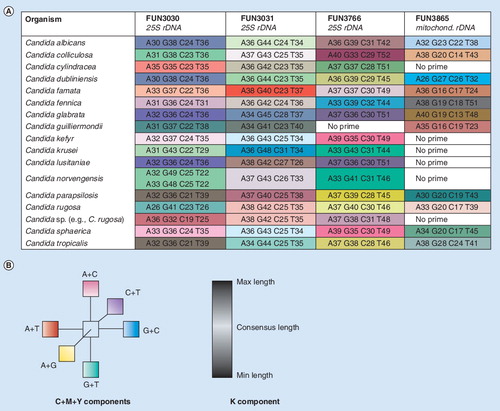
The presence of Candida DNA is determined with three primer pairs targeting ribosomal DNA that can identify and differentiate members of Candida spp., including Candida albicans, Candida glabrata, Candida tropicalis, Candida parapsilosis, Candida lusitaniae, Candida guilliermondii, Candida dubliniensis and Candida krusei. These broad-spectrum primer pairs were selected by careful alignment of fungal and human rRNA gene sequences and were designed to target regions where the 3´ ends of primers hybridize to regions substantially different from the human rRNA gene sequence. In addition, the assay includes a primer pair that targets the mitochondrial small subunit ribosomal RNA gene of the C. albicans group of Candida spp. and provides confirmation of and speciation of C. albicans, C. tropicalis, C. arapsilosis, C. dubliniensis, C. guilliermondii and Candida kefyr.
Triangulation of base composition analysis of multiple targets for microbial identification
In contrast to sequencing, base composition analyses and its ability to distinguish microbial species is not intuitive. With the PCR/ESI-MS method, the base composition, rather than the sequence, of the amplicon is determined and compared with a database of calculated base compositions derived from a curated database of sequences. Thus, the order of the nucleotides is not known. Obviously the base composition is less information-rich than the sequence. The PCR/ESI-MS method makes up for the lower information content of base composition compared with sequencing by amplifying multiple regions of the microbial genome instead of one and by using the aggregate information derived from these PCR reactions to identify the microbe.
Both sequencing and base composition analysis share the common feature of conducting a comparison with a curated database of sequences and the conventions established for curation of databases used for analysis of sequence are directly applicable to the data obtained using PCR/ESI-MS. The Clinical Laboratory Standards Institute recently published approved guidelines for the identification of bacteria and fungi by DNA target sequencing Citation[67]. These guidelines address the issues of the quality of the sequences used in the database, sequence redundancy, periodic updates, organism nomenclature errors and other issues of concern when using a database comparison to identify organisms. The PCR/ESI-MS database is curated in accordance with these guidelines. The PCR/ESI-MS database was generated and is maintained and kept up-to-date as follows. For each primer pair in the assay, a reference sequence for the target gene from the best available published reference sequence is chosen. Additional sequences of the target gene are aligned to the reference sequence. Using the primer sequences for the selected assay, the bacterial section of GenBank is analyzed computationally for entries that would be amplified by the primer pairs. The resulting sequences are curated using bioinformatic tools by comparison with the reference sequence and the expected base compositions for the target region are calculated and used to populate the database. These computationally generated base compositions are augmented with experimentally measured base compositions from reference isolates of the target organisms that have been certified based on established methods. Experimentally observed base compositions from each primer pair target region are compared with the PCR/ESI-MS database of base compositions. The information on base counts from each primer pair provides a signature for the organism.
A visualization of the ability of base composition to identify and speciate Candida is shown in . The columns represent the four amplification target regions for the three chromosomal and the mitochondrial rDNA targets. Each row of four adjacent colored cells represents the base composition signature of each Candida spp. Within each column, cells are color-coded to represent with similar hue signatures that are close to each other in base composition. The intensity of the color in each cell was computed to visually reflect the distinctiveness of its signature, with pale or pastel colors indicating base compositions closer to the overall consensus and more saturated colors representing the most divergent base counts. Although there is occasional overlap of base compositions where two or more Candida spp. might have the same base composition in one target region, the use of multiple target regions and triangulation of the signatures assures correct speciation, yet allows for expected biological variation, such as mutations resulting in differences in two isolates of the same species. As with a sequence similarity search, analysis of base compositions enables identification of variants from the database based on the closest match.
A visualization of the base composition signatures obtained for 332 phylogenetically diverse bacteria in the PCR/ESI-MS database using the same color-coding schema as in is shown in a heat-map format in . These data show the near universal primer coverage of the 16S rDNA primers (first three columns). Although no rDNA primer is completely universal (e.g., the Fusobacteria are not amplified by BCT346 and the Actinomycetales are not amplified by primer pair BCT349), the combination of all nine targets provides universal coverage of the bacterial domain of life. Several of the primer pairs provide specialty functions. For example BCT2249, targeted to the tufB gene, was designed to speciate the Staphylococcaceae, where S. aureus is unambiguously distinguished from the other Staphylococcus spp., but does not prime outside of this genus Citation[63]. Similarly BCT358 (targeting valS) provides high resolving power of the Enterobacteriaceae.
Detection of mixed populations of bacteria in direct blood specimens
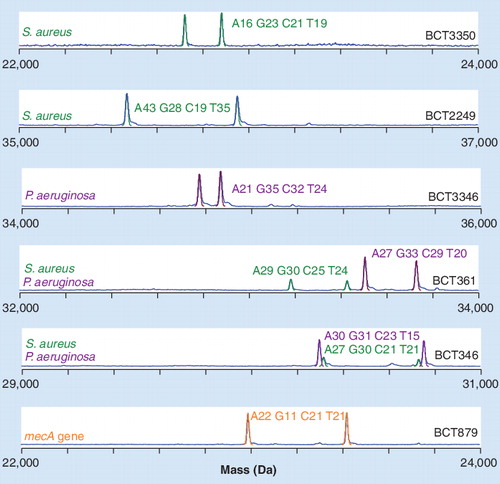
A significant number of bloodstream infections are polymicrobial. The PCR/ESI-MS method can identify mixed populations of bacteria as this technique directly analyzes nucleic acids extracted from blood. A representative result from a blood specimen from a patient infected with both S. aureus and Pseudomonas aeruginosa is shown in . Analysis of individual spectra derived from amplification from the Gram-positive-centric primers (top two spectra) clearly show base compositions that match S. aureus. Spectra from the Gram-negative-centric primers, such as BCT3346 (third spectrum), show base compositions consistent with P. aeruginosa. However, the broad rDNA primers, such as BCT361 and BCT346 (fourth and fifth spectra), show signals and base compositions that correspond to both S. aureus and P. aeruginosa in an approximately 3:1 ratio. In addition, signals from BCT879, targeting the mecA gene are clearly present (bottom spectrum), indicating that the S. aureus present harbor methicillin resistance. Thus, the combination of signals from this blood specimen analyzed without culture indicates an infection that requires antimicrobial therapy that covers both these organisms, including consideration of drug resistance.
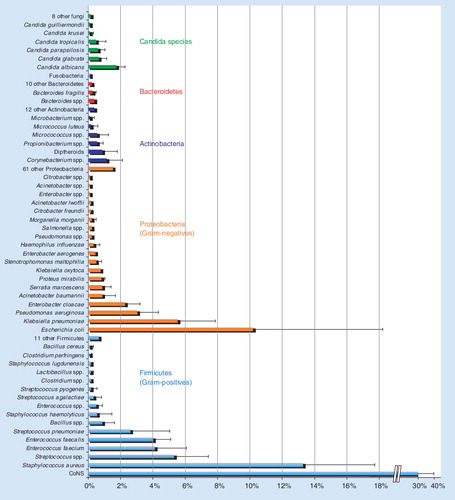
Simultaneous detection of culturable & unculturable organisms
Our view of organisms that cause bloodstream infections is biased because culture is the only method widely used. Since blood cultures are reported to be negative in more than 50% of the cases where true bacterial or fungal sepsis is believed to exist Citation[3], there is a missing component to our understanding of infections. The PCR/ESI-MS method detects pathogens with no bias due to culturability. Aerobic, anaerobic, culturable, fastidious and unculturable organisms are identified in the same way. To get a perspective on the fractional occurrence of culturable organisms, we surveyed samples from seven hospital microbiology laboratories in Cleveland (OH, USA), Nashville (TN, USA), Tuscon (AZ, USA), New York (NY, USA), Baltimore (MD, USA), Salt Lake City (UT, USA) and Barcelona (Spain). To create a metric comparable among the hospitals due to different bed numbers, the positive blood culture counts for each organism category at each site were converted to a percentage of the total blood culture results reported for that hospital. The mean percentages across the seven sites are plotted in with the error bars representing one standard deviation from the mean. Reported organism classes making up less than 1% on average across the sites were binned together. Owing to differences in the reporting precision among various hospital microbiology laboratories (some laboratories report certain organisms at the species level, while others report at the genus level), the data in are reported based on the highest precision reported from all seven hospital laboratories. These combined percentages are displayed by phylum and abundance. Overall, there were 138 unique species identified by culture methods at the seven hospitals. The distribution of pathogens is highly biased in favor of approximately 30 of the most common pathogens, which was fairly consistent across all seven hospitals, as shown by the standard deviations. These data provide a benchmark of culturable organisms, which will be compared with the PCR/ESI-MS analysis of direct patient specimens in the future.
Different patient populations would be expected to vary in the frequency of unculturable organisms. Febrile patients who reported recent tick bites or travel in the wilderness are more likely than other patient populations to be infected with unculturable organisms, such as Ehrlichia, Rickettsia, Bartonella, Anaplasma or other unculturable Proteobacteria, or Spirochaetes, such as Borrelia spp. However, these patients could also be infected with more common pathogens. To investigate pathogens infecting this patient population we used PCR/ESI-MS on whole blood, plasma or cerebrospinal fluid from patients clinically suspected of infection by Ehrlichia or other tick-borne disease organisms Citation[68]. One of the challenges of evaluating new technology that goes beyond the currently available methods is identifying a reference method to compare the results. A combination of culture, antibody-based methods, and broad-range PCR amplification followed by hybridization to specific probes were used in combination to provide a basis for comparison of PCR/ESI-MS results. A two-step, colorimetric microtiter plate PCR assay capable of detecting and differentiating medically important Ehrlichia species was used Citation[69,70]. The first step involved PCR amplification of a genus-specific region in the 16S rRNA gene, which has been described elsewhere Citation[71,72]. In the second step, the PCR amplification product was identified and differentiated to Ehrlichia chaffeensis, Ehrlichia ewingii or Anaplasma phagocytophilum using three species-specific probes. The same PCR enzyme immunoassay (PCR-EIA) format and procedure was used to detect Rickettsia rickettsii by targeting a 154-bp fragment, as described previously Citation[73].
An overview of the results from this study is shown in . Of 213 specimens analyzed, the majority of detected infections in this patient population were E. chaffeensis, but other Ehrlichia and Rickettsia spp. were also identified, consistent with the hypothesis that this patient population is disproportionately infected with unculturable organisms Citation[68]. In comparison to the PCR-EIA, the PCR/ESI-MS sensitivity, specificity, and positive- and negative-predictive values were 95.0, 98.8, 95.0 and 98.8%, respectively. The 38 specimens that were positive for Ehrlichia by both PCR/ESI-MS and PCR-EIA were further characterized to the species level, with 100% agreement between the two assays. In addition, R. rickettsii was detected by PCR/ESI-MS from four specimens, which were confirmed retrospectively by serology and PCR-EIA. In three specimens, the PCR/ESI-MS assay identified P. aeruginosa, Neisseria meningitidis, and S. aureus; these identifications were confirmed by culture and/or clinical diagnosis as being clinically relevant .
The PCR/ESI-MS method is quantitative
An important aspect of PCR/ESI-MS is that the method is quantitative Citation[58,59,61]. As discussed earlier, culture methods are generally not quantitative and little is known regarding the levels of pathogens in bloodstream infections. Even less is known about levels of pathogen nucleic acids in bloodstream infections and there is potential diagnostic value in a quantitative measurement. In each PCR/ESI-MS experiment, an internal calibrant is added to every well of the assay plates. This calibrant acts as a PCR control and a standard for quantification in each well. Essentially, the peak heights in the mass spectrum for amplicons and calibrant are use to determine the relative ratios of calibrant to microbe. As the original concentration of calibrant was known, the concentration of pathogen, measured in genome copy number per milliliters of specimen, can be determined accurately. In experiments with 37 samples that tested positive for Ehrlichia nucleic acid concentrations ranged from near the lower limit of detection to over 500,000, with an average copy number of near 100,000/ml. As described earlier, measurement of CFU per milliliter in bloodstream infections for common culturable organisms ranges from very low (∼10 CFU/ml) to very high, especially in pediatric patients and newborns Citation[7]. A series of clinical trials have been arranged to evaluate PCR/ESI-MS based quantitative measures for a broad variety of pathogens and clinical relevance.
Clinical utility of the PCR/ESI-MS instrument
The PCR/ESI-MS system is available commercially and systems are currently in use for research and disease surveillance at 20 sites in the USA and Europe. With the current system, all steps, including sample collection and preparation, and the automated PCR, MS analysis, signal processing and report generation, can be carried out by clinical laboratory personnel within a 4–6-h period. The mass spectrometer provides data in approximately 30 s, uses no consumable products other than solvents, can analyze approximately 3000 PCR reactions in 24 h in a completely automated fashion. No training in MS is required for personnel who operate the instrument or for those who interpret the results.
Limitations & future improvements
Although broad-range PCR analysis of samples has many advantages, it comes with the difficulty of requiring exquisitely clean reagents that are free from adventitious bacterial and fungal DNA along with work flow-processes that prevent introduction of contamination in all steps leading up to PCR. Efforts focused on this issue have resulted in reliable manufacturing processes and quality-control procedures that successfully avoid contamination. Virtually all commercially obtained materials contain small amounts of bacterial genomic material Citation[74]. This material is introduced even under the most stringent of conditions either by the handling of materials during early production stages or by the contact of the materials with air or surfaces during production. Industrial processes were developed for the production of DNA-free reagents. This includes the production of reagents for PCR, perhaps the most critical reagents from a contamination point of view. Key to the production of contaminant-free reagents was the development of a closed-loop system that allows the ‘cleaning’ of reagents after mixing and then filling of custom intravenous bags for the storage of those reagents. The cleaning of the reagents includes passing them through a 0.2-µm filter to remove organisms, as well as through a special filter designed to remove nucleic acid contaminants. The final kit format for the current PLEX-ID hardware platform is a 96-well plate that can be used in standard liquid-handling robotics, as well as in our automated clean-up procedure. An important source of adventitious DNA is the enzymes used in the PCR reactions. Commercial procedures have been established with a commercial producer of Taq polymerase and large batches of enzyme that meet stringent specifications are regularly obtained. Only lots of enzyme that pass quality-control procedures are used.
An important limitation of molecular methods is that only three genes associated with phenotypic drug resistance are currently interrogated by the BAC Spectrum assay. The current assay is limited to mecA, which is a molecular determinant of antibiotic resistance to β-lactam antibiotics Citation[64], the vanA and vanB genes, which encode resistance to vancomycin Citation[65], and the blaKPC gene, which encodes resistance to the carbapenem class of antibiotics in Klebsiella pneumoniae and other Gram-negative bacilli Citation[66]. Although this is a good start, the molecular mechanisms of resistance to other important antibiotics are either not fully understood or require analysis of too many regions of the microbial genome to be included in the current assay format. Analysis of drug-resistance mechanisms is a highly active area of research and we can expect rapid increases in understanding the molecular underpinnings of resistance. Further improvements in multiplex technology will enable more drug-resistance markers to be analyzed by the PCR/ESI-MS method.
Expert commentary
During the past two decades, technical advances in molecular biology have initiated a revolution in the diagnostic microbiology field Citation[5,6]. Molecular techniques have found their niches in laboratory diagnosis of bloodstream infections by providing rapid and highly discriminatory tools to identify the causal pathogens. The new molecular tools have the potential to improve the sensitivity of current automated blood culture systems by covering unculturable and fastidious pathogens.
Methods to rapidly detect and identify pathogens responsible for bloodstream infections are desperately needed. For the most serious bloodstream infections, such as septic shock, time is of the essence as survival rates drop on an hourly basis if appropriate treatment is delayed. It is important to both identify the infecting organism and determine its sensitivities to antimicrobial agents. The current gold-standard diagnostic methods are based on culture, which is slow, labor intensive, nonquantitative and does not identify over half the cases where true bacterial or fungal sepsis is believed to exist Citation[3].
We are now in the early stages of a new era of molecular diagnostics for bloodstream infections based on advanced technologies, such as MS, next-generation sequencing and other single-molecule detection methods. These analytical methods have the potential to identify pathogens and provide detailed subspecies information. As the molecular mechanisms for antimicrobial resistance become better understood, prediction of drug susceptibilities will be achievable by molecular methods.
Specimen preparation and enrichment of pathogen DNA remains a major challenge. Successful molecular diagnostics for bloodstream infections must integrate technology for front-end specimen collection, lysis, extraction and enrichment of pathogen nucleic acids with a back-end molecular analysis technology that identifies virtually all pathogens. Both components are essential to success, and the lack of a suitable front end has held back progress in the field.
The pioneering efforts of Roche with its SeptiFast product are beginning to show proof of principle that microbial identification by direct molecular analysis of bloodstream infections is achievable. Significant hurdles remain in specimen preparation, and sensitive, comprehensive and rapid analysis of a broad range of pathogens. Automation will be essential to provide speed, throughput and contamination control, and to reduce the labor requirements in molecular analysis of bloodstream infections. Advanced analytical techniques, such as MS, have the potential to change the current paradigm. These are now available and will be evaluated in clinical studies shortly.
The new molecular technologies will also lead to significant new discoveries. For example, microbial causes of many disease states currently not believed to be of infectious origin will be identified. New pathogens will be discovered and the role of microbial infections in broader disease states will be revealed Citation[75–77]. A benefit of determination of microbial drug susceptibility will be that antibiotic stewardship will be enhanced. It will be no longer necessary to treat patients with broad empirical therapy as the correct and optimal antimicrobial therapy can be prescribed immediately based on a rapid, comprehensive and accurate molecular test.
Five-year view
The key objective of molecular diagnostics of bloodstream infections is to identify the pathogen(s) and key genetic determinants of drug resistance as rapidly as possible in order to initiate appropriate antimicrobial therapy. These objectives will be achieved within the next 5 years based on advances in the following three areas. First, specimen preparation, including collection, lysis, extraction and enrichment of pathogen nucleic acids, will undergo a quantum advance based on new approaches. The current preparative methods using columns or magnetic bead-based nucleic acid purification have not fundamentally changed in two decades, are inadequate and are not likely to advance further other than by incremental improvements. We believe that within 5 years a currently unanticipated technology will be developed to address the specimen preparation problem. In the near term, variations of the current specimen approaches will be used to successfully demonstrate the feasibility and value of revolutionary back-end analytical methods. However, the full potential of molecular methods for diagnosis of bloodstream infections will not be reached until a quantum advance in specimen preparation is achieved.
Second, modern analytical methods will provide game-changing advances in measuring the signatures in microbial DNA that simultaneously detect, identify, subspeciate and strain-type pathogen DNA. Advanced MS and next-generation single-molecule sequencing methods have the potential to analyze specimens in a massively parallel fashion and measure molecular details that will enable precise diagnosis of pathogens. Quantitative measurements will demonstrate value once the methods to measure pathogen nucleic acids are widely available.
Third, advances in understanding the molecular nature of microbial drug resistance will provide enabling information to predict which drugs will be most effective without conventional culture-based drug-susceptibility measurements. Whole-genome sequencing methods currently under development will enable massively parallel sequencing of pathogen specimens in various stages of antimicrobial resistance development, providing detailed understanding of the complex process of drug resistance. This will enable the determination of the correct drugs based solely on molecular measurements, which is the last requirement of molecular methods that must be met before culture methods can be eliminated completely.
While the automated blood culture instruments with continuous monitoring will remain the mainstay technique for detection of bacterial organisms causing bloodstream infections, molecular techniques will gradually play a dominant role in rapid identification of detected bloodstream pathogens. With host responses included in the test panel, a functional identification by molecular methods can be performed routinely for virulence factor detection, antibiotic resistance determination and therapy efficacy assessment.
Table 1. Needs and current status of methods to identify bloodstream infections.
Table 2. Current molecular tests for bloodstream infections.
Table 3. Comparison of PCR/electrospray ionization/mass spectrometry results for patients infected with non-Ehrlichia spp. with clinical diagnosis and the results of the laboratory confirmations.
Key issues
• Rapid methods for identification of pathogens responsible for bloodstream infections are in development.
• The ideal test would be rapid, not require culture and would quantitatively identify all pathogens present in a blood sample.
• PCR/electrospray ionization/mass spectrometry can identify pathogens without prior assumptions regarding the type of infection.
• Specimen preparation and enrichment of pathogen DNA remains a major challenge.
Financial & competing interests disclosure
David J Ecker, Rangarajan Sampath, Christian Massire, Heather E Matthews, Donna Toleno, Thomas A Hall, Lawrence B Blyn, Mark W Eshoo and Steven A Hofstadler are employees of Ibis Biosciences, a subsidiary of Abbott Molecular Inc, the commercial manufacturer of the PCR/ESI-MS technology. Yi-Wei Tang is a consultant of Ibis Biosciences. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Related Research Data
References
- Russell JA. Management of sepsis. N. Engl. J. Med.355(16), 1699–1713 (2006).
- Kumar A, Roberts D, Wood KE et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit. Care Med.34(6), 1589–1596 (2006).
- Dellinger RP, Levy MM, Carlet JM et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit. Care Med.36(1), 296–327 (2008).
- Fenollar F, Raoult D. Molecular diagnosis of bloodstream infections caused by non-cultivable bacteria. Int. J. Antimicrob. Agents30(Suppl. 1), S7–S15 (2007).
- Miller MB, Tang YW. Basic concepts of microarrays and potential applications in clinical microbiology. Clin. Microbiol. Rev.22(4), 611–633 (2009).
- Tang YW, Procop GW, Persing DH. Molecular diagnostics of infectious diseases. Clin. Chem.43(11), 2021–2038 (1997).
- Yagupsky P, Nolte FS. Quantitative aspects of septicemia. Clin. Microbiol. Rev.3(3), 269–279 (1990).
- Werner AS, Cobbs CG, Kaye D, Hook EW. Studies on the bacteremia of bacterial endocarditis. JAMA202(3), 199–203 (1967).
- Kreger BE, Craven DE, Carling PC, McCabe WR. Gram-negative bacteremia. III. Reassessment of etiology, epidemiology and ecology in 612 patients. Am. J. Med.68(3), 332–343 (1980).
- Henry NK, McLimans CA, Wright AJ, Thompson RL, Wilson WR, Washington JA 2nd. Microbiological and clinical evaluation of the isolator lysis-centrifugation blood culture tube. J. Clin. Microbiol.17(5), 864–869 (1983).
- Rello J, Lisboa T, Lujan M et al. Severity of pneumococcal pneumonia associated with genomic bacterial load. Chest136(3), 832–840 (2009).
- Carroll KC, Leonard RB, Newcomb-Gayman PL, Hillyard DR. Rapid detection of the staphylococcal mecA gene from BACTEC blood culture bottles by the polymerase chain reaction. Am. J. Clin. Pathol.106(5), 600–605 (1996).
- Chapin K, Musgnug M. Evaluation of three rapid methods for the direct identification of Staphylococcus aureus from positive blood cultures. J. Clin. Microbiol.41(9), 4324–4327 (2003).
- Davis TE, Fuller DD. Direct identification of bacterial isolates in blood cultures by using a DNA probe. J. Clin. Microbiol.29(10), 2193–2196 (1991).
- Fujita S, Senda Y, Iwagami T, Hashimoto T. Rapid identification of staphylococcal strains from positive-testing blood culture bottles by internal transcribed spacer PCR followed by microchip gel electrophoresis. J. Clin. Microbiol.43(3), 1149–1157 (2005).
- Kempf VA, Trebesius K, Autenrieth IB. Fluorescent in situ hybridization allows rapid identification of microorganisms in blood cultures. J. Clin. Microbiol.38(2), 830–838 (2000).
- Louie L, Goodfellow J, Mathieu P, Glatt A, Louie M, Simor AE. Rapid detection of methicillin-resistant staphylococci from blood culture bottles by using a multiplex PCR assay. J. Clin. Microbiol.40(8), 2786–2790 (2002).
- Oliveira K, Procop GW, Wilson D, Coull J, Stender H. Rapid identification of Staphylococcus aureus directly from blood cultures by fluorescence in situ hybridization with peptide nucleic acid probes. J. Clin. Microbiol.40(1), 247–251 (2002).
- Paule SM, Pasquariello AC, Thomson RB Jr, Kaul KL, Peterson LR. Real-time PCR can rapidly detect methicillin-susceptible and methicillin-resistant Staphylococcus aureus directly from positive blood culture bottles. Am. J. Clin. Pathol.124(3), 404–407 (2005).
- Shrestha NK, Tuohy MJ, Hall GS, Isada CM, Procop GW. Rapid identification of Staphylococcus aureus and the mecA gene from BacT/ALERT blood culture bottles by using the LightCycler system. J. Clin. Microbiol.40(7), 2659–2661 (2002).
- Tan TY, Corden S, Barnes R, Cookson B. Rapid identification of methicillin-resistant Staphylococcus aureus from positive blood cultures by real-time fluorescence PCR. J. Clin. Microbiol.39(12), 4529–4531 (2001).
- Parta M, Goebel M, Matloobi M, Stager C, Musher DM. Identification of methicillin-resistant or methicillin-susceptible Staphylococcus aureus in blood cultures and wound swabs by GeneXpert. J. Clin. Microbiol.47(5), 1609–1610 (2009).
- Marlowe EM, Hogan JJ, Hindler JF, Andruszkiewicz I, Gordon P, Bruckner DA. Application of an rRNA probe matrix for rapid identification of bacteria and fungi from routine blood cultures. J. Clin. Microbiol.41(11), 5127–5133 (2003).
- Oliveira K, Brecher SM, Durbin A et al. Direct identification of Staphylococcus aureus from positive blood culture bottles. J. Clin. Microbiol.41(2), 889–891 (2003).
- Goldmeyer J, Li H, McCormac M et al. Identification of Staphylococcus aureus and determination of methicillin resistance directly from positive blood cultures by isothermal amplification and a disposable detection device. J. Clin. Microbiol.46(4), 1534–1536 (2008).
- Stender H. PNA FISH: an intelligent stain for rapid diagnosis of infectious diseases. Expert Rev. Mol. Diagn.3(5), 649–655 (2003).
- Shepard JR, Addison RM, Alexander BD et al. Multicenter evaluation of the Candida albicans/Candida glabrata peptide nucleic acid fluorescent in situ hybridization method for simultaneous dual-color identification of C. albicans and C. glabrata directly from blood culture bottles. J. Clin. Microbiol.46(1), 50–55 (2008).
- Sogaard M, Stender H, Schonheyder HC. Direct identification of major blood culture pathogens, including Pseudomonas aeruginosa and Escherichia coli, by a panel of fluorescence in situ hybridization assays using peptide nucleic acid probes. J. Clin. Microbiol.43(4), 1947–1949 (2005).
- Han J, Swan DC, Smith SJ et al. Simultaneous amplification and identification of 25 human papillomavirus types with Templex technology. J. Clin. Microbiol.44(11), 4157–4162 (2006).
- Tang YW, Kilic A, Yang Q et al. StaphPlex system for rapid and simultaneous identification of antibiotic resistance determinants and Panton–Valentine leukocidin detection of staphylococci from positive blood cultures. J. Clin. Microbiol.45(6), 1867–1873 (2007).
- La Scola B, Raoult D. Direct identification of bacteria in positive blood culture bottles by matrix-assisted laser desorption ionisation time-of-flight mass spectrometry. PLoS ONE4(11), e8041 (2009).
- Seng P, Drancourt M, Gouriet F et al. Ongoing revolution in bacteriology: routine identification of bacteria by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Clin. Infect. Dis.49(4), 543–551 (2009).
- Maes N, De Gheldre Y, De Ryck R et al. Rapid and accurate identification of Staphylococcus species by tRNA intergenic spacer length polymorphism analysis. J. Clin. Microbiol.35(10), 2477–2481 (1997).
- Martineau F, Picard FJ, Ke D et al. Development of a PCR assay for identification of staphylococci at genus and species levels. J. Clin. Microbiol.39(7), 2541–2547 (2001).
- Poyart C, Quesne G, Boumaila C, Trieu-Cuot P. Rapid and accurate species-level identification of coagulase-negative staphylococci by using the sodA gene as a target. J. Clin. Microbiol.39(12), 4296–4301 (2001).
- Jordan JA, Durso MB. Real-time polymerase chain reaction for detecting bacterial DNA directly from blood of neonates being evaluated for sepsis. J. Mol. Diagn.7(5), 575–581 (2005).
- Klouche M, Schroder U. Rapid methods for diagnosis of bloodstream infections. Clin. Chem. Lab. Med.46(7), 888–908 (2008).
- Lehmann LE, Hunfeld KP, Emrich T et al. A multiplex real-time PCR assay for rapid detection and differentiation of 25 bacterial and fungal pathogens from whole blood samples. Med. Microbiol. Immunol.197(3), 313–324 (2008).
- Bloos F, Hinder F, Becker K et al. A multicenter trial to compare blood culture with polymerase chain reaction in severe human sepsis. Intensive Care Med.36(2), 241–247 (2009).
- Dierkes C, Ehrenstein B, Siebig S, Linde HJ, Reischl U, Salzberger B. Clinical impact of a commercially available multiplex PCR system for rapid detection of pathogens in patients with presumed sepsis. BMC Infect. Dis.9, 126 (2009).
- Lehmann LE, Alvarez J, Hunfeld KP et al. Potential clinical utility of polymerase chain reaction in microbiological testing for sepsis. Crit. Care Med.37(12), 3085–3090 (2009).
- Lehmann LE, Hunfeld KP, Steinbrucker M et al. Improved detection of blood stream pathogens by real-time PCR in severe sepsis. Intensive Care Med.36(1), 49–56. (2009).
- Louie RF, Tang Z, Albertson TE, Cohen S, Tran NK, Kost GJ. Multiplex polymerase chain reaction detection enhancement of bacteremia and fungemia. Crit. Care Med.36(5), 1487–1492 (2008).
- Tsalik EL, Jones D, Nicholson B et al. Multiplex PCR to diagnose blood stream infections in patients admitted from the emergency department with sepsis. J. Clin. Microbiol.48(1), 26–33 (2010).
- von Lilienfeld-Toal M, Lehmann LE, Raadts AD et al. Utility of a commercially available multiplex real-time PCR assay to detect bacterial and fungal pathogens in febrile neutropenia. J. Clin. Microbiol.47(8), 2405–2410 (2009).
- Wallet F, Nseir S, Baumann L et al. Preliminary clinical study using a multiplex real-time PCR test for the detection of bacterial and fungal DNA directly in blood. Clin. Microbiol. Infect. DOI: 10.1111/j.1469-0691.2009.02940.x (2009) (Epub ahead of print).
- Westh H, Lisby G, Breysse F et al. Multiplex real-time PCR and blood culture for identification of bloodstream pathogens in patients with suspected sepsis. Clin. Microbiol. Infect.15(6), 544–551 (2009).
- Mancini N, Clerici D, Diotti R et al. Molecular diagnosis of sepsis in neutropenic patients with haematological malignancies. J. Med. Microbiol.57(Pt 5), 601–604 (2008).
- Struelens MJ. Detection of microbial DNAemia: does it matter for sepsis management?. Intensive Care Med.36(2), 193–195 (2009).
- Morata P, Queipo-Ortuno MI, de Dios Colmenero J. Strategy for optimizing DNA amplification in a peripheral blood PCR assay used for diagnosis of human brucellosis. J. Clin. Microbiol.36(9), 2443–2446 (1998).
- Cogswell FB, Bantar CE, Hughes TG, Gu Y, Philipp MT. Host DNA can interfere with detection of Borrelia burgdorferi in skin biopsy specimens by PCR. J. Clin. Microbiol.34(4), 980–982 (1996).
- Peters RP, van Agtmael MA, Gierveld S et al. Quantitative detection of Staphylococcus aureus and Enterococcus faecalis DNA in blood to diagnose bacteremia in patients in the intensive care unit. J. Clin. Microbiol.45(11), 3641–3646 (2007).
- Handschur M, Karlic H, Hertel C, Pfeilstocker M, Haslberger AG. Preanalytic removal of human DNA eliminates false signals in general 16S rDNA PCR monitoring of bacterial pathogens in blood. Comp. Immunol. Microbiol. Infect. Dis.32(3), 207–219 (2009).
- Gebert S, Siegel D, Wellinghausen N. Rapid detection of pathogens in blood culture bottles by real-time PCR in conjunction with the pre-analytic tool MolYsis. J. Infect.57(4), 307–316 (2008).
- Hansen WL, Bruggeman CA, Wolffs PF. Evaluation of new preanalysis sample treatment tools and DNA isolation protocols to improve bacterial pathogen detection in whole blood. J. Clin. Microbiol.47(8), 2629–2631 (2009).
- Wellinghausen N, Kochem AJ, Disque C et al. Diagnosis of bacteremia in whole-blood samples by use of a commercial universal 16S rRNA gene-based PCR and sequence analysis. J. Clin. Microbiol.47(9), 2759–2765 (2009).
- Sachse S, Straube E, Lehmann M, Bauer M, Russwurm S, Schmidt KH. Truncated human cytidylate-phosphate-deoxyguanylate-binding protein for improved nucleic acid amplification technique-based detection of bacterial species in human samples. J. Clin. Microbiol.47(4), 1050–1057 (2009).
- Hofstadler SA, Sampath R, Blyn LB et al. TIGER: the universal biosensor. Int. J. Mass Spectrom.242, 23–41 (2005).
- Ecker DJ, Sampath R, Massire C et al. Ibis T5000: a universal biosensor approach for microbiology. Nat. Rev. Microbiol.6(7), 553–558 (2008).
- Ecker DJ, Drader J, Gutierrez J et al. The Ibis T5000 Universal Biosensor – an automated platform for pathogen identification and strain typing. JALA11, 341–351 (2006).
- Ecker DJ, Sampath R, Blyn LB et al. Rapid identification and strain-typing of respiratory pathogens for epidemic surveillance. Proc. Natl Acad. Sci. USA102(22), 8012–8017 (2005).
- Hall TA, Sampath R, Blyn LB et al. Rapid molecular genotyping and clonal complex assignment of S. aureus isolates by PCR/ESI-MS. J. Clin. Microbiol.47(6), 1733–1741 (2009).
- Wolk DM, Blyn LB, Hall TA et al. Pathogen profiling: rapid molecular characterization of Staphylococcus aureus by PCR/ESI-MS and correlation with phenotype. J. Clin. Microbiol.47(10), 3129–3137 (2009).
- Ubukata K, Nonoguchi R, Matsuhashi M, Konno M. Expression and inducibility in Staphylococcus aureus of the mecA gene, which encodes a methicillin-resistant S. aureus-specific penicillin-binding protein. J. Bacteriol.171(5), 2882–2885 (1989).
- Handwerger S, Perlman DC, Altarac D, McAuliffe V. Concomitant high-level vancomycin and penicillin resistance in clinical isolates of enterococci. Clin. Infect. Dis.14(3), 655–661 (1992).
- Yigit H, Queenan AM, Anderson GJ et al. Novel carbapenem-hydrolyzing β-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob. Agents Chemother.45(4), 1151–1161 (2001).
- Petti CA, Bosshard PP, Brandt ME et al. Interpretive criteria for identification of bacteria and fungi by DNA target sequencing; approved guideline. Clin. Lab. Standards Inst.28, 88 (2008).
- Eshoo MW, Crowder CD, Li H et al. Detection and identification of Ehrlichia species in blood using PCR and electrospray ionization mass spectrometry. J. Clin. Microbiol.48(2), 472–478 (2010).
- Tang YW, Rys PN, Rutledge BJ, Mitchell PS, Smith TF, Persing DH. Comparative evaluation of colorimetric microtiter plate systems for detection of herpes simplex virus in cerebrospinal fluid. J. Clin. Microbiol.36(9), 2714–2717 (1998).
- Thomas LD, Hongo I, Bloch KC, Tang YW, Dummer S. Human ehrlichiosis in transplant recipients. Am. J. Transplant.7(6), 1641–1647 (2007).
- Paddock CD, Folk SM, Shore GM et al. Infections with Ehrlichia chaffeensis and Ehrlichia ewingii in persons coinfected with human immunodeficiency virus. Clin. Infect. Dis.33(9), 1586–1594 (2001).
- Doyle CK, Labruna MB, Breitschwerdt EB et al. Detection of medically important Ehrlichia by quantitative multicolor TaqMan real-time polymerase chain reaction of the dsb gene. J. Mol. Diagn.7(4), 504–510 (2005).
- Eremeeva ME, Dasch GA, Silverman DJ. Evaluation of a PCR assay for quantitation of Rickettsia rickettsii and closely related spotted fever group rickettsiae. J. Clin. Microbiol.41(12), 5466–5472 (2003).
- Peters RP, Mohammadi T, Vandenbroucke-Grauls CM, Danner SA, van Agtmael MA, Savelkoul PH. Detection of bacterial DNA in blood samples from febrile patients: underestimated infection or emerging contamination?. FEMS Immunol. Med. Microbiol.42(2), 249–253 (2004).
- Hong T, Butler WR, Hollis F et al. Characterization of a novel rapidly growing Mycobacterium species associated with sepsis. J. Clin. Microbiol.41(12), 5650–5653 (2003).
- Kilic A, Li H, Mellmann A et al.Acinetobacter septicus sp. nov. association with a nosocomial outbreak of bacteremia in a neonatal intensive care unit. J. Clin. Microbiol.46(3), 902–908 (2008).
- Tang YW, Hopkins MK, Kolbert CP, Hartley PA, Severance PJ, Persing DH. Bordetella holmesii-like organisms associated with septicemia, endocarditis, and respiratory failure. Clin. Infect. Dis.26(2), 389–392 (1998).