
Abstract
Here we describe plasmid vectors and selection protocols developed to allow the construction of recombinant fowlpox viruses (rFPVs) with up to three insertions of foreign DNA in the viral genome. Transient dominant selection allows the construction of recombinant viruses that do not retain the selection markers and can therefore be used for the insertion of additional genes at other sites in the viral genome. A SYBR® Green real-time PCR sequence detection assay was applied to the identification of recombinant viruses from individual plaques, eliminating the need for amplification and hybridization from the transient dominant protocol and resulting in significant savings in time at each round of plaque purification. Dominant selection techniques allow more rapid recombinant virus construction; however, as the markers are retained along with the gene of interest, they can only be used to generate the final recombinant. rFPV vaccines constructed using these techniques have reached preclinical nonhuman primate and phase I human clinical trials in prime/boost vaccination studies as human immunodeficiency virus (HIV) therapeutic and prophylactic vaccines.
A Pox on HIV
Vaccination made its debut with a pox virus, so it's not surprising that these DNA viruses are still being evaluated for immunostimulatory potential. Recombinant pox viruses are appealing for expression of antigens and/or immunomodulatory proteins because they can accommodate longer coding sequences than trimmer retroviral vectors. Avipox viruses, like fowlpox virus, would be of particular interest as vaccine candidates, as their inability to replicate in nonavian cells provides added safety. Unfortunately, for all of their advantages, fowlpox viruses are not easy to engineer. Boyle et al. (p. 104) describe plasmid transfer vectors for production of recombinant fowlpox virus (rFPV). These new tools ease the construction of rFPV, making the process comparable to working with vaccinia or adenovirus recombinants. Significantly, the new transfer vectors permit transient dominant selection, enabling construction of rFPV that are free of extraneous foreign DNA sequences. To show the value of their tools, the authors generate an rFPV containing three human immunodeficiency virus (HIV) antigens, now a vaccine candidate destined for a phase I clinical trial.
Introduction
Prime and boost vaccination strategies, particularly in which the priming is via a DNA vaccine and the boosting is with a recombinant avipox, have been shown to lead to enhanced immune responses (Citation1–4). The resulting interest in such protocols has generated a need to optimize methods for the construction of recombinant avipox vectors. Of particular interest is the ability to express multiple antigens and to coexpress immune modulators within a single vaccine (Citation5,Citation6), which ensures the simultaneous delivery of multiple vaccine components and avoids the duplication of expenses involved in the manufacture and licensing of a number of individual constructs. Here we describe plasmid vectors that allow the insertion of foreign DNA at three sites (separated by more than 65 kb) in the genome of the fowlpox virus (FPV). The widely spaced insertion sites and the short (40 bp) sequence of the FPV promoter PE/L (Citation7), used for expression in each case, minimize concerns regarding the duplication of sequences, which might lead to genome instability. In addition, we have developed protocols in which the selection markers can be excluded from the final construct, enhancing the acceptability of the vaccines to regulatory authorities because the recombinant fowlpox vaccines carry no extraneous foreign DNA sequences other than the vaccine genes of interest.
These plasmids and protocols should significantly facilitate the design and production of complex recombinant fowlpox viruses (rFPVs) suitable for the vaccination of poultry against a range of avian pathogens. Such rFPVs may also have application as vaccines in nonavian species, including humans, in which the abortive replication cycle of avipox viruses in nonavian cells provides an additional level of safety (Citation8). In trials of prophylactic human immunodeficiency virus (HIV) vaccines in nonhuman primates, simultaneous administration of two rFPVs, each expressing a single HIV antigen, have shown significant efficacy (Citation9). Similarly, the coexpression of HIV antigens and human γ-interferon proved safe in macaques and generated enhanced T-cell proliferative responses to Gag antigens (Citation10). To progress rFPV HIV vaccines to human clinical trials in the context of prime-boost vaccination, protocols to enable the construction of rFPVs carrying multiple HIV antigens and immune modulators are required. The plasmid vectors described here facilitate the construction of such complex recombinants. Several rFPV vaccines constructed using these techniques have reached preclinical nonhuman primate and phase I human clinical trials.
Materials and methods
Cells and Viruses
rFPVs were constructed using the FPV M3 strain, a tissue culture-passaged strain derived from the mild vaccine strain (Fort Dodge Pty. Ltd., Sydney, NSW, Australia) (Citation11). Chicken embryo skin (CES) cell cultures were used for the growth and titration of the rFPVs (Citation12).
Plasmid Transfer Vectors
The dominant selection vector pAF09 has been previously described (Citation13).
A transient dominant selection vector, pAFtd, was constructed by removal of the β-galactosidase (β-gal) and Escherichia coli xanthine guanine phosphoribosyl transferase (Ecogpt) markers from pAF09. The β-gal and Ecogpt markers were reinserted into the resulting plasmid outside the FPV sequences. This plasmid allows the insertion of promoter-gene constructs in the intragenic region between the thymidine kinase (TK) gene and the uncharacterized gene (ORF X) immediately downstream using transient dominant selection.
The transient dominant selection vector, pKG10a, was constructed by PCR amplification and cloning of the FPV genomic region containing F6, F7, and F9 (Citation14). Unique restriction enzyme sites for BglII and SalI were inserted in the intragenic region between F7 and F9. The β-gal and Ecogpt markers were inserted within the plasmid backbone external to the FPV regions used for homologous recombination into the FPV genome.
The transient dominant selection vector pCH34 was constructed by PCR amplification and cloning of the FPV genome region, M-1, M-1 3′, and M-2 3′, which flanks the site of naturally occurring insertion of the reticuloendotheliosis provirus (REV) into the FPV genome (Citation15). Unique BamHI and XhoI restriction endonuclease sites were inserted in the intragenic region between M-1 and M-1 3′, and the REV genome long terminal repeat (LTR) was deleted during the manipulation and cloning. Once again, the β-gal and Ecogpt markers were inserted within the plasmid backbone external to the FPV regions used for homologous recombination into the FPV genome. Further detail on the construction of the plasmids is available as supplementary material.
Insertion of Antigen and Immunomodulatory Genes into rFPVs
All genes were amplified by PCR, inserted into subcloning plasmids, sequenced, and inserted into the appropriate FPV transfer vector. Gene sequences were scanned for T5NT motifs, which function as early poxvirus transcription terminator sequences; any instances were removed by PCR mutagenesis to ensure effective early gene expression during the abortive replication cycle of the rFPVs in nonavian cells.
The plasmid vectors were validated by the construction of several series of rFPVs carrying multiple HIV-1, simian immunodeficiency virus (SIV), and human or mouse γ-interferon genes.
Generation of rFPVs by Transient Dominant Selection or Dominant Selection
CES cells were infected with FPV parent strain (FPV M3 or a recombinant carrying genes at one or more locations) and then transfected using Lipofectamine™ (Invitrogen, Carlsbad, CA, USA) with plasmid DNA based on pAFtd, pKG10a, or pCH34. Single crossover recombinants were amplified twice under selective conditions of MX-HAT (2.5 µg/mL mycophenolic acid, 250 µg/mL xanthine, 100 µM hypoxanthine, 0.4 µM aminopterine, and 30 µM thymidine). At the first round of plaque purification, blue plaques (selected by X-gal staining) were picked and replaqued without selection when white plaques were picked and screened for the foreign gene insert using real-time PCR detection. Further rounds of plaque purification (three or four in total) without selection were similarly screened until all of the picked plaques (white) were positive by real-time PCR. Each round of transfection, amplification, and plaque purification required 5–7 days of incubation. Techniques for the selection and generation of rFPVs by dominant selection using plasmid pAF09 have been previously described (Citation11).
Screening of Recombinant Plaques by Real-Time PCR
rFPVs arising from the transient dominant recombination were discriminated from parent FPV/rFPV by real-time PCR screening using a SYBR® Green detection assay run in an ABI Prism® 7700 Sequencer (Applied Biosytems, Foster City, CA, USA). Primer pairs based on the sequence of the gene being inserted were designed using the Primer Express® Software Version 1.5 (Applied Biosystems) with default settings. Individual FPV plaques (white) were picked into 96-well plates containing 50 µL of water, frozen and thawed once, and 1 µL was used with SYBR Green PCR Master Mix (Applied Biosystems) and primers at 50 nM. Default cycling conditions were for 2 min at 50°C, 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. A melting curve analysis was run after the amplification to confirm the specificity of amplification. The plasmid used for gene insertion into the rFPV was used as a positive control for the amplification. Usually, 12–24 plaques were picked for each round of plaque purification, and plaques strongly positive by the SYBR Green PCR assay [cycle threshold (Ct) values less than 30 with the ΔRn set in the mid-log phase of the amplification plot] were replaqued. Alternatively, the plate-read mode at the end of the real-time PCR run was used to select positive plaques. Plaques with values greater than 0.2 corresponding to the Rn/ΔRn ratio were selected (the ratio at the end of 40 or 45 cycles in which Rn is the normalized fluorescence signal of the reporter dye divided by the fluorescence signal of the passive reference dye and ΔRn is the normalized reporter signal minus the baseline signal established in the first few cycles of the PCR). By the third or fourth plaque purification, all picked plaques were positive by the SYBR Green PCR assay. At this stage, all plaques are white when stained with X-gal for β-gal expression.
Confirmation of rFPV Genome Arrangement
For total DNA isolation, 100 µL of cell culture-derived virus stock were extracted with poxvirus DNA extraction buffer (Citation16), followed by proteinase K digestion, two phenol/chloroform extractions, and ethanol precipitation. After airdrying for 1–5 min, the DNA was redissolved in 20 µL TE (Tris-EDTA) with 20 µg/mL RNase. To minimize sheering, wide bore pipettes were used, and mixing was done carefully by hand (not by vortex mixing). For F6, F7, F9, and REV genomic regions, Advantage™ 2 PCR enzyme system (BD Biosciences Clontech, Palo Alto, CA, USA) was used; for the TK genomic region, the GeneAmp® XL PCR kit (Applied Biosystems) was used. shows the primer sequences. For analysis, 5 µL of the PCR product (from a 50-µL reaction) were used for agarose gel electrophoresis with the appropriate size markers. When necessary, the sequence of the inserted gene product was confirmed by nucleotide sequence determination of the PCR product using the FPV genome flanking primers and internal primers as appropriate. Reactions were performed using ABI Prism BigDye™ terminator chemistry (Applied Biosystems) and an ABI Prism 377 DNA sequencer.
Table 1. Genome Analysis of rFPVs by PCR: Primers and Product Sizes from Parent (FPV-M3) and Recombinants
Expression Analysis of Inserted Gene Products
Confluent monolayers of CES cells in 25 cm2 flasks were infected with rFPV at a multiplicity of infection of 5 plaque-forming units (pfu) per cell. For the detection of intracellularly expressed antigens, the cells were harvested by scraping and pelleting by centrifugation at 2000× g at 48–96 h postinfection. The samples were stored at −80°C prior to processing. Cell pellets were solubilized and separated by denaturing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (NuPAGE® Novex Bis-Tris gels; Invitrogen), blotted onto polyvinylidene difluoride (PVDF) membranes, and probed with the appropriate antiserum. Chemiluminescence using peroxidase-labeled secondary antibodies or protein A/G was used for detection. MagicMark™ Western Standards (Invitrogen) were used as size standards. Solublesecreted proteins (e.g., human or mouse γ-interferon) were assayed using ELISA-based kits (R&D Systems, Minneapolis, MN, USA).
Results
To facilitate the insertion of multiple vaccine antigens and immune modulators into the genome of fowlpox, we have developed plasmid transfer vectors suitable for transient dominant selection (). Plasmids pKG10a, pCH34, and pAFtd were used in a transient dominant selection protocol to insert genes sequentially and in any order into the FPV genome, while the dominant selection vector pAF09 was used for the final insertion only when using pKG10a and/or pCH34 for the earlier insertions. The three transient dominant selection plasmids required that the genes be cloned into the plasmid vectors as promoter-gene-terminator fusions (). This was achieved by the PCR addition of promoter and early transcription terminator to the genes of interest using a general approach for primer design and then cloning the promoter-gene-terminator product into the appropriate restriction endonuclease site of the transfer vector. This approach has also been used to insert promoter-gene-terminator fusions into the BglII site of pAF09, allowing the insertion of two genes via pAF09—one in this site and the other as a promoter-gene fusion at the multiple cloning site previously described and utilized (Citation13).
(A) Plasmid vectors for transient dominant (pKG10a, pCH34, and pAFtd) and dominant insertion (pAF09) of antigen and immunomodulatory genes into fowlpox virus (FPV). (B) Primers for the construction of the promoter-gene-terminator cassette for insertion into plasmid vectors. TK ORF X, thymidine kinase (TK) gene and the uncharacterized gene ORF X; REV, reticuloendotheliosis provirus; min, minimum.
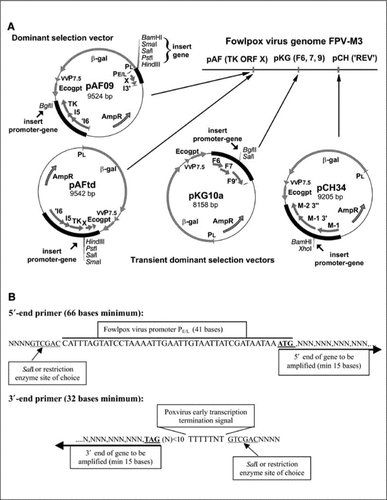
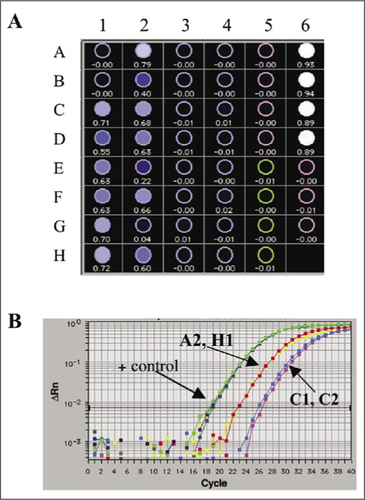
The transient dominant and dominant selection strategies were used to insert the promoter-gene constructs into the FPV genome (). Recombinants generated in cell culture by homologous recombination following infection with FPV and transfection with plasmid were amplified by selection for Ecogpt expression and then plaque-purified on the basis of β-gal expression (dominant selection) or β-gal loss (transient dominant selection). The use of a real-time SYBR Green PCR assay allowed rapid (2–3 h) identification of white plaques carrying the gene of interest and discrimination from parent virus arising from the resolution of the single crossover recombinants generated in the transient dominant selection strategy. provides an example of data obtained using the SYBR Green sequence detection assay during plaque purification of an rFPV. The plate-read mode, which provides RN/ΔRn values at the end of the assay, and the amplification plots were used to select positive plaques as detailed in the Materials and Methods section. Material from individual plaques was used without purification of viral DNA, and compared with the plasmid DNA, which was used for transfection as a positive control and the parental virus (in this case, FPV-M3) as a negative control. In the end-of-assay plate-read mode (), the isolates used in columns 1 and 2 contained recombinant viruses while those in columns 3 and 4 did not. Alternatively, shows amplification plots for controls and four wells positive by Rn/ΔRn (A2, C1, C2, H1), which were selected for further plaque purification. Further confirmation of the specificity of the amplification was obtained by a dissociation analysis of the products at the conclusion of the run (data not shown). Further rounds of plaque purification (Citation2,Citation3) were screened by SYBR Green PCR assay until all picked plaques were positive.
FPV, fowlpox virus; CES, chicken embryo skin; MXHAT, mycophenolic acid-xanthine-hypoxanthine-aminopterine-thymidine selection medium.
(A) Test plate showing Rn/ΔRn (Rn is the ratio of the reporter dye/reference dye fluorescence signals; ΔRn is the normalized reporter signal minus the baseline signal). Eight plaques from four plaque isolates (rows A–H and columns 1–4) were analyzed using primers specific for the inserted gene. Positive controls (+ control) were the plasmid used for insertion into the fowlpox virus (FPV; first four rows of column 6) and the negative controls were the plaques from the parent FPV-M3 and reagent controls (column 5 and the remaining rows of column 6, respectively). (B) Amplification plots for selected wells (A2, H1, C1, and C2) and positive controls (plasmid).
To validate the overall process of rFPV generation using these plasmid vectors, several series of rFPVs have been constructed and characterized for genome arrangement (by PCR analysis of insertion sites). Two of these series are described in this report. For the first series, the HIV-1 gag-pol gene (3.5 kb with extensive modifications to increase safety) was inserted into pKG10a, the env gene (2.1 kb with modifications to increase safety) was inserted into pCH34, and the tat/rev genes (0.8 kb as a fusion protein, fused by a sequence coding for pentahistidine) were inserted into pAF09 at the BglII site. These plasmid constructs were used in the above order to construct rFPVs carrying one and two insertions (produced using plasmids pKG10a and pCH34, respectively) by transient dominant selection, with the third insertion (from plasmid pAF09) achieved by dominant selection. The presence of insertions of the predicted size and at the predicted locations in the viral genome was verified by PCR using primers to the FPV sequences on either side of each insertion site (; ). shows the results of amplifications and gel analysis of intermediate and final recombinants carrying one, two, and three insertions. The results confirmed the complete plaque purification of the intermediate and final recombinants, as judged by the presence of PCR products of the predicted sizes and the absence of PCR products coinciding with those obtained from the parental virus, FPV-M3. DNA sequencing of the PCR products in each case provided further confirmation of correct gene insertion and stability (data not shown). Stability of these recombinants was further assessed by the preparation of viral DNA after each of two additional rounds of passage in CES cells at low multiplicity and PCR specific for each of the three insertion sites. In each case, the presence of a band of the predicted length was observed, and there was no sign of smaller and presumably more readily amplified PCR products expected in the case of loss or rearrangements of inserted DNA (data not shown). The final recombinant (carrying inserts at the three loci) has been subject to good manufacturing practice (GMP) cultivation in preparation for a phase I human clinical trial involving two passages for large-scale cultivation. Stability after GMP manufacture was similarly demonstrated by PCR analysis (data not shown).
Agarose gel electrophoresis of PCR products obtained from rFPVs. (A) PCR products from parent virus (no added genes; lane 0); rFPV containing one gene [human immunodeficiency virus type 1 (HIV-1) gag-pol, lane 1]; fFPV containing two genes (HIV-1 gag-pol and env, lane 2); and rFPV containing three genes (HIV-1 gag-pol, env, and tat/rev, lane 3). PCR amplifications are grouped according to the insertion site: F6, 7, 9 for HIV-1 gag-pol, reticuloendotheliosis provirus (REV) for HIV-1 env, and TK ORF X for HIV-1 tat/rev. (B) PCR products from the parent virus (no added genes, lane 0), rFPV containing HIV-1 tat/rev only (lane 4), rFPV containing simian immunodeficiency virus (SIV) gag-pol only (lane 5), or rFPV containing HIV-1 tat/rev and SIV gag-pol (lane 6). PCR amplifications are grouped according to the insertion site: F6, 7, 9 for SIV gag-pol and TK ORF X for HIV-1 tat/rev. gives the expected fragment.
![Figure 4. Confirmation of recombinant fowlpox virus (rFPV) genomic structure. Agarose gel electrophoresis of PCR products obtained from rFPVs. (A) PCR products from parent virus (no added genes; lane 0); rFPV containing one gene [human immunodeficiency virus type 1 (HIV-1) gag-pol, lane 1]; fFPV containing two genes (HIV-1 gag-pol and env, lane 2); and rFPV containing three genes (HIV-1 gag-pol, env, and tat/rev, lane 3). PCR amplifications are grouped according to the insertion site: F6, 7, 9 for HIV-1 gag-pol, reticuloendotheliosis provirus (REV) for HIV-1 env, and TK ORF X for HIV-1 tat/rev. (B) PCR products from the parent virus (no added genes, lane 0), rFPV containing HIV-1 tat/rev only (lane 4), rFPV containing simian immunodeficiency virus (SIV) gag-pol only (lane 5), or rFPV containing HIV-1 tat/rev and SIV gag-pol (lane 6). PCR amplifications are grouped according to the insertion site: F6, 7, 9 for SIV gag-pol and TK ORF X for HIV-1 tat/rev. Table 1 gives the expected fragment.](/cms/asset/305c703d-cada-4cb2-844a-96e91615bdfd/ibtn_a_12357943_f0004.gif)
Western blot analysis for gene expression in infected cells or ELISA for cytokine expression in culture supernatants were used to demonstrate effective gene expression from the single, double, and triple recombinants of this first series (). As expected, HIV-1 gag gene products were expressed by all three recombinants carrying the HIV-1 gag-pol genes inserted via pKG10a; Env (truncated in size due to deletions in the gene inserted) was expressed by the second and third recombinants while only the triple recombinant expressed the Tat/Rev fusion when probed with antipenta histidine antibody (). Additional recombinants in which human or mouse γ-interferon was inserted with pAF09 instead of the Tat/Rev fusion (as in the recombinants described above) expressed high levels of cytokine—80 to 100,000 pg/mL of human γγ-interferon or approximately 25,000 pg/mL of murine γ-interferon in culture supernatants collected 48 h or later after infection (data not shown).
Lane 0, parent virus; lane 1, rFPV containing one gene [human immunodeficiency virus type 1 (HIV-1) gag-pol]; lane 2, rFPV containing two genes (HIV-1 gag-pol and env); lane 3, rFPV containing three genes (HIV-1 gag-pol, env, and tat/rev). Blots are labeled according to the insertion site: F6, 7, 9 for HIV-1 gag-pol, reticuloendotheliosis provirus (REV) for HIV-1 env; and TK ORF X for HIV-1 tat/rev. Gag gene products were detected with an antibody to p24, env gene products were detected with an antibody raised to recombinant expressed HIV-1 Env, and tat/rev gene products were detected using a monoclonal antibody to the penta-histidine (His5) linker. TK ORF X, thymidine kinase (TK) gene and the unchracterized gene ORF X.
![Figure 5. Western blot analysis of total cell lysates of chicken embryo skin (CES) cells infected with parent virus or recombinant fowlpox virus (rFPV). Lane 0, parent virus; lane 1, rFPV containing one gene [human immunodeficiency virus type 1 (HIV-1) gag-pol]; lane 2, rFPV containing two genes (HIV-1 gag-pol and env); lane 3, rFPV containing three genes (HIV-1 gag-pol, env, and tat/rev). Blots are labeled according to the insertion site: F6, 7, 9 for HIV-1 gag-pol, reticuloendotheliosis provirus (REV) for HIV-1 env; and TK ORF X for HIV-1 tat/rev. Gag gene products were detected with an antibody to p24, env gene products were detected with an antibody raised to recombinant expressed HIV-1 Env, and tat/rev gene products were detected using a monoclonal antibody to the penta-histidine (His5) linker. TK ORF X, thymidine kinase (TK) gene and the unchracterized gene ORF X.](/cms/asset/4c7469fe-43d5-40a8-96ee-1705202942d4/ibtn_a_12357943_f0005.gif)
The transient dominant selection vector pAFtd was validated with a second series of recombinants. In this case, the recombinants were generated using pAFtd carrying an HIV-1 tat/rev fusion and pKG10a containing an unmodified SIV gag-pol. Because both plasmids are transient dominant selection vectors, the order of use does not matter and, in fact, both possibilities were tested. Once again, the presence of insertions of the predicted size and at the predicted locations in the viral genome was determined by PCR using primers to the FPV sequences on either side of each insertion site (; ). shows the results of amplifications and gel analysis of intermediate and final recombinant carrying one or two insertions. The results confirmed the utility of the pAFtd plasmid vector for the insertion of genes into single or double rFPVs by transient dominant selection. Gene expression studies confirmed the expression of the SIV-gag-pol and HIV-1 tat/rev gene products by the single and double recombinants (data not shown).
Discussion
The utility of the avipox vaccine vectors has been hampered by the time taken to construct recombinants in comparison with vaccinia or adenovirus recombinants. However, the large genome size (Citation11) of the avipoxviruses permits the insertion of multiple vaccine antigens, and immune modulators providing suitable plasmid transfer vectors are available. We have developed plasmid transfer vectors and general rFPV construction techniques utilizing transient dominant (Citation17) and dominant selection (Citation18,Citation19) for the insertion of multiple vaccine antigens and immune modulators into the genome of FPV. The use of plasmid vectors carrying both a drug selection marker (Ecogpt) for amplification and a chromogenic substrate marker (β-gal) for the identification of rFPVs greatly facilitates the construction of complex recombinants, even when recombination frequencies are low. The transient dominant selection protocol used for the sequential insertion of genes into the FPV genome involves resolution, after a single crossover recombination event, into either the recombinant virus or the parental strain. The use of real-time PCR detection in conjunction with the transient dominant selection protocol has greatly reduced the time taken to distinguish between these two forms of FPV, which both produce white plaques. Previously, the preparation of viral DNA from material amplified from individual plaques, followed by conventional PCR and/or hybridization with radiolabeled probes, was required to discriminate rFPVs from the parental strain. Using the SYBR Green assay, which can be completed for 80–90 plaques within 2–3 h, we have reduced the time taken for each round of plaque purification by at least 1 week (i.e., a reduction of 3 or more weeks per insertion or 6–9 weeks for a triple recombinant) as well as eliminated the use of radiochemicals from the construction of rFPVs.
We have demonstrated that rFPVs with insertions of foreign DNA at multiple sites have the predicted genome configuration and sequence, that the inserted DNA is maintained and stable over several rounds of amplification in CES cells, including GMP cultivation steps, and that the inserted genes are expressed. The expression of a range of viral antigens [e.g., HIV, hepatitis B, Newcastle disease virus (NDV), infectious bursal disease, duck hepatitis B] has been detected in rFPV-infected avian (CES) or mammalian (e.g., CV-1 and HeLa) cells (data not shown). Similarly, cytokine expression has been shown in the supernatants of rFPV-infected cells of avian and mammalian origin (data not shown). Furthermore, when rFPVs with three insertions were constructed, the differences in the level of expression from the original insertion were not detected following the additional modifications involved in the second and third insertions (data not shown).
Recent interest in the presence of REV sequences or LTR remnants in the FPV genome (Citation15,Citation20) has raised questions concerning the safety of rFPV-based vaccines (particularly for human use) containing retroviral elements. In all the FPV isolates examined thus far, the REV sequences have been found at the same site within the FPV genome. Insertion of foreign DNA using the plasmid vector pCH34 described here has the added advantage of deleting those REV sequences from the FPV genome.
In conclusion, these plasmids and protocols can significantly facilitate the design and production of complex rFPVs suitable for the vaccination of poultry and for development for use as human vaccines. Several recombinants constructed using these techniques have reached phase I human clinical trials.
Supplementary data
Download PDF (141.4 KB)Acknowledgments
Dr. Michael Mather and Ms. Krisanne Gladman provided scientific and technical discussion and assistance with the rFPV constructions and virus characterizations. We are indebted to Catherine Williams, Carol Wykes, and Shea Larkin for the preparation of primary chicken cells and to Tony Pye for DNA sequencing. Parts of these studies were funded by the Australian National Health and Medical Research Council and by NIH NIAID DAIDS HIV Vaccine Development Consortium Contract No. N01-A1-05395 (to D.B.B.).
References
- Kent, S.J., A.Zhao, S.J.Best, J.D.Chandler, D.B.Boyle, and I.A.Ramshaw. 1998. Enhanced T-cell immunogenicity and protective efficacy of a human immunodeficiency virus type 1 vaccine regimen consisting of consecutive priming with DNA and boosting with recombinant fowlpox virus. J. Virol.72:10180–10188.
- Boyle, D.B. 1998. Diversified prime and boost protocols: the route to enhanced immune responses to recombinant DNA based vaccine?Australas. Biotechnol.8:96–97.
- Ramsay, A.J., K.H.Leong, and I.A.Ramshaw. 1997. DNA vaccination against virus infection and enhancement of antiviral immunity following consecutive immunization with DNA and viral vectors. Immunol. Cell Biol.75:382–388.
- Robinson, H.L., D.C.Montefiori, R.P.Johnson, K.H.Manson, M.L.Kalish, J.D.Lifson, T.A.Rizvi, S.Lu, et al.. 1999. Neutralizing antibody-independent containment of immunodeficiency virus challenges by DNA priming and recombinant pox virus booster immunizations. Nat. Med.5:526–534.
- Leong, K.H., A.J.Ramsay, D.B.Boyle, and I.A.Ramshaw. 1994. Selective induction of immune responses by cytokines coexpressed in recombinant fowlpox virus. J. Virol.68:8125–8130.
- Ramsay, A.J., K.H.Leong, D.Boyle, J.Ruby, and I.A.Ramshaw. 1994. Enhancement of mucosal IgA responses by interleukins 5 and 6 encoded in recombinant vaccine vectors. Reprod. Fertil. Dev.6:389–392.
- Kumar, S. and D.B.Boyle. 1990. A poxvirus bidirectional promoter element with early/late and late functions. Virology179:151–158.
- Somogyi, P., J.Frazier, and M.A.Skinner. 1993. Fowlpox virus host range restriction: gene expression, DNA replication, and morphogenesis in nonpermissive mammalian cells. Virology197:439–444.
- Kent, S.J., A.Zhao, S.J.Best, J.D.Chandler, D.B.Boyle, and I.A.Ramshaw. 1998. Enhanced T-cell immunogenicity and protective efficacy of a human immunodeficiency virus type 1 vaccine regimen consisting of consecutive priming with DNA and boosting with recombinant fowlpox virus. J. Virol.72:10180–10188.
- Kent, S.J., A.Zhao, C.J.Dale, S.Land, D.B.Boyle, and I.A.Ramshaw. 2000. A recombinant avipoxvirus HIV-1 vaccine expressing interferon-gamma is safe and immunogenic in macaques. Vaccine18:2250–2256.
- Coupar, B.E., T.Teo, and D.B.Boyle. 1990. Restriction endonuclease mapping of the fowlpox virus genome. Virology179:159–167.
- Silim, A., M.A.El Azhary, and R.S.Roy. 1982. A simple technique for preparation of chicken-embryoskin cell cultures. Avian Dis.26:182–185.
- Heine, H.G. and D.B.Boyle. 1993. Infectious bursal disease virus structural protein VP2 expressed by a fowlpox virus recombinant confers protection against disease in chickens. Arch. Virol.131:277–292.
- Spehner, D., R.Drillien, and J.P.Lecocq. 1990. Construction of fowlpox virus vectors with intergenic insertions: expression of the beta-galactosidase gene and the measles virus fusion gene. J. Virol.64:527–533.
- Hertig, C., B.E.Coupar, A.R.Gould, and D.B.Boyle. 1997. Field and vaccine strains of fowlpox virus carry integrated sequences from the avian retrovirus, reticuloendotheliosis virus. Virology235:367–376.
- Nakano, E., D.Panicali, and E.Paoletti. 1982. Molecular genetics of vaccinia virus: demonstration of marker rescue. Proc. Natl. Acad. Sci. USA79:1593–1596.
- Falkner, F.G. and B.Moss. 1990. Transient dominant selection of recombinant vaccinia viruses. J. Virol.64:3108–3111.
- Boyle, D.B. and B.E.Coupar. 1988. A dominant selectable marker for the construction of recombinant poxviruses. Gene65:123–128.
- Boyle, D.B. and B.E.Coupar. 1988. Construction of recombinant fowlpox viruses as vectors for poultry vaccines. Virus Res.10:343–356.
- Singh, P., W.M.Schnitzlein, and D.N.Tripathy. 2003. Reticuloendotheliosis virus sequences within the genomes of field strains of fowlpox virus display variability. J. Virol.77:5855–5862.
- Afonso, C.L., E.R.Tulman, Z.Lu, L.Zsak, G.F.Kutish, and D.L.Rock. 2000. The genome of fowlpox virus. J. Virol.74:3815–3831.

