Abstract
Discoveries over the last decade have fundamentally transformed the way we define lung cancer. Gone are the days of the simple binary classification system of non-small cell lung cancer (NSCLC) and small cell lung cancer. Today, accurate identification of the histological and molecular subtype of NSCLC is required for selecting standard cytotoxic chemotherapy and targeted therapies. The identification of anaplastic lymphoma kinase (ALK) rearrangements in 5–7% of NSCLC patients and the rapid clinical development of crizotinib for these patients is the most recent clinical example necessitating the proper identification of the molecular characteristics of NSCLC for treatment decisions. The discovery of ALK rearrangements in NSCLC serendipitously coincided with the development of crizotinib for other ALK or MET driven malignancies. The clinical development of crizotinib for ALK-positive NSCLC patients has been an amazing success story of translational medicine that relied on the prior clinical experience of other targeted predecessors (i.e. erlotinib in EGFR mutant NSCLC) and a compound ready for clinical development to gain expedited FDA approval. This review discusses the clinical development and use of crizotinib in NSCLC.
Introduction
Lung cancer is the leading cause of cancer mortality in the US and worldwide.Citation1,Citation2 Over the last decade we have witnessed several discoveries that have fundamentally transformed the way we define lung cancer. Historically, lung cancer histology has predominantly relied on a simplistic binary classification that divided lung cancer cases into either non-small cell lung cancer (NSCLC) or small cell lung cancer. Today, accurate identification of the histological and molecular subtype of NSCLC has become crucial in selecting standard cytotoxic chemotherapy and targeted therapies. This was first demonstrated when the interactions between NSCLC histology and bevacizumab toxicity and pemetrexed efficacy were observed. The development of the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors, erlotinib and gefitinib, and the subsequent identification of activating EGFR mutations, also led to a focused effort to better define the molecular characteristics of NSCLC.Citation3–Citation6 Finally, the recent development of crizotinib for patients with NSCLC and an anaplastic lymphoma kinase (ALK) rearrangement demonstrated the necessity of identifying the molecular characteristics of NSCLC for the development of novel therapeutic agents.Citation7–Citation9 Together, these discoveries have shifted the simplistic binary lung cancer classification system to a refined classification system which defines the histological and molecular subsets of NSCLC.
The ALK gene was originally discovered by cloning a translocation found in a subset of anaplastic large cell lymphomas.Citation10 The presence of ALK rearrangements in NSCLC were first reported in 2007Citation7 and are present in 5%–7% of NSCLC patients.Citation11–Citation14 The activated ALK fusion proteins have been shown to drive oncogenic transformation through several molecular signaling pathways,Citation15 including PI3K/AKT/mTOR, JAK/STAT, and RAS/MEK/ERK (). The discovery of ALK rearrangements in NSCLC serendipitously coincided with the development of crizotinib for other ALK or MET-driven malignancies,Citation16,Citation17 allowing for expedited clinical development () and ultimately approval by the US Food and Drug Administration (FDA). This review will discuss the clinical development and use of crizotinib in NSCLC.
Figure 1 Aberrant ALK signaling cascade.
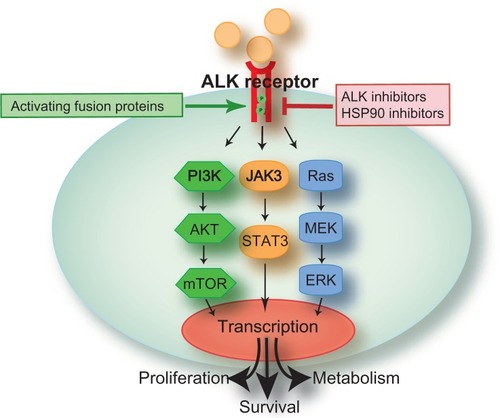
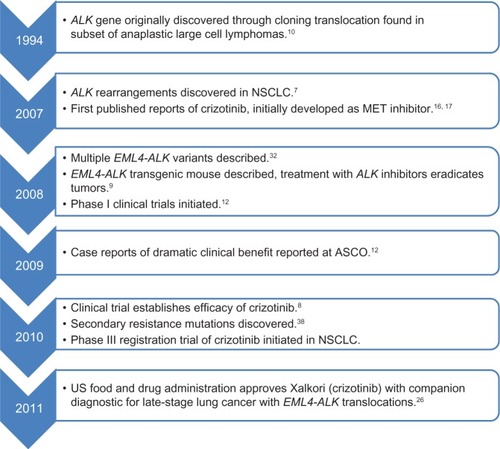
Figure 2 Major events leading to rapid clinical development of crizotinib for ALK-positive NSCLC.
Clinicopathological characteristics of EML4-ALK-positive patients
NSCLC patients with EML4-ALK rearrangements are associated with a specific pattern of patient characteristics.Citation8,Citation13,Citation14 ALK-rearranged adenocarcinomas have a distinct histology that is dominated by a solid tumor growth pattern, signet-ring cells, and intracellular mucin; these features are common among gastric, colon, and breast adenocarcinomas, but rare in NSCLC.Citation13 Patients affected are on average 10–15 years younger than patients lacking an ALK rearrangement and have a history of never having smoked or of former light smoking (≤10 pack-years).Citation8,Citation13,Citation14 ALK rearrangement, EGFR mutation, and KRAS mutation are generally found to occur independently of one another and represent distinct molecular subsets,Citation14 but concomitant ALK rearrangements and EGFR mutations have been observed.Citation18–Citation23
Finally, the presence of ALK rearrangement does not appear to be associated with ethnicity or gender, nor does there appear to be an association with time to progression or overall survival on combined platinum chemotherapy.Citation14 The results evaluating the association with platinum-based chemotherapy will need to be confirmed in a larger study because the current analysis is limited by the presence of only a few patients with ALK rearrangement and a lack of uniformity in the chemotherapy the patients received.
Clinical development of crizotinib for NSCLC
The use of single-agent crizotinib in the treatment of locally advanced or metastatic ALK-positive NSCLC was investigated in two multicenter, single-arm studies (studies A and B).Citation24–Citation26 As defined in the first-in-human Phase I trial of crizotinib, the maximally tolerated dose of 250 mg of crizotinib orally twice dailyCitation12 was used in each of these trials. In study A, ALK-positive NSCLC was identified using the Vysis ALK break-apart fluorescence in situ hybridization (FISH) probe kit (Abbott Molecular Inc, Des Plaines, IL, USA), while study B identified ALK-positive patients using a number of local clinical trial assays. The primary efficacy endpoint in both studies was objective response rate using RECIST (Response Evaluation Criteria in Solid Tumors). In addition to objective response rate, both studies evaluated duration of response. At the time of regulatory submission, results from 136 patients were available from study A (Profile 1005), a global, multicenter, open-label, single-arm Phase II trial.Citation25,Citation26 All patients enrolled in study A had received prior systemic therapy. Of the 136 patients, there was one complete response and 67 partial responses for an objective response rate of 50% (95% confidence interval [CI] 42–59, ). The investigators also reported a median duration of treatment of 22 weeks and a median duration of response of 41.9 weeks. Since the initial results were reported to the FDA, Profile 1005 has continued to enroll patients. As of June 2011, 439 patients were evaluable for safety and 255 were evaluable for tumor response.Citation27 At that time, the objective response rate was 53% (95% CI 47–60), median duration of treatment was 25 weeks (77% still ongoing), median duration of response was 43 weeks, and progression-free survival was 8.5 months (95% CI 6.2–9.9).Citation27 At the time of data cutoff for regulatory submission, study B had enrolled 119 patients with locally advanced or metastatic ALK-positive NSCLC, of whom all but 15 patients had received prior systemic therapy. Of the 119 enrolled patients, there were two complete responses and 69 partial responses for an objective response rate of 61% (95% CI 52–70, ). The median duration of treatment was 32 weeks and the median duration of response was 48.1 weeks.Citation24,Citation26 Study B has continued to enroll patients, and updated data from 143 response-evaluable patients showed 87 objective responses (61%, 95% CI 52–69), including three complete responses and 84 partial responses.Citation28 Median progression-free survival was 9.7 months (95% CI 7.7–12.8). While median overall survival data are not mature at this time, the estimated overall survival at 6 and 12 months was 87.9% (95% CI 81.3–92.3) and 74.8 (95% CI 66.4–81.5), respectively.
Table 1 Efficacy data for approval by the US Food and Drug AdministrationCitation24–Citation26
Based on the response rates from studies A and B, crizotinib was granted accelerated approval by the FDA for the treatment of patients with locally advanced or metastatic NSCLC which is ALK-positive by an FDA-approved test.Citation26 As a condition of the accelerated approval, further post-marketing testing to evaluate clinical outcomes and survival are required. There are now two randomized Phase III studies (Profile 1007 and Profile 1014) evaluating progression-free survival as the primary endpoint and overall survival as a secondary endpoint, while Profile 1007 is comparing crizotinib with pemetrexed or docetaxel as second-line therapy. Profile 1014 is comparing crizotinib with a platinum-pemetrexed combination in newly diagnosed ALK-positive patients with NSCLC.
An interim analysis of data from Profile 1007 was reported at the 2012 Congress of the European Society for Medical Oncology in Vienna, Austria.Citation29 The study included 347 patients with ALK-positive lung cancer who had already been treated with chemotherapy. Patients were randomized to receive crizotinib or standard chemotherapy with pemetrexed or docetaxel. Crizotinib demonstrated a prolonged median progression-free survival of 7.7 months compared with 3 months among those patients who received chemotherapy. The objective response rate was also significantly higher in patients treated with crizotinib (65%) compared with those treated with chemotherapy (20%). Because of the nature of the early analysis, a statistically significant difference in overall survival was not yet seen. Because of significant crossover in the study, where patients in the chemotherapy arm who experienced disease progression were allowed to cross over to receive crizotinib, it may be difficult to see an overall survival benefit even after the data have matured. Finally, while investigators reported that crizotinib was associated with more adverse events than chemotherapy, patients treated with crizotinib reported improved quality of life compared with patients treated with chemotherapy. These results are very promising and suggest that crizotinib should be considered the new standard of care for patients with ALK-positive NSCLC.
Clinical diagnostic techniques
Unlike traditional chemotherapeutic agents, molecularly targeted therapies face the additional challenges of needing an understanding of the molecular target and having a validated test to detect the specific molecular alteration. For crizotinib, ALK rearrangements were identified as the molecular targetCitation7,Citation8 and the Vysis break-apart FISH probe kit was concurrently approved by the FDA as the companion diagnostic.Citation30 In support of the clinicopathological data, the National Comprehensive Cancer Network (NCCN) guidelines for NSCLC now recommend ALK testing concurrently with EGFR mutation testing for adenocarcinoma, large cell carcinoma, and not otherwise specified histological subtypes.Citation30 The current guidelines do not recommend testing in NSCLC patients with squamous cell carcinoma.Citation30
The Vysis ALK break-apart FISH probe kit is the only FDA-approved companion diagnostic to identify ALK-positive NSCLC patients. This test was the only assay used to identify and enroll patients with ALK rearrangements into crizotinib clinical trials prospectively, and therefore is the only assay validated to correlate with crizotinib response. The break-apart FISH probe kit has been shown to be both highly sensitive and specific when using a cutoff of >15% of cells and counting 60 cells.Citation22 In addition to these features, the break-apart assay can be performed on formalin-fixed paraffin-embedded tissue, making it widely applicable, because almost all NSCLC tissue is formalin-fixed paraffin-embedded. Another advantage of this method is that it will detect all ALK rearrangements and is not specific for any particular fusion partner or variant. Despite all of these positive features, the ALK FISH test has several disadvantages compared with other methods of detection. In a normal sample, the 5′ and 3′ ends of the ALK gene are differently labeled with red and green fluorescent probes and are in close proximity to one another. However, in the presence of an ALK rearrangement, the signals “break apart” from one another. The ability to detect the subtle change resulting from chromosomal inversion on chromosome 2p that produces EML4-ALK fusion requires technical expertise, experience, and precise measurement. Without such experience and expertise, the altered probe hybridization patterns may be difficult to discern, leading to false negative results.Citation13 In addition, the test is expensive and is not amenable to high throughput screening that would be ideal for testing the large number of samples needed to identify the few ALK-positive patients.
While the break-apart assay can confirm the presence of an ALK rearrangement, it is not capable of defining the fusion partner or the precise fusion variant. However, realtime polymerase chain reaction is a highly specific method of defining the type of translocation present in a given patient sample.Citation31 In addition to being the most sensitive method of detecting ALK rearrangements, it also has the advantage of requiring limited material for analysis,Citation32 is relatively easy to perform, and is less labor-intensive than FISH. However, the inability to extract sufficient quantity and quality RNA from formalin-fixed paraffin-embedded tissue is a major limitation of this methodology. In addition, even if an adequate RNA can be obtained, all known fusion partnersCitation33 must be known so that all primer sets are included in the analysis to ensure that no ALK-rearrangements are missed.
A third approach to identifying ALK-positive patients is immunohistochemistry. Because immunohistochemistry is routinely performed in every clinical pathology laboratory, ALK immunohistochemistry analysis could easily be incorporated into the normal staining performed during diagnosis and histological subtyping of NSCLC. As a low-cost and routine methodology, immunohistochemistry represents the ideal screen to identify the small subset of NSCLC patients harboring an ALK rearrangement. The major hurdle facing the routine use of ALK immunohistochemistry is the availability of a reliable antibody. A commercially available ALK1 antibody (Dako, Glostrup, Denmark) is currently being used to identify ALK rearrangements in anaplastic large cell lymphoma. However, the expression of ALK in NSCLC appears to be approximately five-fold lower than in anaplastic large cell lymphoma,Citation34 possibly due to differences in the transcriptional activity of the promoter regions of their respective fusion partners (EML4 versus nucleophosmin). As a result, standard staining procedures for the ALK1 antibody have been shown to have a high rate of false negative results for identifying ALK-positive NSCLC.Citation13,Citation34,Citation35 Amplification techniques to enhance the signal of ALK1 staining have been shown to improve immunohistochemistry detection of known ALK-positive samples.Citation13,Citation34,Citation36 Alternative ALK antibodies, such as D5F3, have been shown to have higher sensitivity than the ALK1 antibodyCitation34,Citation36 and have demonstrated complete concordance with genetic data.Citation34 While several alternative ALK antibodies have been tested,Citation34,Citation36 it appears that the D5F3 antibody is the front runner to gain acceptance as a cost-effective and rapid screening tool for identifying ALK-positive NSCLC patients. In early 2012, Ventana Medical Systems Inc (Tucson, AZ, USA) established a collaboration with Pfizer (New York, NY, USA) and a license agreement with Cell Signaling Technology (Beverly, MA, USA) to develop D5F3 as the first fully automated and standardized immunohistochemistry companion diagnostic test for ALK gene rearrangements in NSCLC patients.
Adverse events and precautions
In clinical trials leading to FDA approval, the most common adverse reactions were vision disorders, nausea, diarrhea, vomiting, edema, and constipation ().Citation26 Of these, visual disturbances were seen in 60%–65% of cases and were primarily associated with the transition between light and dark. The most common disturbance includes transient light flashes in the peripheral vision lasting just a few seconds. Onset is generally seen within 2 weeks after initiating therapy, generally decreases in frequency with continued crizotinib use, and ceases following discontinuation of the drug. While not as frequent as the aforementioned adverse effects, other common side effects include sinus bradycardia () and hematological toxicity (). Grade 1–2 sinus bradycardia has occurred in 5% of patients (12 of 255) treated as part of studies A and B.Citation24–Citation26 It is generally profound (heart rate ≤ 45) but asymptomatic and appears to be a pharmacodynamic effect of crizotinib exposure.Citation37 Grade 3 or 4 lymphopenia, neutropenia, and thrombocytopenia occurred in 11.4%, 5.2%, and 0.4% of patients, respectively.Citation26 If a patient experiences grade 3 or 4 hematological toxicity, crizotinib should be withheld until complete blood cell counts return to grade ≤ 2 (). For patients experiencing grade 4 myelosuppression, the dose should be reduced to 200 mg twice daily when treatment is resumed. In the case of recurrent hematological toxicity, crizotinib should be withheld again until recovery to grade ≤ 2, then resume at 250 mg once daily. Crizotinib should be permanently discontinued in the event of further grade 4 recurrence.
Table 2 Common adverse events reported in studies A and B
Table 3 Serious adverse events requiring crizotinib dose modification
Although rare, three more serious adverse events have led to additional labeling under “warnings and precautions” on the package insert.Citation26 First, crizotinib-induced hepatotoxicity, as measured by alanine aminotransferase increases, has been observed in 13% of patients, with grade 3 and 4 alanine aminotransferase increases occurring in 5% of patients (). While fatal hepatotoxicity is rare, occurring in less than 1% of patients in clinical trials, patients should be counseled to report to their physician immediately any symptoms of hepatotoxicity, such as weakness, fatigue, anorexia, nausea, vomiting, abdominal pain (especially right upper quadrant abdominal pain), jaundice, dark urine, generalized pruritus, and bleeding diathesis, especially in combination with fever and rash. Concurrent elevations in alanine aminotransferase greater than three times the upper limit of normal and total bilirubin greater than two times the upper limit of normal, with normal alkaline phosphatase, occurred in less than 1% of patients in clinical trials. Elevation in alanine aminotransferase greater than five times the upper limit of normal occurred in 7% of patients in study A and in 4% of patients in study B. These laboratory findings were generally asymptomatic and reversible upon interruption of treatment. Most patients were able to resume treatment at a lower dose without recurrence of elevated liver enzymes; however, three patients from study A (2%) and one patient from study B (less than 1%) required permanent discontinuation of treatment. If grade 3 or 4 alanine aminotransferase or aspartate aminotransferase elevations are observed with concurrent grade ≤ 1 bilirubin, then crizotinib should be held until laboratory investigations return to grade ≤ 1 or baseline (). At that time, crizotinib can be restarted at 200 mg twice daily. For more severe hepatotoxicity (grade 2–4 alanine aminotransferase or aspartate aminotransferase and grade 2–4 bilirubin), crizotinib should be permanently discontinued. Routine monitoring with liver function tests including alanine aminotransferase and total bilirubin should be performed once a month and as clinically indicated. More frequent repeat testing should be performed in patients with increased liver transaminases, alkaline phosphatase, or total bilirubin, who develop further transaminase elevations.
Crizotinib has been associated with severe, life-threatening, or fatal treatment-related pneumonitis in 1.6% (4 of 255) of patients across studies A and B ().Citation24–Citation26 Similar to other severe toxicities, all of the cases occurred within 2 months after initiation of treatment. Patients should be monitored for pulmonary symptoms indicative of pneumonitis, and crizotinib should be permanently discontinued in patients diagnosed with treatment-related pneumonitis.
QTc prolongation, while rare (1.3%, 4 of 308 patients), has been observed. Therefore, crizotinib should be avoided in patients with congenital long QT syndrome. Monitoring at baseline and periodically thereafter with electrocardiography and electrolytes should be performed in patients with congestive heart failure, bradyarrhythmias, or electrolyte abnormalities, and in patients taking medications that are known to prolong the QT interval. Crizotinib should be permanently discontinued in patients who develop grade 4 QTc prolongation (). If a patient experiences a grade 3 QTc prolongation, the drug should be held until recovery to grade ≤ 1. Once recovered, crizotinib may be resumed at 200 mg twice daily. In case of recurrence of grade 3 QTc prolongation, withhold crizotinib until recovery to grade ≤ 1, then resume crizotinib at 250 mg once daily. In the event that grade 3 QTc prolongation recurs following dose reduction, crizotinib should be permanently discontinued.
Resistance
Despite dramatic clinical responses following initiation of crizotinib therapy in patients with ALK-positive NSCLC, most of these patients will ultimately experience crizotinib failure and progressive disease within a year of starting treatment because of acquired drug resistance. Several mechanisms of crizotinib resistance have been described, including secondary ALK mutations,Citation18,Citation38–Citation42 ALK gene amplification,Citation39–Citation41 and activation of alternative oncogenes (EGFR, KIT, KRAS) via mutational activation, gene amplification, or autocrine signalingCitation39,Citation41,Citation43 (). Secondary mutations within and around the kinase domain ATP-binding site are the most common mechanism of acquired resistance to tyrosine kinase inhibitors.Citation44–Citation46 This mechanism of drug resistance has been documented for inhibitors of BCR-ABL, EGFR, FLT3, KIT, and platelet-derived growth factor receptor.Citation47 The most common mutation among these kinases takes place at the gatekeeper residue, where the size of the side chain of the amino acid at this position regulates the accessibility of the hydrophobic pocket located at the back of the Adenosine-5’-triphosphate (ATP)-binding site.Citation47 Similar to the threonine at position 315 in ABL and at position 790 in EGFR,Citation44–Citation46 the ALK L1196M mutation corresponds to this well described gatekeeper residue. Thus, all three of these mutations have been demonstrated to interfere with the binding of their respective tyrosine kinase inhibitors.Citation38,Citation44–Citation47 The other ALK resistance mutations are located around the conformationally sensitive C-helix and activation loop, and may affect kinase activity and inhibitor binding through alterations in the structure or stability of these elements. For instance, substitution of a bulkier alanine at the glycine 1269 residue positioned at the end of the narrow ATP-binding pocket of ALK is thought to reduce the binding affinity of crizotinib as a result of steric hindrance.Citation39 Further, while the L1152R and C1156Y mutations are not in contact with the ATP-binding cleft itself, they are in close proximity to it and adjacent to the C-helix.Citation18 Therefore, these mutations may alter drug contact and/or create steric hindrance.
Table 4 Mechanisms of acquired crizotinib resistance
In addition to secondary mutations, several other mechanisms of acquired resistance have been described. In a series of 14 ALK-positive NSCLC patients with documented disease progression while on crizotinib, of whom 11 had material for molecular analysis, only four were found to have secondary mutations.Citation39 Two patients were found to have ALK copy number gain (one of which also had a secondary mutation). Mutations in EGFR and KRAS were identified in one and two patients, respectively. In total, this series was able to identify mechanisms of resistance in eight of 11 patients. A second series of 18 ALK-positive NSCLC patients with documented disease progression while on crizotinib identified an array of secondary mutations in four patients and ALK gene amplification in one patient.Citation41 This study also confirmed the proposed EGFR-mediated resistance to crizotinib seen in preclinical models,Citation41,Citation43 by comparing pre-crizotinib and post-crizotinib tissue samples from nine patients.Citation41 Using immunohistochemical staining for phospho-EGFR, EGFR activation was increased following the development of crizotinib resistance in four of nine samples. Finally, one patient’s post-crizotinib tumor was found to have KIT gene amplification, increased KIT expression, and increased expression of the KIT ligand (stem cell factor), and a second patient’s tumor was also shown to have KIT amplification.Citation41
As new mechanisms of resistance continue to emerge, investigators have begun to identify novel approaches to treating patients who have developed crizotinib-resistant disease. There is a large group of second-generation ALK inhibitors in preclinical and clinical development (), some of which have been shown to be effective against specific common secondary mutationsCitation40,Citation48,Citation49 (). A second therapeutic approach to treating ALK-rearranged lung cancers with or without secondary mutations is through inhibition of the molecular chaperone, heat shock protein 90. Heat shock protein 90 inhibitors bind in the ATP-binding pocket of the enzyme, and prevent it from regulating the activation and stability of its client proteins, including ALK. Inhibition of heat shock protein 90 has been shown to result in reduced expression of EML4-ALK,Citation40,Citation50 possibly through proteasome-mediated degradation.Citation51 This treatment approach is effective in both crizotinib-sensitive and crizotinib-resistant (due to secondary mutations) ALK-positive cell lines.Citation40,Citation41 While several heat shock protein 90 inhibitors () have entered clinical development, there has not yet been a clinical trial that has selected patients with ALK-positive disease. Instead, clinical trials to date have tested heat shock protein 90 inhibitors in NSCLC patients with heterogeneous molecular subtypes. These trials have assessed common NSCLC genetic aberrations, including KRAS and EGFR mutations, as well as ALK-rearrangements. In subset analysis of ALK-positive NSCLC patients (most of whom were crizotinib-naïve), heat shock protein 90 inhibitors have shown promising results.Citation52–Citation54 For instance, in a Phase II trial of the heat shock protein 90 inhibitor, ganetespib, four of eight (50%) patients with advanced ALK-positive NSCLC experienced objective responses while receiving treatment with ganetespib monotherapy.Citation54 In addition, the responses were durable, lasting an average of approximately one year. Finally, seven of eight patients (88%) experienced disease control. Whether this approach will be as effective in patients with acquired crizotinib resistance remains to be seen and represents an important area of investigation. However, based on earlier results, the CHIARA trial (CHaperone Inhibition in ALK Rearranged lung cAncer) was initiated to evaluate ganetespib monotherapy in up to 110 patients with stage IIIB/IV NSCLC harboring an ALK gene rearrangement and who have not been previously treated with a direct ALK inhibitor (ie, crizotinib, clinicaltrials.gov, NCT01562015). Other potential therapeutic approaches to mitigate the development of acquired resistance or treat crizotinib-resistant disease are through dual inhibition of critical signaling pathways. For instance, a clinical trial evaluating the dual use of ganetespib and crizotinib in ALK-positive patients is currently enrolling patients (clinicaltrials.gov, NCT01579994). It will be interesting to see if this approach is more effective than either therapy alone and whether dual treatment prevents development of acquired resistance.
Table 5 ALK-targeted therapies in development
Other approaches include the combination of ALK inhibitors, such as crizotinib, with therapies targeting EGFR (erlotinib, clinicaltrials.gov, NCT00965731), PAN-EGFR family (dacomitinib, PF-00299804, clinicaltrials.gov, NCT01121575), or KIT. Finally, targeting downstream effector pathways, such as PI3K/AKT/mTOR and MEK/ERK, represents another therapeutic approach for treating tumors which have developed resistance to targeted receptor tyrosine kinase inhibitors, such as crizotinib. In a lapatinib-resistant HER2-driven breast cancer model, persistent PI3K/AKT/mTOR and MEK/ERK signaling is maintained, and dual inhibition of these pathways has been shown to be an effective treatment strategy.Citation55 While there is evidence that these pathways represent important signaling pathways involved in crizotinib resistance, it remains unclear how effective these various combinations will be and whether they should be used in the first-line setting or saved for use once crizotinib resistance has emerged.
Crizotinib for other mutations (ROS1 and MET)
In addition to ALK-positive NSCLC, crizotinib may also be an effective treatment option for other molecular subsets of NSCLC. ROS1 is a receptor tyrosine kinase of the insulin receptor family. Chromosomal rearrangements involving the ROS1 gene were first described in glioblastoma and cholangiocarcinoma,Citation56–Citation58 and more recently in NSCLC.Citation59–Citation61 Multiple ROS1 fusion partners have been indentified in NSCLC (TPM3, SDC4, SLC34A2, CD74, EZR, LRIG3, and FIGCitation60,Citation61) and shown to fuse at exons 32, 34, 35, or 36 of ROS1. These chimeric proteins maintain constitutive ROS1 kinase activity, leading to persistent downstream signaling and transforming ability via enhanced cell growth, proliferation, and decreased apoptosis.
ROS1 rearrangements have been found in approximately 1%–2% of NSCLC patients,Citation61,Citation62 thereby representing 2000–4000 new cases of ROS1-positive NSCLC each year in the US. ROS1 rearrangements share several clinicopathological features with patients possessing ALK rearrangements. Like ALK-positive NSCLC patients, ROS1-positive NSCLC patients tend to be younger (median age, 49.5 years), never-smokers, and have a histological diagnoses of adenocarcinoma.Citation62 Additionally, ROS1 rearrangements are not found to overlap with other common NSCLC mutations, including EGFR mutations, KRAS mutations, or ALK rearrangements.Citation62
Preclinical studies of cell lines harboring ROS1 rearrangements are sensitive to tyrosine kinase inhibitors,Citation59,Citation63 including the ALK inhibitor TAE684Citation64 and the dual ALK/MET inhibitor crizotinib.Citation62 These observations were initially confirmed by a partial response in a single CD74-ROS1-positive NSCLC patient treated with crizotinib.Citation62 However, a larger cohort of ROS1-positive NSCLC patients treated with crizotinib showed an objective response rate of 54% (7/13, one complete response and six partial responses), with six responses achieved by the first restaging scan at 7–8 weeks after the initiation of therapy.Citation65 The disease control rate (partial response + stable disease + complete response) at 8 weeks was 85% (11/13). Median duration of treatment was 20 weeks (range 4+ to 59+), and at the time of data cutoff for the report all responses were continuing on crizotinib.Citation65
Patients with MET (mesenchymal-epithelial transition) mutations or amplifications represent another molecular subtype of NSCLC that may be responsive to crizotinib therapy. MET is a receptor tyrosine kinase, which signals via RAS, PI3K/AKT, and STAT to promote mitosis, survival, angiogenesis, migration, and invasion.Citation66,Citation67 While MET mutations are rare, MET amplification has been documented in 4%–11% of NSCLCs,Citation68–Citation72 making it an attractive drug target. Several MET-specific therapies are currently in clinical trials, such as tivantinib (ARQ 197) and MetMab.Citation71 However, while the clinical development of crizotinib initially led to its approval by the FDA for ALK-positive NSCLC, it too was originally developed as a MET inhibitor and has demonstrated excellent in vitro activity, with an IC50 (half maximal inhibitory concentration) of 8 nM against MET. Although comprehensive clinical data are lacking to support its efficacy in treating MET-driven NSCLC, a recent case reportCitation72 has demonstrated a rapid and durable response to crizotinib in an NSCLC patient with de novo MET amplification and lacking an ALK rearrangement. While this report only represents one patient, the findings are intriguing and suggest that crizotinib may be effective in MET-amplified NSCLC, as has been shown for other MET-amplified cancers.Citation73,Citation74
Summary
The clinical development of crizotinib has been an amazing success story in translational medicine that relied on the prior clinical experience of other targeted predecessors (ie, erlotinib in EGFR-mutant NSCLC) and a compound ready for clinical development to gain expedited FDA approval. While we await completion of the Phase III clinical trials, early results suggest crizotinib should be considered the standard of care for the first-line treatment of ALK-positive NSCLC patients,Citation29 and possibly NSCLC patients with other genetic aberrations (ROS1 rearrangements or MET amplification). While crizotinib has demonstrated a dramatic clinical response in patients with ALK-positive NSCLC, patients will ultimately develop acquired resistance to crizotinib. Therefore, additional therapeutic approaches to prevent acquired resistance to crizotinib or effectively treat crizotinib-resistant disease are greatly needed.
Acknowledgment
This work was supported by the American Cancer Society (PF-10-239-01-TBG).
Disclosure
The author reports no conflicts of interest in this work.
References
- JemalABrayFCenterMMFerlayJWardEFormanDGlobal cancer statisticsCA Cancer J Clin2011612699021296855
- JemalASiegelRXuJWardECancer statistics, 2010CA Cancer J Clin201060527730020610543
- KimESHirshVMokTGefitinib versus docetaxel in previously treated non-small-cell lung cancer (INTEREST): a randomised phase III trialLancet200837296521809181819027483
- LynchTJBellDWSordellaRActivating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinibN Engl J Med2004350212129213915118073
- PaezJGJännePALeeJCEGFR mutations in lung cancer: correlation with clinical response to gefitinib therapyScience200430456761497150015118125
- ShepherdFARodrigues PereiraJCiuleanuTErlotinib in previously treated non-small-cell lung cancerN Engl J Med2005353212313216014882
- SodaMChoiYLEnomotoMIdentification of the transforming EML4-ALK fusion gene in non-small-cell lung cancerNature2007448715356156617625570
- KwakELBangYJCamidgeDRAnaplastic lymphoma kinase inhibition in non-small-cell lung cancerN Engl J Med2010363181693170320979469
- SodaMTakadaSTakeuchiKA mouse model for EML4-ALK-positive lung cancerProc Natl Acad Sci USA200810550198931989719064915
- MorrisSWKirsteinMNValentineMBFusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphomaScience19942635151128112848122112
- KrisMGJohnsonBEKwiatkowskiDJIdentification of driver mutations in tumor specimens from 1,000 patients with lung adenocarcinoma: the NCI’s Lung Cancer Mutation Consortium (LCMC)J Clin Oncol201129Suppl Abstr CRA7506
- KwakELCamidgeDRClarkJClinical activity observed in a phase I dose escalation trial of an oral c-met and ALK inhibitor, PF-02341066J Clin Oncol200927 Suppl148s
- RodigSJMino-KenudsonMDacicSUnique clinicopathologic features characterize ALK rearranged lung adenocarcinoma in the western populationClin Cancer Res200915165216522319671850
- ShawATYeapBYMino-KenudsonMClinical features and outcome of patients with non small-cell lung cancer who harbor EML4-ALKJ Clin Oncol200927264247425319667264
- ChiarleRVoenaCAmbrogioCPivaRInghiramiGThe anaplastic lymphoma kinase in the pathogenesis of cancerNat Rev Cancer200881112318097461
- ChristensenJGZouHYArangoMECytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphomaMol Cancer Ther2007612 Pt 13314332218089725
- ZouHYLiQLeeJHAn orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanismsCancer Res20076794408441717483355
- SasakiTKoivunenJOginoAA novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitorsCancer Res201171186051606021791641
- TiseoMGelsominoFBoggianiDEGFR and EML4-ALK gene mutations in NSCLC: A case report of erlotinib-resistant patient with both concomitant mutationsLung Cancer201171224124321168933
- WallanderMLGeiersbachKBTrippSRLayfieldLJComparison of reverse transcription-polymerase chain reaction, immunohistochemistry, and fluorescence in situ hybridization methodologies for detection of echinoderm microtubule-associated proteinlike 4-anaplastic lymphoma kinase fusion-positive non-small cell lung carcinoma: Implications for optimal clinical testingArch Pathol Lab Med2012136779680322742552
- YangJZhangXSuJConcomitant EGFR mutation and EML4-ALK gene fusion in non-small cell lung cancerJ Clin Oncol201129Suppl 1510517
- CamidgeDRKonoSAFlaccoAOptimizing the detection of lung cancer patients harboring anaplastic lymphoma kinase (ALK) gene rearrangements potentially suitable for ALK inhibitor treatmentClin Cancer Res201016225581559021062932
- ZhangXZhangSYangXFusion of EML4 and ALK is associated with development of lung adenocarcinomas lacking EGFR and KRAS mutations and is correlated with ALK expressionMol Cancer2010918820624322
- CamidgeDRBangYKwakELProgression-free survival (PFS) from a Phase I study of crizotinib (PF-02341066) in patients with ALKpositive non-small cell lung cancer (NSCLC)J Clin Oncol201129Suppl 152501
- CrinoLCrinòLKimDInitial phase II results with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC): Profile 1005J Clin Oncol201129Suppl Abstr 7514
- Xalkori [crizotinib, package insert]New York, NYPfizer Inc2011
- KimDWAhnMJShiYResults of a global phase II study with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC)J Clin Oncol201230Suppl Abstr 7533
- CamidgeDRBangYJKwakELActivity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a Phase 1studyLancet Oncol201213101011101922954507
- ShawATKimDWNakagawaKPhase III study of crizotinib versus pemetrexed or docetaxel chemotherapy in patients with advanced ALKpositive non-small cell lung cancer (NSCLC) (PROFILE 1007)Abstract LBA1 presented at the Annual Congress of the European Society for Medical OncologyVienna, AustriaSeptember 28–October 2, 2012
- ShawATSolomonBKenudsonMMCrizotinib and testing for ALKJ Natl Compr Canc Netw20119121335134122157554
- TakeuchiKChoiYLSodaMMultiplex reverse transcription-PCR screening for EML4-ALK fusion transcriptsClin Cancer Res200814206618662418927303
- NakajimaTKimuraHTakeuchiKTreatment of lung cancer with an ALK inhibitor after EML4-ALK fusion gene detection using endobronchial ultrasound-guided transbronchial needle aspirationJ Thorac Oncol20105122041204321102268
- ChoiYLTakeuchiKSodaMIdentification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancerCancer Res200868134971497618593892
- Mino-KenudsonMChirieacLRLawKA novel, highly sensitive antibody allows for the routine detection of ALK-rearranged lung adenocarcinomas by standard immunohistochemistryClin Cancer Res20101651561157120179225
- MartelliMPSozziGHernandezLEML4-ALK rearrangement in non-small cell lung cancer and non-tumor lung tissuesAm J Pathol2009174266167019147828
- TakeuchiKChoiYLTogashiYKIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancerClin Cancer Res20091593143314919383809
- OuSHAzadaMDyJStiberJAAsymptomatic profound sinus bradycardia (heart rate ≤ 45) in non-small cell lung cancer patients treated with crizotinibJ Thorac Oncol20116122135213722088989
- ChoiYLSodaMYamashita Y, et al; ALK Lung Cancer Study Group. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitorsN Engl J Med2010363181734173920979473
- DoebeleRCPillingABAisnerDLMechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancerClin Cancer Res20121851472148222235099
- KatayamaRKhanTMBenesCTherapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALKProc Natl Acad Sci USA2011108187535754021502504
- KatayamaRShawATKhanTMMechanisms of acquired crizotinib resistance in ALK-rearranged lung cancersSci Transl Med20124120120ra17
- SasakiTOkudaKZhengWThe neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancersCancer Res20107024100381004321030459
- TanizakiJOkamotoIOkabeTActivation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancerClin Cancer Res201218226219622622843788
- GorreMEMohammedMEllwoodKClinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplificationScience2001293553187688011423618
- PaoWMillerVAPolitiKAAcquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domainPLoS Med200523e7315737014
- KwakELSordellaRBellDWIrreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinibProc Natl Acad Sci USA2005102217665767015897464
- ZhangJYangPLGrayNSTargeting cancer with small molecule kinase inhibitorsNat Rev Cancer200991283919104514
- LovlyCMHeuckmannJMde StanchinaEInsights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitorsCancer Res201171144920493121613408
- SakamotoHTsukaguchiTHiroshimaSCH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutantCancer Cell201119567969021575866
- ChenZSasakiTTanXInhibition of ALK, PI3K/MEK, and HSP90 in murine lung adenocarcinoma induced by EML4-ALK fusion oncogeneCancer Res201070239827983620952506
- NeckersLWorkmanPHsp90 molecular chaperone inhibitors: are we there yet?Clin Cancer Res2012181647622215907
- GossGDThe GALAXY trial (NCT01348126): a randomized IIB/III study of ganetespib (STA-9090) in combination with docetaxel versus docetaxel alone in subjects with stage IIIB or IV NSCLCJ Clin Oncol201230Suppl 15TPS7613
- SequistLVGettingerSSenzerNNActivity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancerJ Clin Oncol201028334953496020940188
- WongKKoczywasMGoldmanJWAn open-label Phase II study of the Hsp90 inhibitor ganetespib (STA-9090) as monotherapy in patients with advanced non-small cell lung cancer (NSCLC)J Clin Oncol201129Suppl Abstr 7500
- RobertsPJUsaryJEDarrDBCombined PI3K/mTOR and MEK inhibition provides broad antitumor activity in faithful murine cancer modelsClin Cancer Res201218195290530322872574
- CharestALaneKMcMahonKFusion of FIG to the receptor tyrosine kinase ROS in a glioblastoma with an interstitial del(6) (q21q21)Genes Chromosomes Cancer2003371587112661006
- GuTLDengXHuangFSurvey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinomaPLoS One201161e1564021253578
- SharmaSBirchmeierCNikawaJO’NeillKRodgersLWiglerMCharacterization of the ros1-gene products expressed in human glioblastoma cell linesOncogene Res198952911002691958
- RikovaKGuoAZengQGlobal survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancerCell200713161190120318083107
- RimkunasVMCrosbyKELiDAnalysis of receptor tyrosine kinase ROS1-positive tumors in non-small cell lung cancer: identification of a FIG-ROS1 fusionClin Cancer Res201218164449445722661537
- TakeuchiKSodaMTogashiYRET, ROS1 and ALK fusions in lung cancerNat Med201218337838122327623
- BergethonKShawATOuSHROS1 rearrangements define a unique molecular class of lung cancersJ Clin Oncol201230886387022215748
- YasudaHPreclinical rationale for use of the clinically available multitargeted tyrosine kinase inhibitor crizotinib in ROS1-translocated lung cancerJ Thorac Oncol2012771086109022617245
- McDermottUGenomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitorsCancer Res20086893389339518451166
- ShawATClinical activity of crizotinib in advanced non-small cell lung cancer (NSCLC) harboring ROS1 gene rearrangementASCO Meeting Abstracts20123015_suppl7508
- FengYThiagarajanPSMaPCMET signaling: novel targeted inhibition and its clinical development in lung cancerJ Thorac Oncol20127245946722237263
- Ma PC, et al. c-Met: structure, functions and potential for therapeutic inhibitionCancer Metastasis Rev200322430932512884908
- ChenYTClinical implications of high MET gene dosage in non-small cell lung cancer patients without previous tyrosine kinase inhibitor treatmentJ Thorac Oncol20116122027203522052229
- DziadziuszkoRCorrelation between MET gene copy number by silver in situ hybridization and protein expression by immunohistochemistry in non-small cell lung cancerJ Thorac Oncol20127234034722237262
- ParkSHigh MET copy number and MET overexpression: poor outcome in non-small cell lung cancer patientsHistol Histopathol201227219720722207554
- VincentMDBiomarkers that currently affect clinical practice: EGFR, ALK, MET, KRASCurr Oncol201219Suppl 1S33S4422787409
- OuSHActivity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplificationJ Thorac Oncol20116594294621623265
- ChiASClinical improvement and rapid radiographic regression induced by a MET inhibitor in a patient with MET-amplified glioblastomaASCO Meeting Abstracts20122915_suppl2072
- LennerzJKMET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinibJ Clin Oncol201129364803481022042947