
Abstract
Molecular dynamics simulations have evolved into a mature technique that can be used effectively to understand macromolecular structure-to-function relationships. Present simulation times are close to biologically relevant ones. Information gathered about the dynamic properties of macromolecules is rich enough to shift the usual paradigm of structural bioinformatics from studying single structures to analyze conformational ensembles. Here, we describe the foundations of molecular dynamics and the improvements made in the direction of getting such ensemble. Specific application of the technique to three main issues (allosteric regulation, docking, and structure refinement) is discussed.
Introduction
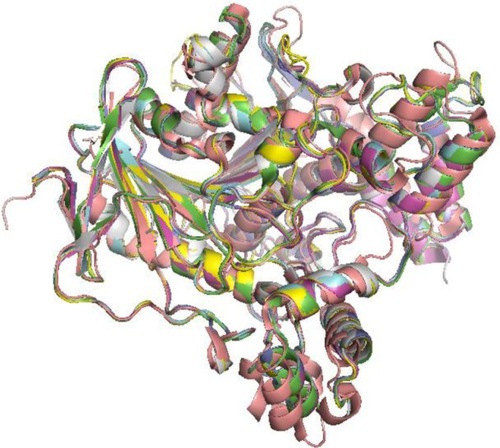
The study of the macromolecular structure is a key point in the understanding of biology. Biological function is based on molecular interactions, and these are a consequence of macromolecular structures. Since initial structure determinations in the 50s, both in the protein and in the nucleic acid worlds, the increase in the knowledge of how macromolecular structures are built has been continuous. At present, protein data bank (PDB)Citation1 holds more than 110,000 entries, including more than 100,000 proteins, 2,800 nucleic acids, alone or forming complexes, and approximately 20,000 small molecules complexed to macromolecules. Molecular recognition rules as defined by such structural knowledge powers the understanding of basic biological phenomena, like enzyme mechanisms and regulation, transport across membranes, the building of large structures like ribosomes, or viral capsids, or how DNA is read and transcription is controlled. The study and prediction of protein–protein interaction networks is one of the growing fields in modern systems biology. On a more practical note, protein three-dimensional (3D) structures are the basis for structure-based drug design. The simple visual analysis of 3D structures of protein or nucleic acids, as obtained from the experiments, has driven large number of successful studies in biochemistry. However, despite their enormous utility, structures stored at the PDB provide only a partial view of 3D structure. Both protein and nucleic acids are flexible entities, and dynamics can play a key role in their functionality. Proteins undergo significant conformational changes while performing their function. As a rule, any complex made by any protein implies some structural rearrangement. This can be easily checked just by comparing a series of PDB entries that just differ in a small ligand bound to a given protein. shows a superimposition of experimental acetylcholinesterase structures. There are no changes in the overall fold, just small rearrangements in the structure; however, these differences are large enough to fool ligand-docking algorithms. Larger conformational changes are also present in the known protein structures.Citation2–Citation6 Conformational changes are a common part of an enzymes’ catalytic cycle.Citation7 For instance, loop or domain closures contribute to isolate the active site from solvent and, in so doing, alter the chemical environment around substrates, or trigger the catalytic event by bringing essential partners together.Citation8–Citation10 Also, allostery is the most common enzyme regulation strategy. Allosteric regulation is entirely based on the possibility of a given protein to coexist in two or more conformations of comparable stability. Binding to ligands (allosteric regulators), or simply protein concentration, or crowding, may switch stabilities among conformations and trigger the shape transition. Additionally, some features of protein function can be understood only when dynamic properties are taken into account. For instance, diffusion of small substrates through heme-dependent enzyme molecules requires the transient appearance of channels in the protein structure.Citation11–Citation18 Also, cavities have transient phenomena that in some cases can only be revealed or analyzed following its dynamics.Citation19,Citation20
Figure 1 Structure variability within a protein family.
In the case of nucleic acids, conformational changes are even more complex. Standard B-DNA has a relatively simple structure in comparison with protein or complex RNAs; however, it is an extremely plastic molecule that undergoes large conformational changes to adapt to its interaction partners. Binding of transcription factors to DNA, for example, is not only dependent on DNA sequence recognition, but also a direct consequence of the ability of the DNA molecule to adapt to the protein surface.Citation21,Citation22
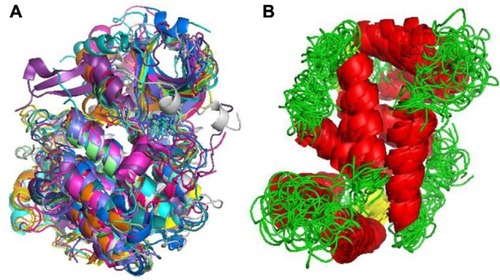
The traditional approach to understand conformation influence on macromolecular function is to cumulate experimental structures covering the conformational space. This has led to the generation of crystal structures for macromolecules in several environments, or macromolecules complexed with different molecules, and contributes to the enormous redundancy seen in the PDB. Examples of this approach are the 87 structures for CK2 homologues (), where a common fold is maintained, and different degrees of conformational variation are clearly visible, especially in loop regions. A single experiment could generate conformational ensembles as those taken from nuclear magnetic resonance experiments (). In the latter case, the source of the variability found is rather a consequence of the lack of experimental data in some specific regions of the structure. Indeed, the study of PDB as source for molecular flexibility has been exploited in some extent.Citation18,Citation23–Citation26 Taking such “experimental ensembles” a partial view of the macromolecule flexibility can be obtained, although PDB composition is necessarily biased. Theoretical techniques appear as the most convenient way to obtain a picture of macromolecular dynamic properties. Recent advances in the performance of simulation algorithms, including specific strategies to increase the conformational sampling, have popularized the concept of “conformational ensemble”, as the alternative to the analysis of PDB’s single structures. Ensembles can be analyzed to derive thermodynamic properties of the system, like entropy or free energy.Citation27 If properly built, ensembles can also be used to reconstruct complex conformational transitions or even folding events.Citation27–Citation29 Ensembles are also a better reference to reproduce experimental results, as experiments measure averages of properties over a real ensemble.Citation30,Citation31
Figure 2 Experimental ensembles.
Abbreviations: NMR, nuclear magnetic resonance; PDB, protein data bank.
Molecular dynamics simulation
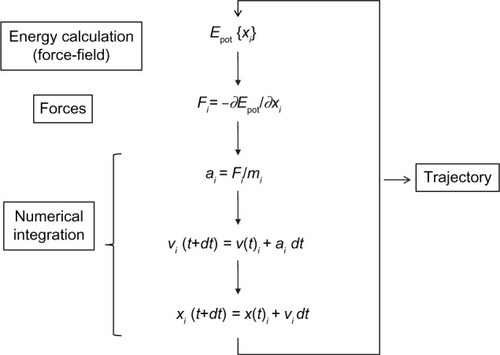
Molecular dynamics (MD) simulation, first developed in the late 70s,Citation32,Citation33 has advanced from simulating several hundreds of atoms to systems with biological relevance, including entire proteins in solution with explicit solvent representations, membrane embedded proteins, or large macromolecular complexes like nucleosomesCitation34,Citation35 or ribosomes.Citation36,Citation37 Simulation of systems having ~50,000–100,000 atoms are now routine, and simulations of approximately 500,000 atoms are common when the appropriate computer facilities are available. This remarkable improvement is in large part a consequence of the use of high performance computing (HPC), and the simplicity of the basic MD algorithm (). An initial model of the system is obtained from either experimental structures or comparative modeling data. The simulated system could be represented at different levels of detail. Atomistic representation is the one that leads to the best reproduction of the actual systems. However, coarse-grained representations are becoming very popular when large systems or long simulations are required (see Orozco et alCitation38 for a review of such strategies). Solvent representation is a key issue in system definition. Several approaches have been assayedCitation39–Citation47 but, again, the most effective is the simplest one, the explicit representation of solvent molecules, although at the expense of increasing the size of the simulated systems. Explicit solvent is able to recover most of the solvation effects of real solvent including those from entropic origin like the hydrophobic effect. Once the system is built, forces acting on every atom are obtained by deriving equations, the force-fields, where potential energy is deduced from the molecular structure.Citation48–Citation53 Force-fields are complex equations, but they are easy to calculate. The simplicity of the force-field representation of molecular features: springs for bond length and angles, periodic functions for bond rotations and Lennard–Jones potentials, and the Coulomb’s law for van der Waals and electrostatic interactions, respectively, assures that energy and force calculations are extremely fast even for large systems. Force-fields currently used in atomistic molecular simulations differ in the way they are parameterized. Parameters are not necessarily interchangeable, and not all force-fields allow to represent all molecule types, but simulations conducted using modern force-fields are normally equivalent.Citation54,Citation55 Once the forces acting on individual atoms are obtained, classical Newton’s law of motion is used to calculate accelerations and velocities and to update the atom positions. As integration of movement is done numerically, to avoid instability, a time step shorter than the fastest movements in the molecule should be used. This ranks normally between 1 and 2 fs for atomistic simulations, and is the major bottleneck of the simulation procedure. Microsecond-long simulations, barely scratching the time scales of biological processes, require iterating over this calculation cycle 109 times. This is one of the strengths of coarse-grained strategies. As a more simplified representation of the system is used, much larger time steps are possible, and therefore the effective length of the simulations is dramatically extended. Of course, this can be obtained at the expense of the accuracy of the simulation ensemble. Algorithmic advances, that include fine-tuning of energy calculations, parallelization, or the use of graphical processing units (GPUs), have largely improved the performance of MD simulations.
Figure 3 Molecular dynamics basic algorithm.
Abbreviations: Epot, potential energy; t, simulation time; dt, iteration time; For each spatial coordinate of the N simulated atoms (i): x, atom coordinate; F, forces component; a, acceleration; m, atom mass; v, velocity.
The present generation of computers takes benefit of parallelism and accelerators to speed-up the process. The most popular simulation codes (AMBER,Citation56 CHARMM,Citation57 GROMACS,Citation58 or NAMDCitation59) have long been compatible with the messaging passing interface (MPI). When a large number of computer cores can be used simultaneously, MPI can greatly reduce the computation time. To benefit the locality of interactions, the general strategy is to distribute the system to simulate among processors. This strategy is called spatial decomposition. Only a small fragment of the system has to be simulated in each processor. The most efficient division is not based in the list of particles, but in their position in space. Each processor deals with a region of space irrespective of which particles are present there. Communication between processors is also reduced, as only those simulating neighboring regions have to share information (see Larsson et alCitation60 for a review). As stated, the use of accelerators, mainly GPU, has become a major breakthrough in simulation codes. Originally designed to handle computer graphics, GPUs have evolved into general-purpose, fully programmable, high-performance processors and represent a major technical improvement to perform atomistic MD. Most major MD codes have already been prepared for GPUs, and even MD codes written specifically to be used on GPUs have been developed (ACEMDCitation61). Simulation on GPUs alone or combined with MPI is, at present, the default strategy for high-throughput MD simulations. Remarkably, while simulations have been the most popular use of HPC in life sciences, the increasing power and sophistication of GPUs is leading to a greater use of personal workstations with a comparable performance.
Strategies to improve ensemble generation
Pure computational brute force, just making longer simulations, is not enough to extend the conformational sampling in biomolecular systems. The complex shape of the free energy landscape makes most of the simulations explore just a small region around the energy minimum closest to the initial conformation. With the availability of the present HPC systems, an obvious strategy is to perform a series of parallel simulations with several starting conformations. Although this could be efficient, it requires a specific knowledge of the system to simulate, and cannot be applied as a general strategy. This approach is particularly useful when several crystal structures are available (for instance in the case of allosterically regulated enzymes). A second problem that appears when collections of parallel simulations are calculated, is the generation of a usable ensemble out of the trajectories obtained. Recently the Markov state model (MSM) theory has been used to this end.Citation62–Citation65 MSM theory discretizes the conformational ensemble in a collection of states, and constructs a matrix with the transition probabilities among them. The analysis of such a matrix would allow reconstruction of the global behavior of the system. Since the transition rates converge more rapidly than the population of the involved states, this approach has the advantage that the collection of simulations is not required to be especially long. This approach has been used mainly in the study of folding processes,Citation28,Citation66 but also in the kinetic characterization of the formation of ligand–protein complexes.Citation67 Other approaches have been designed to increase the sampling space in single simulations, like metadynamics,Citation68–Citation71 for instance, where already visited conformations are penalized; weighted ensembles, where additional simulations are started when new conformational spaces are visited;Citation72 or accelerated MD where energy barriers are artificially reduced.Citation73–Citation75 However, even with a perfect sampling, MD simulations cannot surmount barriers in the energy landscape higher than the total energy added to the system. The obtained ensemble with a single simulation is limited to those states that are accessible at the simulation temperature. Simulations at high temperatures were common in the origins of MD, but they lead to unrealistic trajectories, and hence should be combined to room temperature runs. This approach, called simulated annealing, has been largely replaced by replica exchange methods.Citation76,Citation77 Such methods launch parallel simulations in different conditions. The most common variation is simulation temperature. The sampling ability of the simulation increases with temperature. Higher temperature simulations can surmount energy barriers and explore new regions of the ensemble. Periodically, energies of the different simulations are compared and structures are swapped according to its energy rank. The resulting simulation has sampled a larger conformational space, due to high temperature simulations, and retains the ability to represent the low-temperature states of the system. The main difference with the simulated annealing approach is that a realistic ensemble of the system is obtained and thermodynamic information can be derived from simulations. The idea has been extended to other simulation strategies. Most remarkably, replicas based on differences in the Hamiltonian replica exchange, including alchemical free energy calculationsCitation78 or constant-pH simulations,Citation79–Citation83 are becoming popular.
Tools to popularize MD
Preparation for simulation implies the following of a series of operations that are far from being just routine. First, the initial structure comes from the experiment. Expected issues include nonstructured or missing regions or residues, nonstandard ligands, or even structures bearing errors in the interpretation of experimental data. When a single system is simulated, all the effort in the preparation of the system is worth, as it assures the quality of the simulation result. Such setup is usually done manually, with a considerable human effort. A standard procedure to set up a system implies a number of well-known procedures: fixing structure errors; ionization of titrable amino acids; addition of structural water molecules, counter ions, and solvent; and energy minimization and equilibration of the system at the desired temperature. An expert modeler normally carries out these procedures using a set of helper programs. Such an expert has the necessary knowledge to surmount specific problems that may arise. For instance, the workflow used in the MoDEL projectCitation84 was programmed to run automatically, but a nonnegligible fraction of over 1,500 proteins prepared failed at some point of the process. With this scenario, for newcomers to MD simulation, even a single system setup could represent an unaffordable problem. Even worse, non-expert users tend to blindly use default procedures leading easily to artifactual trajectories, which are hard to distinguish from the correct ones. This strongly contributes to the lack of popularity of biomolecular simulations among the bioinformatics or the biochemical community. MD simulations have been restricted to those research groups bearing the necessary expertise. Solving this issue requires an automatic setup of the simulation system. We would be looking for a clever black box for the nonexperts, but also for a robust software suite that can account for a large set of unrelated protein structures. All major MD codesCitation56–Citation59 come with a set of accompanying programs, which perform most steps of the preparation. Additionally, a number of initiatives, combining those tools with a user-friendly interface, have come into the scene to address this problem. CHARMM-GuiCitation85 and CHARMMing,Citation86 for CHARMM, or Guimacs,Citation87 Gromita,Citation88 and jSimMacs,Citation89 for GROMACS, provide automatic setup functionality. VMDCitation90 provides a number of plug-ins that allow to launch simulations with NAMD. Most of these tools provide a friendly environment to prepare systems for simulation without the need of a deep knowledge of the underlying operations, thus facilitating the access to the field for the newcomers. Unfortunately, due to the lack of a standard for the representation of molecular simulation data, most helper applications are restricted to a single MD package, and data is not easily interchangeable. Besides, although most use some kind of embedded scripting language, automation of procedures is not a straightforward task. Lessons learned in the preparation of the MoDEL database, by our group, leaded to the generation of a new set of tools, MDMoby and MDWebCitation91 that try to cover both aspects of the problem. On one hand, MDMoby provides a full set of web services, covering all setup, simulation, and analysis operations. The modular nature of such collection of web services allows incorporating them as a tool kit to the design of complex setup protocols and to run them programmatically. In turn, MDWeb, a web-based interface, provides a user-friendly bench where user can check for the quality of the input structure, tailor their own setup protocols, or use a collection of predefined ones.
Application: understanding allostery
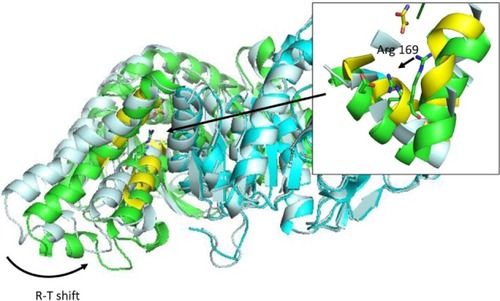
Most regulation phenomena in proteins are explained within conformational transitions. The concept of allostery that translates conformational dynamics in functional implications has been analyzed since the early times of protein biochemistry.Citation92–Citation94 Conformation shift involved in allostery spans from small rearrangements to large quaternary shifts as those originally accounted by the Monod model. In any case, there is a general agreement that conformational shifts involved in allosteric transitions are simple in terms of collective movements.Citation95 For this reason, molecular simulations could be a natural tool to understand allostery. However, the ability of free atomistic simulation algorithms to follow a complete transition path is limited. Most of the traditional reports of simulations in this field use simplified frameworks, like discrete MDCitation96 or Go-Models,Citation97 or even popular nonsimulation equivalents like elastic network models,Citation98–Citation101 and seek to find the transition path between known experimental structures. With full atom representations, it is usual to trick the algorithm by using targeted,Citation102–Citation104 or supervised MDCitation105 where the simulation is artificially driven to the desired conformation. In this case, the analysis of the path could give insight into the energetics and details of the allosteric transition. For those cases where allosteric regulation is known to occur, but one of the ends is unknown, long simulations (aloneCitation106,Citation107 or with enhanced conformational sampling) are required.Citation108,Citation109 The direct use of conformational ensembles without any conditioning is still out of the routine possibilities of present MD simulations; however, specific cases with well-defined collective motions could be feasible. shows an example where only 50 ns of simulation allowed for a conformational shift in Bacillus stearothermophilus lactate dehydrogenase (PDB code: 1LDN). This enzyme is known to exist in two states: a fully active, tetrameric state and a less active, dimeric one. Fructose-1,6-bisphosphate is a known allosteric regulator that allows the tetrameric state to be formed.Citation110,Citation111 The construction of stable dimeric state by site-directed mutagenesis allowed for the analysis of the less active form in more detail and confirmed that a significant conformational shift occurs when the tetrameric state is formed.Citation112,Citation113 The simulation of a B. stearothermophilus lactate dehydrogenase dimer protein starting with the conformation of the tetrameric one, as obtained from the experimental structure, reveals a significant intersubunit movement compatible with the experimental behavior of the enzyme (). In this case, no computational bias was introduced; however, protein setup was done mimicking experimental conditions where the conformational shift is known to occur. The use of experimental restrains (but not necessarily the target structure) is being exploited to guide the simulation.Citation31,Citation114 Alternatively, free long simulations may be replaced by shorter simulations and analyzed with MSMs to provide quantitative insight.Citation63–Citation65 However target structure should be at some point explored to allow reconstructing the full process. The power of molecular simulations to uncover allosteric regulations is not in any doubt; however, there is still a long way until it could be routinely applied to all cases.
Figure 4 R-T transition on Bacillus stearothermophilus lactate dehydrogenase after 50 ns simulation in explicit solvent.
Abbreviation: R-T, relaxed and tense states (as defined by Monod’s model).
Application: molecular docking and drug design
One of the most practical application of the concept of molecular recognition are docking strategies, either small molecule or protein docking. To understand how a ligand, typically a substrate or a regulator, binds to its macromolecular counterpart is a key issue in the understanding of function itself, and it is the basis of structurally driven drug design. The recognition process is by nature dynamic.Citation115 Molecules are flexible entities, and the recognition process itself implies structural rearrangements, and this shape adjustment is part of the binding process not only from the structural point of view, but also from the energetics. Although this is a generally well-accepted idea, docking algorithms are far from considering dynamic effects as a routine. Most docking or virtual screening codes work on rigid structures as obtained from the PDB. shows a traditional cross-docking experiment where a collection of acetylcholinesterase ligands are docked back in the same set of receptor structures. Protein structures correspond to the ones shown in . In this experiment, all receptor structures correspond to the same protein, but crystallized with a different ligand; and all ligands are known to bind the receptor in the same place and pose. In these conditions, the experiment just measures the impact of small receptor rearrangements caused by ligand binding, on the docking efficiency. The usual result, as the one shown in , is that even though the protein does not change, a different PDB structure implies poorer docking results. Even docking of a ligand back on its original PDB structure (diagonal results in ) tends to fail due to the usual overcompression of structures derived from X-ray crystallography. This problem is especially relevant in protein–protein docking where considerable differences are found between bound and unbound structures. For ligand-docking methods, ligand flexibility could be largely recovered by using conformer families.Citation116–Citation118 In the case of protein flexibility, solutions are not so extended. Most of them use algorithms to select from a limited alternative set of protein conformations, either precomputed or simulated.Citation119–Citation121 It is also possible to introduce flexibility a posteriori as a refinement process.Citation122 It is still not clear whether structural adjustment comes from selection of available conformations or it is induced by the binding process itself,Citation123,Citation124 but in any case the use of structural ensembles instead of single structures can be expected to improve the binding prediction.Citation125–Citation127 The concept of “ensemble docking” usually requires the selection of a representative set of snapshots coming from a simulation and uses them as targets in normal docking procedures.Citation128–Citation130 Integration of docking and simulation in a single calculation is less popular, but some examples have been reported.Citation16,Citation67,Citation121,Citation131
Figure 5 Cross-docking experiment with selected acetylcholinesterase structures from PDB.
Abbreviations: PDB, protein data bank; RMSD, root-mean-square deviation.
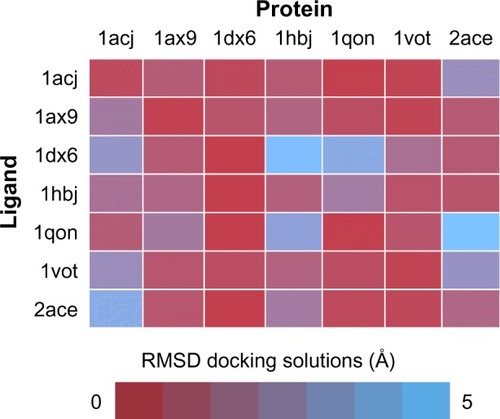
The use of simulations for the improvement of virtual screening or docking processes has a clear advantage. However, due to the speed requirements of docking, most methods based on traditional atomistic simulations are too slow to be considered, when used in a real scenario. Coarse-grained methods or any sort of accelerated MD could be a way to take benefit of simulation in a near future.
Application: refining structure predictions
Structure prediction has been one of the most ancient problems addressed in structural bioinformatics. MD, including the longest simulations performed, has been extensively used for ab initio protein structure prediction,Citation132–Citation136 aiming to simulate protein folding from scratch, although this is not the preferred strategy to obtain theoretical model of protein structure. Instead, template-based modeling is the most efficient technique.Citation137–Citation144 In template-based modeling, one or several 3D structures of protein showing a reasonable degree of similarity to the protein of interest are taken as templates. Irrespective of the modeling algorithm, the end result is a model bearing the new amino acid sequence and a structure averaging the used templates. In most cases, the last step of the prediction procedure implies relaxation of the structure using normally molecular mechanics. In others, restrained simulations are used throughout all the process.Citation144 The use of MD simulations looks like an obvious step in refining such models.Citation145 Simulation would allow the structure to adapt to the new sequence, and in theory give a more realistic model. Although this point is reasonable as a concept, MD simulations require systems to be close to their equilibrium (native) conformation. Otherwise, significant and difficult to detect artifacts may occur. Critical assessment of protein structure prediction contests,Citation139 where prediction algorithms face problems with known but nonpublic 3D structures, provide an excellent dataset to test this issue. Applying different MD approaches to the refinement of such predictions has led to a number of conclusions. The most naive approach, a single simulation starting from the predicted conformation, tends to deviate significantly from the desired structure.Citation145–Citation147 Apparently, inaccuracies in the model have a large impact in the quality of the simulation results. Instead, results clearly indicate that deviation from the original structure is directly correlated with the loss of quality of the model. A second conclusion is that the ensemble of structures taken from the simulations is a closer representation of the target structure, thus indicating that the native and original structures both lie within the conformational space of the simulation.Citation146
Conclusion
MD simulations have already more than 40 years of history. However, it was not until the recent years that MD has achieved time scales that begin to be compatible with biological processes. At present, when routine simulations are approaching the microsecond scale, conformational changes, or ligand binding can be effectively simulated. The improvement of the computational equipment, especially the use of GPUs, and the improvements made in the optimization of MD algorithms, including coarse-grained ones, allow us to move from the analysis of single structures, the basis of the molecular modeling as we know it, to the analysis of conformational ensembles. Conformational ensembles are a much better representation of real macromolecules, as they account for flexibility and dynamic properties (including all thermodynamic information) and ease the match with experimental results. Although the shift in concept is clear, and the technology is coming along, there is still a long way until biomolecular simulations, the generation of conformational ensembles, would become a routine. Tools exist that make the setup of a macromolecular system much easier, and even allow the nonexperts to enter the simulation world. However, lack of representation standards, much less optimized analysis tools, and even the difficulties in simply storing and transmitting the huge amount of trajectory data that is generated are still issues that remain to be solved. In any case, MD is already a valuable tool in helping to understand biology.
Disclosure
The authors report no conflicts of interest in this work.
References
- BermanHMBattistuzTBhatTNThe protein data bankActa Crystallogr D Biol Cryst20025889990712037327
- FloresSEcholsNMilburnDThe database of macromolecular motions: new features added at the decade markNucleic Acids Res200634Database issueD296D30116381870
- BhabhaGBielJTFraserJSKeep on moving: discovering and perturbing the conformational dynamics of enzymesAcc Chem Res201548242343025539415
- CockrellGMKantrowitzERViewMotions rainbow: a new method to illustrate molecular motions in proteinsJ Mol Graph Model201340485323353585
- GersteinMKrebsWA database of macromolecular motionsNucleic Acids Res19982618428042909722650
- GohCSMilburnDGersteinMConformational changes associated with protein-protein interactionsCurr Opin Struct Biol200414110410915102456
- KokkinidisMGlykosNMFadouloglouVEProtein flexibility and enzymatic catalysisAdv Protein Chem Struct Biol20128718121822607756
- StevensRCLipscombWNAllosteric control of quaternary states in E. coli aspartate transcarbamylaseBiochem Biophys Res Commun19901713131213182222446
- ShinodaTAraiKShigematsu-IidaMDistinct conformation-mediated functions of an active site loop in the catalytic reactions of NAD-dependent D-lactate dehydrogenase and formate dehydrogenaseJ Biol Chem200528017170681707515734738
- KarplusMRole of conformation transitions in adenylate kinaseProc Natl Acad Sci U S A201010717E71 author reply E7220424124
- KalkoSGGelpiJLFitaIOrozcoMTheoretical study of the mechanisms of substrate recognition by catalaseJ Am Chem Soc2001123399665967211572688
- JakopitschCDroghettiESchmuckenschlagerFFurtmullerPGSmulevichGObingerCRole of the main access channel of catalase-peroxidase in catalysisJ Biol Chem200528051424114242216244360
- Bidon-ChanalAMartiMACrespoALigand-induced dynamical regulation of NO conversion in Mycobacterium tuberculosis truncated hemoglobin-NProteins200664245746416688782
- HaraIIchiseNKojimaKRelationship between the size of the bottleneck 15 A from iron in the main channel and the reactivity of catalase corresponding to the molecular size of substratesBiochemistry2007461112217198371
- Bidon-ChanalAMartiMAEstrinDALuqueFJDynamical regulation of ligand migration by a gate-opening molecular switch in truncated hemoglobin-N from Mycobacterium tuberculosisJ Am Chem Soc2007129216782678817488073
- GuallarVLuCBorrelliKEgawaTYehSRLigand migration in the truncated hemoglobin-II from Mycobacterium tuberculosis: the role of G8 tryptophanJ Biol Chem200928453106311619019831
- DaigleRGuertinMLaguePStructural characterization of the tunnels of Mycobacterium tuberculosis truncated hemoglobin N from molecular dynamics simulationsProteins200975373574719003999
- AhalawatNMurarkaRKConformational changes and allosteric communications in human serum albumin due to ligand bindingJ Biomol Struct Dyn2015113
- BarbanyMMeyerTHospitalAMolecular dynamics study of naturally existing cavity couplings in proteinsPLoS One2015103e011997825816327
- BowmanGRBolinERHartKMMaguireBCMarquseeSDiscovery of multiple hidden allosteric sites by combining Markov state models and experimentsProc Natl Acad Sci U S A201511292734273925730859
- BouvierBZakrzewskaKLaveryRProtein-DNA recognition triggered by a DNA conformational switchAngew Chem Int Ed Engl201150296516651821626631
- ZakrzewskaKLaveryRTowards a molecular view of transcriptional controlCurr Opin Struct Biol201222216016722296921
- PerezANoyALankasFLuqueFJOrozcoMThe relative flexibility of B-DNA and A-RNA duplexes: database analysisNucleic Acids Res200432206144615115562006
- Leo-MaciasALopez-RomeroPLupyanDZerbinoDOrtizARAn analysis of core deformations in protein superfamiliesBiophys J20058821291129915542556
- Velazquez-MurielJARuedaMCuestaIPascual-MontanoAOrozcoMCarazoJMComparison of molecular dynamics and superfamily spaces of protein domain deformationBMC Struct Biol20099619220918
- MichelettiCComparing proteins by their internal dynamics: exploring structure-function relationships beyond static structural alignmentsPhys Life Rev201310112623199577
- BaxaMCHaddadianEJJumperJMFreedKFSosnickTRLoss of conformational entropy in protein folding calculated using realistic ensembles and its implications for NMR-based calculationsProc Natl Acad Sci U S A201411143153961540125313044
- NoeFSchuetteCVanden-EijndenEReichLWeiklTRConstructing the equilibrium ensemble of folding pathways from short off-equilibrium simulationsProc Natl Acad Sci U S A200910645190111901619887634
- NaganathanANOrozcoMThe native ensemble and folding of a protein molten-globule: functional consequence of downhill foldingJ Am Chem Soc201113331121541216121732676
- CandottiMEsteban-MartinSOrozcoMAtomistic insights on protein-urea structural organization from MD simulations of a chemically denatured protein ensemble determined by NMRFEBS J2012279531531
- Bryn FenwickREsteban-MartinSSalvatellaXUnderstanding biomolecular motion, recognition, and allostery by use of conformational ensemblesEur Biophys J201140121339135522089251
- McCammonJAGelinBRKarplusMDynamics of folded proteinsNature19772675612585590301613
- WarshelALevittMTheoretical studies of enzymic reactions – dielectric, electrostatic and steric stabilization of carboniumion in reaction of lysozymeJ Mol Biol19761032227249985660
- RoccatanoDBarthelAZachariasMStructural flexibility of the nucleosome core particle at atomic resolution studied by molecular dynamics simulationBiopolymers2007855–640742117252562
- SharmaSDingFDokholyanNVMultiscale modeling of nucleosome dynamicsBiophys J20079251457147017142268
- TinocoIJrWenJDSimulation and analysis of single-ribosome translationPhys Biol20096202500619571367
- BrandmanRBrandmanYPandeVSA-site residues move independently from P-site residues in all-atom molecular dynamics simulations of the 70S bacterial ribosomePLoS One201271e2937722235290
- OrozcoMOrellanaLHospitalACoarse-grained representation of protein flexibility. Foundations, successes, and shortcomingsChristovCAdvances in Proteins Chemistry and Structural Biology85Computational Chemistry Methods in Structural BiologyWaltham, MAAcademic Press2011183215
- LazaridisTKarplusMEffective energy function for proteins in solutionProteins199935213315210223287
- RouxBSimonsonTImplicit solvent modelsBiophys Chem1999781–212017030302
- OrozcoMLuqueFJTheoretical methods for the description of the solvent effect in biomolecular systemsChem Rev2000100114187422611749344
- BashfordDCaseDAGeneralized born models of macromolecular solvation effectsAnn Rev Phys Chem20005112915211031278
- HaberthuerUCaflischAFACTS: fast analytical continuum treatment of solvationJ Comput Chem200829570171517918282
- LuchkoTGusarovSRoeDRThree-dimensional molecular theory of solvation coupled with molecular dynamics in AmberJ Chem Theory Comput20106360762420440377
- VorobjevYNAdvances in implicit models of water solvent to compute conformational free energy and molecular dynamics of proteins at constant pHAdv Protein Chem Struct Biol20118528132221920327
- KleinjungJFraternaliFDesign and application of implicit solvent models in biomolecular simulationsCurr Opin Struct Biol20142512613424841242
- AnandakrishnanRDrozdetskiAWalkerRCOnufrievAVSpeed of conformational change: comparing explicit and implicit solvent molecular dynamics simulationsBiophys J201510851153116425762327
- HermansJBerendsenHJCVangunsterenWFPostmaJPMA consistent empirical potential for water-protein interactionsBiopolymers198423815131518
- MackerellADWiorkiewiczkuczeraJKarplusMAn all-atom empirical energy function for the simulation of nucleic-acidsJ Am Chem Soc1995117481194611975
- OttKHMeyerBParametrization of GROMOS force field for oligosaccharides and assessment of efficiency of molecular dynamics simulationsJ Comput Chem199617810681084
- MacKerellADBashfordDBellottMAll-atom empirical potential for molecular modeling and dynamics studies of proteinsJ Phys Chem B1998102183586361624889800
- CornellWDCieplakPBaylyCIA 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic-moleculesJ Am Chem Soc19951171951795197
- KaminskiGAFriesnerRATirado-RivesJJorgensenWLEvaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptidesJ Phys Chem B20011052864746487
- RuedaMFerrer-CostaCMeyerTA consensus view of protein dynamicsProc Natl Acad Sci U S A2007104379680117215349
- PerezALankasFLuqueFJOrozcoMTowards a molecular dynamics consensus view of B-DNA flexibilityNucleic Acids Res20083672379239418299282
- CaseDADardenTACheathamTE IAMBER 12San Francisco, CAUniversity of California2012
- BrooksBRBrooksCL3rdMackerellADJrCHARMM: the biomolecular simulation programJ Comput Chem200930101545161419444816
- HessBKutznerCvan der SpoelDLindahlEGROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulationJ Chem Theory Comput20084343544726620784
- NelsonMTHumphreyWGursoyANAMD: a parallel, object oriented molecular dynamics programInt J Supercomput Appl High Perform Comput1996104251268
- LarssonPHessBLindahlEAlgorithm improvements for molecular dynamics simulationsWiley Interdiscip Rev Comput Mol Sci20111193108
- HarveyMJGiupponiGDe FabritiisGACEMD: accelerating biomolecular dynamics in the microsecond time scaleJ Chem Theory Comput2009561632163926609855
- PanACRouxBBuilding Markov state models along pathways to determine free energies and rates of transitionsJ Chem Phys2008129606410718715051
- BowmanGRHuangXPandeVSUsing generalized ensemble simulations and Markov state models to identify conformational statesMethods200949219720119410002
- ChoderaJDNoeFMarkov state models of biomolecular conformational dynamicsCurr Opin Struct Biol20142513514424836551
- DaLTSheongFKSilvaDAHuangXApplication of Markov state models to simulate long timescale dynamics of biological macromoleculesAdv Exp Med Biol2014805296624446356
- VoelzVABowmanGRBeauchampKPandeVSMolecular simulation of ab initio protein folding for a millisecond folder NTL9(1–39)J Am Chem Soc201013251526152820070076
- BuchIGiorginoTDe FabritiisGComplete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulationsProc Natl Acad Sci U S A201110825101841018921646537
- MichelettiCLaioAParrinelloMReconstructing the density of states by history-dependent metadynamicsPhys Rev Lett2004921717060115169135
- LaioARodriguez-ForteaAGervasioFLCeccarelliMParrinelloMAssessing the accuracy of metadynamicsJ Phys Chem B2005109146714672116851755
- RaiteriPLaioAGervasioFLMichelettiCParrinelloMEfficient reconstruction of complex free energy landscapes by multiple walkers metadynamicsJ Phys Chem B200611083533353916494409
- BarducciABonomiMParrinelloMMetadynamicsWiley Interdiscip Rev Comput Mol Sci201115826843
- DicksonABrooksCLWExplore: hierarchical exploration of high-dimensional spaces using the weighted ensemble algorithmJ Phys Chem B2014118133532354224490961
- Abdul-WahidBFengHRajanDAWE-WQ: fast-forwarding molecular dynamics using the accelerated weighted ensembleJ Chem Inf Model201454103033304325207854
- DoshiUHamelbergDTowards fast, rigorous and efficient conformational sampling of biomolecules: advances in accelerated molecular dynamicsBiochim Biophys Acta20151850587888825153688
- MiaoYFeixasFEunCMcCammonJAAccelerated molecular dynamics simulations of protein foldingJ Comput Chem201536201536154926096263
- NymeyerHGnanakaranSGarciaAEAtomic simulations of protein folding, using the replica exchange algorithmMethods Enzymol200438311914915063649
- KubitzkiMBde GrootBLMolecular dynamics simulations using temperature-enhanced essential dynamics replica exchangeBiophys J200792124262427017384062
- MengYDashtiDSRoitbergAEComputing alchemical free energy differences with Hamiltonian replica exchange molecular dynamics (H-REMD) simulationsJ Chem Theory Comput2011792721272722125475
- BurgiRKollmanPAVan GunsterenWFSimulating proteins at constant pH: an approach combining molecular dynamics and Monte Carlo simulationProteins200247446948012001225
- SternHAMolecular simulation with variable protonation states at constant pHJ Chem Phys20071261616411217477594
- BaptistaAMMartelPJPetersenSBSimulation of protein conformational freedom as a function of pH: constant-pH molecular dynamics using implicit titrationProteins19972745235449141133
- GohGBHulbertBSZhouHBrooksCLConstant pH molecular dynamics of proteins in explicit solvent with proton tautomerismProteins20148271319133124375620
- ItohSGDamjanovicABrooksBRpH replica-exchange method based on discrete protonation statesProteins201179123420343622002801
- MeyerTD’AbramoMHospitalAMoDEL (molecular dynamics extended library): a database of atomistic molecular dynamics trajectoriesStructure201018111399140921070939
- JoSKimTIyerVGImWCHARMM-GUI: a web-based graphical user interface for CHARMMJ Comput Chem200829111859186518351591
- MillerBTSinghRPKlaudaJBHodoscekMBrooksBRWoodcockHL3rdCHARMMing: a new, flexible web portal for CHARMMJ Chem Inf Model20084891920192918698840
- KotaPGUIMACS – a Java based front end for GROMACSIn Silico Biol200771959917688433
- SellisDVlachakisDVlassiMGromita: a fully integrated graphical user interface to gromacs 4Bioinform Biol Insights200939910220140074
- RoopraSKnappBOmasitsUSchreinerWjSimMacs for GROMACS: a Java application for advanced molecular dynamics simulations with remote access capabilityJ Chem Inf Model200949102412241719852516
- HumphreyWDalkeASchultenKVMD: visual molecular dynamicsJ Mol Graph Model19961413338
- HospitalAAndrioPFenollosaCCicin-SainDOrozcoMGelpiJLMDWeb and MDMoby: an integrated web-based platform for molecular dynamics simulationsBioinformatics20122891278127922437851
- MonodJChangeuxJPJacobFAllosteric proteins and cellular control systemsJ Mol Biol19636430632913936070
- MonodJWymanJChangeuxJPOn nature of allosteric transitions – a plausible modelJ Mol Biol19651218811814343300
- KoshlandDENemethyGFilmerDComparison of experimental binding data and theoretical models in proteins containing subunitsBiochemistry1966513653855938952
- Henzler-WildmanKAThaiVLeiMIntrinsic motions along an enzymatic reaction trajectoryNature2007450717183884418026086
- SfrisoPEmperadorAOrellanaLHospitalALluis GelpiJOrozcoMFinding conformational transition pathways from discrete molecular dynamics simulationsJ Chem Theory Comput20128114707471826605625
- SfrisoPHospitalAEmperadorAOrozcoMExploration of conformational transition pathways from coarse-grained simulationsBioinformatics201329161980198623740746
- KimMKJerniganRLChirikjianGSEfficient generation of feasible pathways for protein conformational transitionsBiophys J20028331620163012202386
- BaharIRaderAJCoarse-grained normal mode analysis in structural biologyCurr Opin Struct Biol200515558659216143512
- OrellanaLRuedaMFerrer-CostaCLopez-BlancoJRChaconPOrozcoMApproaching elastic network models to molecular dynamics flexibilityJ Chem Theory Comput2010692910292326616090
- OrellanaLOrozcoMUnderstanding protein dynamics with coarse-grained models: from structures to diseaseFEBS J2012279528528
- SchlitterJEngelsMKrugerPJacobyEWollmerATargeted molecular-dynamics simulation of conformational change – application to the T-R transition in insulinMol Simul1993102–6291308
- KrugerPVerheydenSDeclerckPJEngelborghsYExtending the capabilities of targeted molecular dynamics: simulation of a large conformational transition in plasminogen activator inhibitor 1Protein Sci200110479880811274471
- PerdihAKotnikMHodoscekMSolmajerTTargeted molecular dynamics simulation studies of binding and conformational changes in E. coli MurDProteins200768124325417427948
- DeganuttiGCuzzolinACiancettaAMoroSUnderstanding allosteric interactions in G protein-coupled receptors using supervised molecular dynamics: a prototype study analysing the human A3 adenosine receptor positive allosteric modulator LUF6000Bioorg Med Chem201523144065407125868747
- ArkhipovAShanYDasRArchitecture and membrane interactions of the EGF receptorCell2013152355756923374350
- KlepeisJLLindorff-LarsenKDrorROShawDELong-timescale molecular dynamics simulations of protein structure and functionCurr Opin Struct Biol200919212012719361980
- AndersonJSMustafiSMHernándezGLeMasterDMStatistical allosteric coupling to the active site indole ring flip equilibria in the FK506-binding domainBiophys Chem2014192414825016286
- BoczekEEReefschlägerLGDehlingMConformational processing of oncogenic v-Src kinase by the molecular chaperone Hsp90Proc Natl Acad Sci U S A201511225E3189E319826056257
- WigleyDBMuirheadHGamblinSJHolbrookJJCrystallization of a ternary complex of lactate-dehydrogenase from Bacillus stearothermophilusJ Mol Biol19882044104110433065514
- WigleyDBGamblinSJTurkenburgJPStructure of a ternary complex of an allosteric lactate-dehydrogenase from Bacillus stearothermophilus at 2.5 A resolutionJ Mol Biol199222313173351731077
- CameronADRoperDIMoretonKMMuirheadHHolbrookJJWigleyDBAllosteric activation in Bacillus stearothermophilus lactate-dehydrogenase investigated by an x-ray crystallographic analysis of a mutant designed to prevent tetramerization of the enzymeJ Mol Biol199423846156258176749
- JacksonRMGelpiJLCortesAConstruction of a stable dimer of Bacillus stearothermophilus lactate dehydrogenaseBiochemistry19923135830783141525168
- Esteban-MartinSBryn FenwickRSalvatellaXRefinement of ensembles describing unstructured proteins using NMR residual dipolar couplingsJ Am Chem Soc2010132134626463220222664
- KoshlandDEApplication of a theory of enzyme specificity to protein synthesisProc Natl Acad Sci U S A19584429810416590179
- HawkinsPCDNichollsAConformer generation with OMEGA: learning from the data set and the analysis of failuresJ Chem Inf Model201252112919293623082786
- IshikawaYA script for automated 3-dimentional structure generation and conformer search from 2-dimentional chemical drawingBioinformation201391998899224391363
- KlettJCortes-CabreraAGil-RedondoRGagoFMorrealeAALFA: Automatic ligand flexibility assignmentJ Chem Inf Model201454131432324392957
- FriesnerRABanksJLMurphyRBGlide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracyJ Med Chem20044771739174915027865
- NabuursSBWagenerMDe VliegJA flexible approach to induced fit dockingJ Med Chem200750266507651818031000
- BorrelliKWVitalisAAlcantaraRGuallarVPELE: protein energy landscape exploration. A novel Monte Carlo based techniqueJ Chem Theory Comput2005161304131126631674
- EmperadorASolernouASfrisoPEfficient relaxation of protein-protein interfaces by discrete molecular dynamics simulationsJ Chem Theory Comput2013921222122926588765
- CsermelyPPalotaiRNussinovRInduced fit, conformational selection and independent dynamic segments: an extended view of binding eventsTrends Biochem Sci2010351053954620541943
- TobiDBaharIStructural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound stateProc Natl Acad Sci U S A200510252189081891316354836
- FukunishiYStructural ensemble in computational drug screeningExp Opin Drug Metab Toxicol201067835849
- IvetacAMcCammonJAA molecular dynamics ensemble-based approach for the mapping of druggable binding sitesMethods Mol Biol2011819312
- OkamotoYKokuboHTanakaTLigand docking simulations by generalized-ensemble algorithmsKarabenchevaChristovaTDynamics of Proteins and Nucleic Acids92Waltham, MAAcademic Press20136391
- FenollosaCOtonMAndrioPCortesJOrozcoMGoniJRSEABED: Small molEcule activity scanner weB servicE baseDBioinformatics201531577377525348211
- Maria NovoaERibas de PouplanaLBarrilXOrozcoMEnsemble docking from homology modelsJ Chem Theory Comput2010682547255726613506
- OsguthorpeDJShermanWHaglerATExploring protein flexibility: incorporating structural ensembles from crystal structures and simulation into virtual screening protocolsJ Phys Chem B2012116236952695922424156
- ChaudhuriRCarrilloOLaughtonCAOrozcoMApplication of drug-perturbed essential dynamics/molecular dynamics (ED/MD) to virtual screening and rational drug designJ Chem Theory Comput2012872204221426588953
- DornME SilvaMBBuriolLSLambLCThree-dimensional protein structure prediction: methods and computational strategiesComput Biol Chem201453251276
- Lindorff-LarsenKPianaSDrorROShawDEHow fast-folding proteins foldScience2011334605551752022034434
- PianaSLindorff-LarsenKShawDEProtein folding kinetics and thermodynamics from atomistic simulationProc Natl Acad Sci U S A201210944178451785022822217
- PianaSLindorff-LarsenKShawDEAtomic-level description of ubiquitin foldingProc Natl Acad Sci U S A2013110155915592023503848
- BonneauRBakerDAb initio protein structure prediction: progress and prospectsAnn Rev Biophys Biomol Struct20013017318911340057
- LanceBKDeaneCMWoodGRExploring the potential of template-based modellingBioinformatics201026151849185620525820
- JooKLeeJLeeSSeoJHLeeSJLeeJHigh accuracy template based modeling by global optimizationProteins2007698389
- MoultJA decade of CASP: progress, bottlenecks and prognosis in protein structure predictionCurr Opin Struct Biol200515328528915939584
- RoyAKucukuralAZhangYI-TASSER: a unified platform for automated protein structure and function predictionNat Protoc20105472573820360767
- ZhangYProtein structure prediction: when is it useful?Curr Opin Struct Biol200919214515519327982
- GinalskiKElofssonAFischerDRychlewskiL3D-jury: a simple approach to improve protein structure predictionsBioinformatics20031981015101812761065
- MisuraKMSBakerDProgress and challenges in high-resolution refinement of protein structure modelsProteins2005591152915690346
- SaliABlundellTLComparative protein modeling by satisfaction of spatial restraintsJ Mol Biol199323437798158254673
- RavalAPianaSEastwoodMPDrorROShawDERefinement of protein structure homology models via long, all-atom molecular dynamics simulationsProteins20128082071207922513870
- MirjaliliVFeigMProtein structure refinement through structure selection and averaging from molecular dynamics ensemblesJ Chem Theory Comput2013921294130323526422
- ChenJBrooksCLIIICan molecular dynamics simulations provide high-resolution refinement of protein structure?Proteins200767492293017373704
- PronkSPallSSchulzRGROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkitBioinformatics201329784585423407358