Abstract
Leukodystrophies are white matter disorders that are genetic in nature. In the young, they represent an important cause of progressive neurological disability. They are frequently recognized on MRI, but their identification remains a challenge. Their diagnosis is important for prognostication, palliative and experimental treatment, as well as family screening. The diagnostic strategy rests upon clinical clues and MRI patterns, complemented by appropriately selected electrophysiological and laboratory testing. Considerable overlap exists between white and gray matter disease, as neuronal degeneration will result in myelin loss. An understanding of the pathophysiology and natural disease evolution is necessary to understand the risks and benefits of experimental and palliative treatments.
Reproduced with permission from Citation[74]. Copyright © 2005, Wolters Kluwer Health.
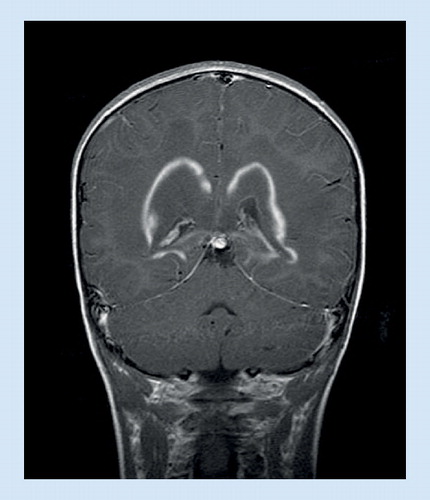
![Figure 1. Hypodontia in hypomyelination, hypodontia, hypogonadotropic hypogonadism (HHHH syndrome).Reproduced with permission from Citation[74]. Copyright © 2005, Wolters Kluwer Health.](/cms/asset/9f9045ff-8691-48dc-a665-918118c3d848/iern_a_11212636_f0001_b.jpg)
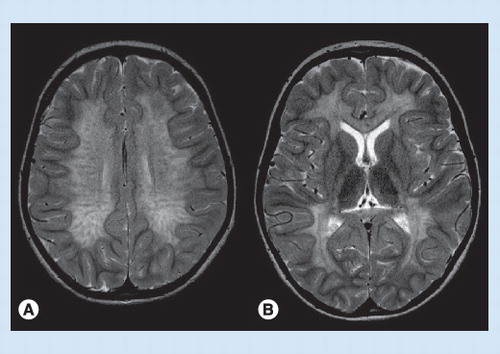
(A) The central white matter shows signal hyperintensity and a streaky pattern, while the subcortical U fibers are spared. (B) Involvement of corpus callosum and posterior limb of internal capsule.
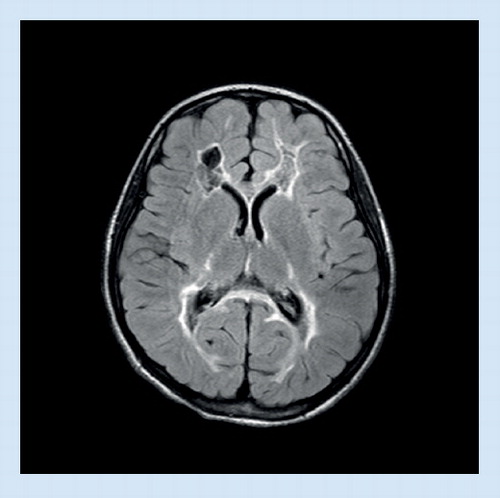
(A) Signal abnormalities in the medulla, (B) the hilus of the dentate nucleus, and (C) a thin periventricular rim. (D & E) Peculiar garland-like structures are present along the ventricular wall. (F) Swelling and prominent signal abnormality of the cervical spinal cord.
Reproduced with permission from Citation[75]. Copyright © 2006, Wolters Kluwer Health.
![Figure 3. MRI in juvenile Alexander disease.(A) Signal abnormalities in the medulla, (B) the hilus of the dentate nucleus, and (C) a thin periventricular rim. (D & E) Peculiar garland-like structures are present along the ventricular wall. (F) Swelling and prominent signal abnormality of the cervical spinal cord.Reproduced with permission from Citation[75]. Copyright © 2006, Wolters Kluwer Health.](/cms/asset/9f3a2b45-a032-4c74-8142-3779b871d54d/iern_a_11212636_f0003_b.jpg)
Contrast enhancement around a bilateral zone of white matter.
(A) Signal changes in the bilateral thalami, (B) optic nerve enlargement.
Reproduced with permission from Citation[76].
![Figure 6. MRI in globoid cell leukodystrophy (Krabbe disease).(A) Signal changes in the bilateral thalami, (B) optic nerve enlargement.Reproduced with permission from Citation[76].](/cms/asset/6fab3c77-7891-400e-9ea7-15aedeac6def/iern_a_11212636_f0006_b.jpg)
The term ‘leukodystrophy’ refers to deterioration of white matter of the brain. The deterioration coincides with clinical regression of skills, and in the most severe cases neurological devastation. Leukodystrophies are genetic diseases with degeneration of myelin sheaths in the CNS and sometimes also in peripheral nerves. Their basic defect is directly related to the synthesis and maintenance of myelin membranes. Defects causing secondary myelin damage are called leukoencephalopathies. Most leukodystrophies manifest themselves during childhood or adolescence, are incurable and have a progressive course, leading to premature death. Diagnosis is important as palliative or experimental therapies may offer benefits, for reproductive counseling and family screening of currently unaffected individuals. The few treatments available are more effective in the early stages.
Making the diagnosis of a leukodystrophy requires knowledge of clinical features and neuroimaging. Familiarity with the typical age of onset of various phenotypes of leukodystrophies, as well as heightened vigilance to brain MRI patterns, is invaluable in the diagnostic algorithm. In addition, electrophysiological findings, specific laboratory tests and other special investigations may be needed to arrive at a precise diagnosis. The list of defined leukodystrophies and genetic leukoencephalopathies to consider in the young is extensive and is still growing. In this article, we review the current knowledge on recognition and management of these disorders from a pediatric viewpoint. In view of the complex designations of the disorders, we have resorted to using abbreviations that are increasingly found in the literature.
Clinical features & the diagnosis of a leukodystrophy
Age of onset
The leading symptoms of a leukodystrophy are neurological and appear, with few exceptions, in previously healthy children. The clinical onset is frequently insiduous, and symptoms then usually progress slowly, with possible periods of stagnation. Seemingly vague (and otherwise unexplained) progressive motor or mental symptoms in a young person may direct suspicion towards a leukodystrophy.
is a near exhaustive list of leukodystrophies and genetic leukoencephalopathies occurring in the young. A single one of these genetically defined diseases can have widely variable phenotypes that may manifest, depending on the severity of mutations, at any age between infancy and adulthood. This bewildering variety of diagnostic possibilities requires a clinical strategy. Starting from the age of the patient at the onset of symptoms, certain types of leukodystrophies can be considered .
General physical features
Most patients with leukodystrophies do not have any physical abnormality. Some have a large head (Alexander disease, Canavan disease, megalencephalic leukodystrophy with cysts and vanishing white matter disease). Very rarely (fucosidosis and metachromatic leukodystrophy [MLD] with multiple sulfatase deficiency) they have dysmorphic features and skeletal abnormalities that resemble those seen in mucopolysaccharidoses. Dental abnormalities are seen in some forms of hypomyelination .
Neurologic features
Neurological symptoms of leukodystrophies consist of progressive motor symptoms (mostly spasticity) and changes in cognition and language. Peripheral nerve involvement is present in certain forms (MLD, globoid cell leukodystrophy and some hypomyelinating disorders) and may lead to the unusual combination of spasticity and reduced muscle stretch reflexes. Early or recalcitrant seizures can occur, but are an unusual feature of disorders mainly affecting brain white matter.
Neuroimaging & the diagnosis of a leukodystrophy
An MRI of the head is the most important ancillary test in a patient suspected of having a leukodystrophy. The minimal requirement for a standard investigation is T1- and T2-weighted and fluid-attenuated inversion-recovery (FLAIR) images. The usefulness of other techniques depends on individual circumstances and is discussed below.
The correlation of MRI results with clinical findings can vary widely. The evaluation of certain patterns and localizations of lesions is crucial in differentiating the metabolic/degenerative characteristics of a disorder from purely inflammatory, tumorous or vascular etiologies. In infancy, the rapid changes in myelination must be taken into account. A comprehensive MRI-based approach to the diagnosis of white matter disorders has recently been published Citation[1]. Its authors suggest a stepwise image analysis using the following discriminators:
First, is hypomyelination (delayed myelination or permanent hypomyelination) or some other brain white matter pathology present? In young infants, the deposition of myelin can best be followed on T1-weighted images. A child over 1.5 years of age has accumulated enough myelin to let the white matter appear dark on T2-weighted images. A high signal on T2-weighted images is, therefore, abnormal for cerebral white matter after this age. The important differentiation between delayed myelination and permanent hypomyelination (and also the recognition of ongoing demyelination) can be made on two MRIs with a significant time interval.
Second, are the white matter abnormalities confluent and bilateral, essentially symmetric, as is typical of genetic white matter disorders, or multifocal and asymmetric as frequently seen in acquired disorders?
Third, if confluent white matter abnormalities are present, where is their predominant localization? The major preferential localizations are frontal, parieto-occipital, periventricular, subcortical, diffuse cerebral and in the posterior fossa, each of which are associated with certain forms of leukodystrophy.
A rough classification of leukodystrophies according to their presentation on neuroimaging is shown in Box 1.
Certain typical MRI patterns are seen in several different leukodystrophies, for example, the streaky ‘tigroid’ appearance of central white matter in MLD and globoid cell leukodystrophy , or the sparing of the subcortical U-fibers as seen, for example, in X-linked adrenoleukodystrophy (X-ALD) and MLD. Streaky patterns are related to the anatomy of the Virchow–Robin spaces in the brain and the accumulation of storage material or fluid around blood vessels. Other patterns are so characteristic as to practically allow a diagnosis. This may be possible, in such cases as juvenile Alexander disease (AD) and vanishing white matter disease. Hypomyelination is observed in a growing number of genetic disorders that have frequently been classified by recognition of their characteristic MRI pattern Citation[2].
Contrast enhancement is seen in leukodystrophies with an inflammatory component, particularly in X-ALD . Cystic lesions are best detected using FLAIR studies . Magnetic resonance spectroscopy (MRS) is helpful for detecting elevated N-acetyl aspartate (NAA) in Canavan disease, or lactate in leukoencephalopathy with brainstem and spinal cord involvement and elevated lactate Citation[3], and other mitochondrial disorders Citation[4]. A decreased NAA peak indicates neuronal involvement occurring in primary white matter disease. Massive swelling of the optic nerves may be seen in infantile globoid cell leukodystrophy . Peripheral nerves can also be thickened in this disorder, and spinal roots may show contrast enhancement. When detecting calcifications, CT is superior to MRI.
A quantitative scoring of MRI lesions has been devised for X-ALD and is widely used to assess eligibility for treatments such as hematologic stem cell transplantation Citation[5,6]. Analogous scores have also been used to describe the lesions in patients with globoid cell leukodystrophy Citation[7] and MLD Citation[8].
Careful image analysis, making use of standard works on myelination disorders Citation[9] and sometimes asking for an expert opinion, will save much time and unnecessary expensive laboratory studies.
Electrophysiology & leukodystrophies
Evoked potentials and nerve conduction velocities, particularly after the first decade, often reveal symmetric involvement of long spinal tracts and peripheral nerves, and are thus helpful in differentiating leukodystrophies from other demyelinating disorders. Furthermore, the presence of peripheral nerve involvement on nerve conduction studies (NCS) can prove valuable in differentiating certain leukodystrophies from others. For instance, patients with X-ALD show normal nerve conduction velocities, while patients with metachromatic or globoid cell leukodystrophy commonly display abnormalities.
Nerve conduction studies
In Krabbe disease, the severity of abnormalities in NCS appears to correlate with clinical severity Citation[10,11]. In general, NCS more frequently reveals abnormalities in symptomatic patients than evoked responses. In general, the degree of MRI abnormality correlates with that seen on NCS and clinical severity. Thus NCS, in combination with neuroimaging studies, appear to be the most sensitive laboratory measures to help assess the severity and the phenotypic classification of globoid cell leukodystrophy.
Evoked potentials
For boys with X-ALD, brainstem auditory evoked responses (BAER) are usually normal in the first decade of life. BAER later become abnormal in the course of the disease when demyelinating lesions extend in the brainstem and spinal cord. Visual-evoked potentials (VEP) in X-ALD become abnormal once there are extensive demyelinating lesions in the occipital white matter, somatosensory-evoked potentials and motor-evoked responses even later in the course of the disease. Even patients with a normal MRI may have an abnormal neurophysiologic pattern identical to that seen in adrenomyeloneuropathy (AMN) patients (evidently milder in term of abnormalities); usually BAER are first to be abnormal, then somatosensory-evoked potential of the lower limbs and then motor-evoked responses to lower limbs Citation[12].
Laboratory tests in leukodystrophies
Laboratory tests ordered prior to a thorough evaluation of clinical and imaging findings are frequently of low yield and cause high costs. There is no routine all-encompassing laboratory protocol for a suspected leukodystrophy. The utility of individual tests is established through the findings on a focused clinical exam and suspicion arising from an analysis of the imaging pattern. Tests used to establish or confirm a specific diagnosis are listed in (last column). A number of tests that may be useful to do relatively early in the diagnostic process include those listed below .
Evaluation of other organ function in leukodystrophies
Ophthalmology
Cataracts can be seen in association with cerebrotendinous xanthomatosis and certain forms of hypomyelination . On retinal examination, the presence of a ‘cherry red spot’ can help distinguish infantile and/or macrocephalic patients from infantile GM2 gangliosidosis (Tay–Sachs and Sandhoff disease).
Endocrinology
X-linked adrenoleukodystrophy can also manifest as Addison’s disease without evidence of neurological involvement Citation[13,14]. Such patients are referred to as the ‘Addison-only’ phenotype. More than 70% of all male patients will have adrenal insufficiency. Usually glucocorticoids are more affected than mineralocorticoids. X-ALD is estimated to be the cause of adrenal insufficiency in approximately 35% of patients with idiopathic Addison’s disease Citation[15]. Prior studies have demonstrated Addisonian crisis as a common cause of acute presentation of childhood X-ALD.
Besides adrenal dysfunction, other endocrine systems, such as that of the ovaries, can be affected in leukodystrophy patients. Primary or secondary ovarian failure can be seen to be associated with all degrees of neurologic severity in vanishing white matter disease Citation[16]. In rare severe variants, growth failure, cataracts, hepatosplenomegaly, pancreatitis and kidney hypoplasia can occur Citation[17].
GI issues
The association of gallbladder papillomatosis with MLD is well known. Less well known is the occurrence of intestinal bleeding in this context. Recently, a child with hemobilia was reported who required cholecystectomy Citation[18]. Caution regarding this life-threatening condition is necessary. Feeding and swallowing issues are a common feature of all advanced leukodystrophy patients and require evaluation by a multidisciplinary team.
Therapeutics in the leukodystrophies
While the prognosis for leukodystrophy patients is often dismal, this should not lead to therapeutic negligence and defeatism. There remains a valuable role in addressing both symptomatic management versus disease-modifying treatment with patients with leukodystrophies and their families. Many physicians recoil when confronted with a rare disorder and attribute all symptoms to the underlying disease and do not look for treatable causes.
Medical management
The prevention of secondary complications, such as infections and aspirations, are tantamount to preserving quality of life in leukodystrophy patients. Several studies have demonstrated improved survival in leukodystrophy patients over the last 50 years Citation[19]. This may be attributed to improved medical management, such as the prudent use of antibiotics and gastric tube placement. Careful evaluation with swallowing studies should be a cornerstone in monitoring patients with advancing brain disease. Gallbladder-associated pain and vomiting in MLD may be alleviated using ursodeoxycholic acid.
The use of antispasmodics and pain medication can further relieve muscle spasms and unnecessary suffering in children that are often nonverbal and unable to localize pain or voice complaints. Unexplained irritability should always prompt a thorough evaluation for the source of pain (muscle spasms, latent fracture and visceral pain) Citation[20]. A wide range of analgesics are available, but few careful studies. One study reported that caregivers stated improved irritability after use of gabapentin Citation[21].
More than 70% of male X-ALD patients have adrenocrotical insufficiency. As the insufficiency can be latent, adrenocorticotropic hormone levels need to be followed routinely during childhood. As adrenal replacement can be life saving, families should be educated about the importance of stress dose steroids. Beyond glucocorticoid replacement, some patients require fludrocortisone and, in the case of hypogonadism, androgens.
Enzyme replacement & metabolic correction
Enzyme replacement has been successful in ameliorating disease in animal models of metachromatic and globoid cell leukodystrophy, but has not so far been successful in humans Citation[22,23]. Both the larger volume of the human brain and the challenges of overcoming the BBB pose significant obstacles.
Lorenzo’s oil is a combination of erucic and oleic acid that is taken orally and lowers levels of plasma very-long-chain fatty acids in X-ALD patients Citation[24]. This is of value in asymptomatic boys, but unfortunately does not arrest progression once brain demyelination has set in. In the latter case, even aggressive immune suppression has failed and only timely bone marrow transplantation can stabilize patients.
Cell-based therapies
Bone marrow transplantation has been efficacious in certain genetic conditions for many years and is able to halt progression in the early stages of cerebral X-ALD Citation[25–27]. The procedure seems less efficacious for MLD Citation[28], as well as other leukodystrophies, and carries significant risks and hazards Citation[29].
Less than two-thirds of males with X-ALD will ever develop cerebral disease and a minority of patients with early cerebral disease may even arrest spontaneously. As a result, bone marrow transplantation should not be regarded as a therapy that all asymptomatic boys with X-ALD should undergo. The success of bone marrow transplantation in the early stages of the disease has not been demonstrated in boys with more advanced disease.
Gene therapy using a lentiviral vector for the correction of autologous stem cells has recently been developed as a therapeutic method for X-ALD Citation[30] and MLD. The use of autologous cells mobilized from peripheral blood reduces the risk of graft-versus-host complications. However, a risk remains of the most relevant hematopoietic stem cells not being corrected. Further close monitoring is warranted as there are reports of the integration of viral vectors causing cancer.
Definitive diagnosis of a leukodystrophy
Although leukodystrophies currently cannot be cured, it is of great importance to arrive at a definitive diagnosis in an individual patient. The diagnosis will put an end to a frequently long and distressing search for the cause of a child’s abnormalities. It may allow the family to focus on palliative care or more aggressively pursue experimental treatments. All in all, the diagnosis can facilitate a more realistic view of the patient’s further course, and appropriate family genetic counseling.
The initial sections of this article will facilitate the path to diagnosis by narrowing the spectrum of suspected disorders. It allows the physician and family to economize efforts and resources by ordering a relatively small number of diagnostic tests. At this point it may be useful to consult . An initial classification of a patient’s findings as ‘white matter disease’, as opposed to gray matter disease, may be useful in many instances, but considerable overlap exists among these two groups: certain neuronal storage disorders can impress as ‘leukodystrophy-like’ on imaging because degeneration of neurons will also result in a loss of myelin.
Expert commentary
The field of leukodystrophies is growing owing to advances in MR diagnostics and an explosion of gene discovery over the last decade. We have outlined a clinical approach that remains indispensable, even with advances in technology. Leukodystrophies are caused by mutations in single genes and follow Mendelian genetics. Currently, genes underlying Mendelian traits are being discovered at a threefold higher rate compared with those for complex traits Citation[31]. Therefore, we expect the number of identifiable leukodystrophies to increase in the coming years, as well as their burden on the healthcare system and society at large.
A clinical diagnosis is necessary: despite the array of new MRI and gene sequencing technologies, the clinical diagnosis remains tantamount. The age of onset and the pattern of neurological involvement dictates the workup and allows for a successful diagnosis. The diagnostic test alone is meaningless if not placed into a clinical context. Despite solid biochemical assays being available for most classic leukodystrophies, these tests do not distinguish between different phenotypes (e.g., the level of plasma very-long-chain fatty acids does not predict childhood cerebral adrenoleukodystrophy vs AMN). In other cases, the clinical description will help resolve false-positive and false-negative tests.
The diagnosis has consequences for clinical management: not unlike most other neurological disorders there is little chance for a cure in leukodystrophy patients. Yet treatments are available and the single gene mutations that impact specific enzyme and protein function allow for potential interventions (e.g., chenodeoxycholic acid in cerebrotendinous xanthomatosis). Symptomatic treatments for pain, spasticity and seizures are widely used but lack controlled studies in the field of leukodystrophies. A vast array of supplements are being utilized without good justification and with considerable cost burden to families or insurance schemes. In rare cases, the response to trials such L-dopa have been useful and revealed another diagnosis Citation[32]. However, the success of treatments for disorders outside the CNS such as Gaucher and Pompe disease, remains out of reach. While often undervalued, the diagnosis and phenotypic designation can guide medical management and help families to make decisions on gastric tube placement and other life-prolonging procedures.
Quality of life often improves with diagnosis: in the early stages of disability the diagnosis can be horrifying and families are at times bewildered, incredulous and devastated. However, more often the diagnosis brings relief, particularly to those families who have been seeking a diagnosis for a long time. Although the disorder remains incurable and often not treatable, it brings closure and allows families to focus on palliative care and quality of life for their loved ones. There now exists a wide range of palliative care options Citation[20,33,34]. These include medical, psychiatric, psychological, educational and other options. Their impact will often depend on a thorough understanding of the nature of the disorder.
Opportunities for experimental treatments: while experimental treatments for leukodystrophies are in their infancy, one aspect is clear: the therapeutic window is often limited to early manifestations of the disease. In unaffected patients, the risk may not be justified. In more advanced patients, the disease may be too far gone and aggravated by the side effects of the intervention. Again, clinical judgment and the weighing of risks and potential benefits will guide the individual patient. As gene therapy and enzyme replacement emerge, physicians will increasingly face difficult questions that require an understanding of the pathogenesis, disease course and training in clinical trials. Ethical dilemmas are bound to occur.
Do not think ‘rare’. Collectively the leukodystrophies rival the incidence of other demyelinating disorders, such as multiple sclerosis. Yet the relative rarity has shaped the mindset of many physicians and patients lament the fact that they have to educate residents and physicians in emergency rooms (‘why do I have such a rare disease that not even my doctors know about?’). Many patients avoid hospitals because of a lack of trust that the medical establishment will be aware of their diagnosis. Certainly our age will increasingly depend on electronic services and the internet to disseminate information. Networks of expert healthcare providers will partner with disease foundations and serve patients and families, as well as general practitioners, as no one alone can be expected to keep up with the growth of information.
Five-year view
Research to date on leukodystrophies suggests several different avenues for study in the next 5 years. Advances in gene sequencing technologies, improved design of clinical trials, the development of new biomarkers and genetic animal models, improved identification of susceptibility factors and more efficient drug delivery are just a few of the reasons for progress.
Currently, out of nearly 7000 suspected or known Mendelian disorders identified based on clinical features, less than half have been linked to a gene Citation[35]. In the leukodystrophies, more than a dozen clinically defined disorders exist whose genetic etiology has not been found. With the advances in next generation sequencing technology we expect this to change, as large volumes of sequence data will be delivered at low cost Citation[36]. Even rare diseases in individual families will thereby have access to gene identification. Bioinformatics tools have been standardized for DNA sequencing and are making progress in the field of RNA.
The study of leukodystrophies remains challenging owing to the limited number of study subjects for clinical trials. The nature of leukodystrophies lends itself to the use of MRI as biomarker. Yet alternative study designs even without imaging should be considered (factorial and n-of-1 studies Citation[37]). The prudent choice of outcome measures can allow study questions to be answered with fewer subjects. This can also be achieved by monitoring trials during their conduct.
Susceptibility factors for some leukodystrophies, such as head trauma and fever for vanishing white matter disease, have long been known. We expect natural history studies in the coming years to add more detail to these environmental factors, as well as experimental studies to elucidate genetic modifiers.
Overall we expect these advances to improve the prospects to forestall onset of illness and clinical decline in the growing number of leukodystrophies.
Table 1. Childhood leukodystrophies and genetic leukoencephalopathies†.
Table 2. Leukodystrophies to consider in relation to age of patient at onset.
Table 3. Some laboratory tests useful in diagnosing a leukodystrophy.
Box 1. Childhood leukodystrophies according to major neuroimaging patterns.
Disorders with confluent MRI lesions
• Alexander disease†
• Canavan disease
• Globoid cell leukodystrophy
• Leukoencephalopathy with metaphyseal chondrodysplasia
• Metachromatic leukodystrophy
• Metachromatic leukodystrophy with multiple sulfatase deficiency
• Mitochondrial disorders
• X-linked adrenoleukodystrophy
Disorders with cavitating MRI lesions
• Cystical leukoencephalopathy without megalencephaly
• Glycine leukoencephalopathy
• Leukoencephalopathy with calcifications and cysts
• Megalencephalic leukodystrophy with cysts
• Progressive cavitating leukoencephalopathy
Disorders with hypomyelination
• Fucosidosis
• Folate receptor defect
• Hypomyelination with atrophy of the basal ganglia and cerebellum
• Hypomyelination and congenital cataract
• Hypomyelination, hypodontia, hypogonadotropic hypogonadism
• Hypomyelination with monocarboxylate transporter-8 deficiency
• Pelizaeus–Merzbacher disease
• Pelizaeus–Merzbacher-like disease
• Sialic acid storage disorder
• Tremor-ataxia with central hypomyelination
Disorders with Calcifications
• Aicardi–Goutières syndrome
• Cerebrotendinous xanthomatosis
• Leukoencephalopathy with calcifications and cysts
Key issues
• Brain white matter abnormalities are a frequent cause of CNS symptoms in children.
• A great variety of gene defects contribute to brain white matter abnormalities of the young.
• Defects within the myelin sheath cause leukodystrophies, those outside cause metabolic leukoencephalopathies. They may resemble each other.
• Brain white matter and gray matter diseases may overlap.
• The disorders are incurable and most lead to progressive motor and mental disability.
• Suspicion for such a disorder is usually raised by MRI, but the definitive diagnosis is a challenge.
• A timely diagnosis is required for family counseling and optimizing palliative care and experimental treatments.
• Diagnosis starts from clues on physical examination and an awareness of MRI algorithm, and is complemented by targeted laboratory testing.
• Management is multidisciplinary. It involves pediatric disciplines, including child psychiatry and social support of families.
Financial & competing interests disclosure
Alfried Kohlschütter receives financial support from LeukoTreat (http://leukotreat.eu), a project of the EU to develop treatment strategies for leukodystrophies. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Notes
†Infantile type.
References
- Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology72(8), 750–759 (2009).
- Steenweg ME, Vanderver A, Blaser S et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain133(10), 2971–2982 (2010).
- Lin J, Faria EC, Da Rocha AJ et al. Leukoencephalopathy with brainstem and spinal cord involvement and normal lactate: a new mutation in the DARS2 gene. J. Child Neurol.25(11), 1425–1428 (2010).
- Jose RA, Tulio BF, Carlos MMA Jr et al. Lactate detection by MRS in mitochondrial encephalopathy: optimization of technical parameters. J. Neuroimaging18(1), 1–8 (2008).
- Loes DJ, Fatemi A, Melhem ER et al. Analysis of MRI patterns aids prediction of progression in X-linked adrenoleukodystrophy. Neurology61(3), 369–374 (2003).
- Beam D, Poe MD, Provenzale JM et al. Outcomes of unrelated umbilical cord blood transplantation for X-linked adrenoleukodystrophy. Biol. Blood Marrow Transplant.13(6), 665–674 (2007).
- Provenzale JM, Peddi S, Kurtzberg J, Poe MD, Mukundan S, Escolar M. Correlation of neurodevelopmental features and MRI findings in infantile Krabbe’s disease. AJR Am. J. Roentgenol.192(1), 59–65 (2009).
- Eichler F, Grodd W, Grant E et al. Metachromatic leukodystrophy: a scoring system for brain MR imaging observations. AJNR Am. J. Neuroradiol.30(10), 1893–1897 (2009).
- Van der Knaap MS, Valk J. Magnetic Resonance of Myelination and Myelin Disorders. Birkhäuser (Ed.). Springer, Berlin. Germany (2005).
- Husain AM, Altuwaijri M, Aldosari M. Krabbe disease: neurophysiologic studies and MRI correlations. Neurology63(4), 617–620 (2004).
- Siddiqi ZA, Sanders DB, Massey JM. Peripheral neuropathy in Krabbe disease: electrodiagnostic findings. Neurology67(2), 263–267 (2006).
- Aubourg P, Adamsbaum C, Lavallard-Rousseau MC et al. Brain MRI and electrophysiologic abnormalities in preclinical and clinical adrenomyeloneuropathy. Neurology42(1), 85–91 (1992).
- Moser HW, Raymond GV, Dubey P. Adrenoleukodystrophy: new approaches to a neurodegenerative disease. JAMA294(24), 3131–3134 (2005).
- Dubey P, Raymond GV, Moser AB, Kharkar S, Bezman L, Moser HW. Adrenal insufficiency in asymptomatic adrenoleukodystrophy patients identified by very long-chain fatty acid screening. J. Pediatr.146(4), 528–532 (2005).
- Laureti S, Casucci G, Santeusanio F, Angeletti G, Aubourg P, Brunetti P. X-linked adrenoleukodystrophy is a frequent cause of idiopathic Addison’s disease in young adult male patients. J. Clin. Endocrinol. Metab.81(2), 470–474 (1996).
- van der Knaap MS, Pronk JC, Scheper GC. Vanishing white matter disease. Lancet Neurol.5(5), 413–423 (2006).
- van der Knaap MS, van Berkel CG, Herms J et al. eIF2B-related disorders: antenatal onset and involvement of multiple organs. Am. J. Hum. Genet.73(6), 1199–1207 (2003).
- Garavelli L, Rosato S, Mele A et al. Massive hemobilia and papillomatosis of the gallbladder in metachromatic leukodystrophy: a life-threatening condition. Neuropediatrics40(6), 284–286 (2009).
- Mahmood A, Berry J, Wenger DA et al. Metachromatic leukodystrophy: a case of triplets with the late infantile variant and a systematic review of the literature. J. Child Neurol.25(5), 572–580 (2009).
- Klick JC, Hauer J. Pediatric palliative care. Curr. Probl. Pediatr. Adolesc. Health Care40(6), 120–151 (2010).
- Hauer JM, Wical BS, Charnas L. Gabapentin successfully manages chronic unexplained irritability in children with severe neurologic impairment. Pediatrics119(2), e519–e522 (2007).
- Matzner U, Lullmann-Rauch R, Stroobants S et al. Enzyme replacement improves ataxic gait and central nervous system histopathology in a mouse model of metachromatic leukodystrophy. Mol. Ther.17(4), 600–606 (2009).
- Kondo Y, Wenger DA, Gallo V, Duncan ID. Galactocerebrosidase-deficient oligodendrocytes maintain stable central myelin by exogenous replacement of the missing enzyme in mice. Proc. Natl Acad. Sci. USA102(51), 18670–18675 (2005).
- Moser HW, Raymond GV, Lu SE et al. Follow-up of 89 asymptomatic patients with adrenoleukodystrophy treated with Lorenzo’s oil. Arch. Neurol.62(7), 1073–1080 (2005).
- Aubourg P, Blanche S, Jambaque I et al. Reversal of early neurologic and neuroradiologic manifestations of X-linked adrenoleukodystrophy by bone marrow transplantation. N. Engl. J. Med.322(26), 1860–1866 (1990).
- Peters C, Charnas LR, Tan Y et al. Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999. Blood104(3), 881–888 (2004).
- Escolar ML, Poe MD, Provenzale JM et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N. Engl. J. Med.352(20), 2069–2081 (2005).
- Biffi A, Lucchini G, Rovelli A, Sessa M. Metachromatic leukodystrophy: an overview of current and prospective treatments. Bone Marrow Transplant.42(Suppl. 2), S2–S6 (2008).
- Orchard PJ, Tolar J. Transplant outcomes in leukodystrophies. Semin. Hematol.47(1), 70–78 (2010).
- Benhamida S, Pflumio F, Dubart-Kupperschmitt A et al. Transduced CD34+ cells from adrenoleukodystrophy patients with HIV-derived vector mediate long-term engraftment of NOD/SCID mice. Mol. Ther.7(3), 317–324 (2003).
- Glazier AM, Nadeau JH, Aitman TJ. Finding genes that underlie complex traits. Science298(5602), 2345–2349 (2002).
- Thibert R, Hyland K, Chiles J, Steinberg S, Eichler F. Levodopa response reveals sepiapterin reductase deficiency in a female heterozygote with adrenoleukodystrophy. J. Inherit. Metab. Dis. (2011) (In press).
- Hauer J. Identifying and managing sources of pain and distress in children with neurological impairment. Pediatr. Ann.39(4), 198–205 (2010).
- Malcolm C, Forbat L, Anderson G, Gibson F, Hain R. Challenging symptom profiles of life-limiting conditions in children: a survey of care professionals and families. Palliat. Med.25(4), 357–364 (2011).
- Kaiser J. Human genetics. Affordable ‘exomes’ fill gaps in a catalog of rare diseases. Science330(6006), 903 (2010).
- Metzker ML. Sequencing technologies – the next generation. Nat. Rev. Genet.11(1), 31–46 (2010).
- Nikles J, Mitchell GK, Schluter P et al. Aggregating single patient (n-of-1) trials in populations where recruitment and retention was difficult: the case of palliative care. J. Clin. Epidemiol.64(5), 471–480 (2011).
- Davison JE, Davies NP, English MW et al. Magnetic resonance spectroscopy in the diagnostic evaluation of brainstem lesions in Alexander disease. J. Child Neurol.26(3), 356–360 (2011).
- Orcesi S, Pessagno A, Biancheri R et al. Aicardi–Goutières syndrome presenting atypically as a sub-acute leukoencephalopathy. Eur. J. Paediatr. Neurol.12(5), 408–411 (2008).
- Orcesi S, La Piana R, Fazzi E. Aicardi–Goutières syndrome. Br. Med. Bull.89, 183–201 (2009).
- Zano S, Malik R, Szucs S, Matalon R, Viola RE. Modification of aspartoacylase for potential use in enzyme replacement therapy for the treatment of Canavan disease. Mol. Genet. Metab.102(2), 176–180 (2011).
- Hartley J, Westmacott R, Decker J, Shroff M, Yoon G. Childhood-onset CADASIL: clinical, imaging, and neurocognitive features. J. Child Neurol.25(5), 623–627 (2010).
- Bjorkhem I, Hansson M. Cerebrotendinous xanthomatosis: an inborn error in bile acid synthesis with defined mutations but still a challenge. Biochem. Biophys. Res. Commun.396(1), 46–49 (2010).
- Lancaster JL, Cody JD, Andrews T, Hardies LJ, Hale DE, Fox PT. Myelination in children with partial deletions of chromosome 18q. AJNR Am. J. Neuroradiol.26(3), 447–454 (2005).
- Jaeken J, Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu. Rev. Genomics Hum. Genet.8, 261–278 (2007).
- Henneke M, Diekmann S, Ohlenbusch A et al. RNASET2-deficient cystic leukoencephalopathy resembles congenital cytomegalovirus brain infection. Nat. Genet.41(7), 773–775 (2009).
- Steinfeld R, Grapp M, Kraetzner R et al. Folate receptor α defect causes cerebral folate transport deficiency: a treatable neurodegenerative disorder associated with disturbed myelin metabolism. Am. J. Hum. Genet.85(3), 354–363 (2009).
- Mamourian AC, Hopkin JR, Chawla S, Poptani H. Characteristic MR spectroscopy in fucosidosis: in vitro investigation. Pediatr. Radiol.40(8), 1446–1449 (2010).
- Tazir M, Nouioua S, Magy L et al. Phenotypic variability in giant axonal neuropathy. Neuromuscul. Disord.19(4), 270–274 (2009).
- Srinivasan J, Coleman L, Kornberg AJ. Juvenile onset globoid cell leukodystrophy masquerading as XL-adrenoleukodystrophy. J. Paediatr. Child Health44(7–8), 459–461 (2008).
- Del Toro M, Arranz JA, Macaya A et al. Progressive vacuolating glycine leukoencephalopathy with pulmonary hypertension. Ann. Neurol.60(1), 148–152 (2006).
- van der Knaap MS, Linnankivi T, Paetau A et al. Hypomyelination with atrophy of the basal ganglia and cerebellum: follow-up and pathology. Neurology69(2), 166–171 (2007).
- Rossi A, Biancheri R, Zara F et al. Hypomyelination and congenital cataract: neuroimaging features of a novel inherited white matter disorder. Am. J. Neuroradiol.29(2), 301–305 (2008).
- Orcesi S, Tonduti D, Uggetti C, Larizza D, Fazzi E, Balottin U. New case of 4H syndrome and a review of the literature. Pediatr. Neurol.42(5), 359–364 (2010).
- Vaurs-Barriere C, Deville M, Sarret C et al. Pelizaeus–Merzbacher-like disease presentation of MCT8 mutated male subjects. Ann. Neurol.65(1), 114–118 (2009).
- Bernard G, Thiffault I, Tetreault M et al. Tremor-ataxia with central hypomyelination (TACH) leukodystrophy maps to chromosome 10q22.3–10q23.31. Neurogenetics11(4), 457–464 (2010).
- Carrilho I, Santos M, Guimaraes A et al. Infantile neuroaxonal dystrophy: what’s most important for the diagnosis? Eur. J. Paediatr. Neurol.12(6), 491–500 (2008).
- Briggs TA, Abdel-Salam GM, Balicki M et al. Cerebroretinal microangiopathy with calcifications and cysts (CRMCC). Am. J. Med. Genet.146A(2), 182–190 (2008).
- Ferreira M, Torraco A, Rizza T et al. Progressive cavitating leukoencephalopathy associated with respiratory chain complex I deficiency and a novel mutation in NDUFS1. Neurogenetics12(1), 9–17 (2011).
- Neubauer BA, Stefanova I, Hubner CA et al. A new type of leukoencephalopathy with metaphyseal chondrodysplasia maps to Xq25-q27. Neurology67(4), 587–591 (2006).
- van der Knaap MS, Lai V, Kohler W et al. Megalencephalic leukoencephalopathy with cysts without MLC1 defect. Ann. Neurol.67(6), 834–837 (2010).
- Naidu S, Bibat G, Lin D et al. Progressive cavitating leukoencephalopathy: a novel childhood disease. Ann. Neurol.58(6), 929–938 (2005).
- Lopez-Hernandez T, Ridder MC, Montolio M et al. Mutant glialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am. J. Hum. Genet.88(4), 422–432 (2011).
- Lerman-Sagie T, Leshinsky-Silver E, Watemberg N, Luckman Y, Lev D. White matter involvement in mitochondrial diseases. Mol. Genet. Metab.84(2), 127–136 (2005).
- Timothy J, Geller T. SURF-1 gene mutation associated with leukoencephalopathy in a 2-year-old. J. Child Neurol.24, 1206–1301 (2009).
- Biancheri R, Rossi D, Cassandrini D, Rossi A, Bruno C, Santorelli FM. Cavitating leukoencephalopathy in a child carrying the mitochondrial A8344G mutation. AJNR Am. J. Neuroradiol.31(9), E78–E79 (2010).
- Artigalas OA, da Silva LR, Burin M et al. Multiple sulfatase deficiency: clinical report and description of two novel mutations in a Brazilian patient. Meta. Brain Dis.24(3), 493–500 (2009).
- Steenweg ME, Jakobs C, Errami A et al. An overview of L-2-hydroxyglutarate dehydrogenase gene (L2HGDH) variants: a genotype–phenotype study. Hum Mutat31(4), 380–390 (2010).
- Henneke M, Combes P, Diekmann S et al.GJA12 mutations are a rare cause of Pelizaeus–Merzbacher-like disease. Neurology70(10), 748–754 (2008).
- Feinstein M, Markus B, Noyman I et al. Pelizaeus–Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am. J. Hum. Genet.87(6), 820–828 (2010).
- Mochel F, Engelke UF, Barritault J et al. Elevated CSF N-acetylaspartylglutamate in patients with free sialic acid storage diseases. Neurology74(4), 302–305 (2010).
- Harreld JH, Smith EC, Prose NS, Puri PK, Barboriak DP. Trichothiodystrophy with dysmyelination and central osteosclerosis. Am. J. Neuroradiol.31(1), 129–130 (2010).
- van der Lei HDW, van Berkel CGM, van Wieringen WN et al. Genotype–phenotype correlation in vanishing white matter disease. Neurology75(17), 1555–1559 (2010).
- Wolf NI, Harting I, Boltshauser E. Leukoencephalopathy with ataxia, hypodontia, and hypomyelination. Neurology64(8), 1461–1644 (2006).
- van der Knaap MS, Ramesh V, Schiffmann R et al. Alexander disease: ventricular garlands and abnormalities of the medulla and spinal cord. Neurology66(4), 494–498 (2006).
- Krishnamoorthy KS, Eichler FS, Goyal NA, Small JE, Snuderl M. Case records of the Massachusetts General Hospital. Case 3-2010. A 5-month-old boy with developmental delay and irritability. N. Engl. J. Med.362(4), 346–356 (2010).
- Dastych M, Gottwaldova J, Pohludka M, Prikryl P, Benovska M. Determination of asialotransferrin in the cerebrospinal fluid with the HPLC method. Scand. J. Clin. Lab. Invest.70(2), 87–91 (2010).
- Callahan JW, Skomorowski MA. Diagnosis of Krabbe disease by use of a natural substrate. Methods Mol. Biol.347, 321–330 (2006).
- Gorg M, Wilck W, Granitzny B et al. Stabilization of juvenile metachromatic leukodystrophy after bone marrow transplantation: a 13-year follow-up. J. Child Neurol.22(9), 1139–1142 (2007).