
Abstract
Chronic inflammation has been identified as an important risk factor for the development of malignancy, and knowledge about its molecular and cellular mechanisms is increasing. Several chronic inflammatory diseases of the gastrointestinal tract are important as risk factors for malignancy and have been studied in detail. In this review, we summarize important molecular mechanisms in chronic inflammation and highlight established and potential links between chronic inflammation and gastrointestinal cancer. In addition, we present the role of chronic inflammation in numerous tumors within the gastrointestinal tract as well as the relevant pathways or epidemiologic observations linking the pathogenesis of these tumors to inflammation.
Different entities of gastrointestinal cancer
Despite advances in cancer therapy, including targeted therapies, many malignancies are still associated with high mortality. Several cancers with a particularly poor prognosis, such as pancreatic ductal adenocarcinoma (PDAC) or gastric carcinoma, are located within the gastrointestinal (GI) tract. According to the GLOBOCAN database, neoplastic disorders of the GI tract account for more than 37% of cancer-related deaths worldwide, and the incidence of some GI malignancies, such as esophageal adenocarcinoma (EAC), has even been increasing over the last decade.Citation1,Citation2
GI cancers are a very heterogeneous group, as they can derive from various cell types. There are lymphoproliferative disorders like mucosa-associated lymphoid tissue (MALT) lymphoma and neuroendocrine tumors, predominantly located in the small intestine and pancreas. In addition to hepatocellular carcinoma (HCC), cholangiocarcinomas also occur in the liver, and stromal neoplasias such as GI stromal tumors (GIST) and squamous cell carcinomas can be found in the stomach and esophagus. However, most GI cancers are adenocarcinomas.
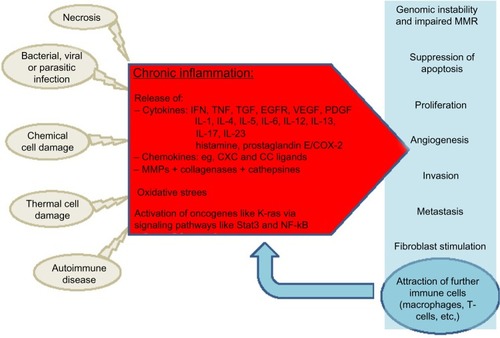
As a barrier to the environment on the one hand, and as the main organ system for digestion and absorption on the other, the GI tract is exposed to many substances, microorganisms, and irritants. Some of these, such as alcohol, Helicobacter pylori, and arsenic are well established risk factors for GI cancers.Citation3–Citation5 The common link between most of these carcinogens is chronic inflammation ().
Table 1 Gastrointestinal tumors that have been associated with chronic inflammation
VirchowCitation6 first described an infiltration of leukocytes in cancer in 1881. In the meantime, inflammation is considered to be one of the hallmarks of carcinogenesis and has become a main focus of research.
Processes of chronic inflammation
The immune system acts as a defense against bacterial, viral, or parasitic infections. It is also responsible for identification, destruction, and phagocytosis of damaged, apoptotic, or neoplastic cells. Local activation of the immune system results in inflammation. The presentation of antigens from pathogens, mechanical or chemical cell irritation, necrotic cells, or autoimmune processes can induce an immune response and inflammation.
In general, the immune system can be distinguished into an innate and an adaptive immune system. Natural killer cells, mast cells, monocytes, macrophages, dendritic cells, and granulocytes usually evoke the first immune reaction and initiate inflammation. They activate specialized T- and B-lymphocytes as part of the adaptive immune system that play an important role in the regulation of inflammation and in the pathogen-specific immune response. However, the latest research has revealed a group of innate lymphoid cells that resemble T-helper (TH) cells in function, and are presumably involved in the initiation of the immune response and inflammation.Citation7,Citation8
Accordingly, acute inflammation is characterized by activation and infiltration of innate immune cells. After interaction with pathogens or through attraction by chemokines, such as transforming growth factor (TGF)-β or platelet-derived growth factor (PDGF) from damaged epithelial cells, neutrophils, macrophages, and mast cells secrete proinflammatory cytokines as well as more chemokines. This signaling results in invasion and activation of additional immune cells. Infiltrating granulocytes produce cytotoxic oxygen and nitrogen radicals to fight pathogens.
Interleukin (IL)-1 from mononuclear phagocytes induces an autocrine activation. Together with tumor necrosis factor (TNF), it enhances the presentation of adhesion molecules and stimulates macrophages to segregate metalloproteinases (MMPs) and collagenases, leading to connective tissue damage. In addition TNF, histamine, and prostaglandins synergistically increase endothelial permeability, which facilitates leukocyte infiltration.
TNF also induces fibroblast growth and differentiation as well as apoptosis of affected epithelial cells, but it also stimulates IL-1 and IL-6 release. It can act as a pro-angiogenic at the site of inflammation, but can also act as an anti-angiogenic at high concentrations.Citation9
After phagocytosis of damaged cells or pathogens, the adaptive immune response is mediated by antigen-presenting cells (APCs) such as dendritic cells. An infiltration with T- and B-lymphocytes is typical for sites of chronic inflammation. According to their IL secretion, TH cells are divided into TH1, TH2, and TH17 cells.Citation10,Citation11 TH1 cells produce interferon (IFN)-γ, TNF-α, and IL-12 and thus control cellular immunity. TH2 cells produce IL-4, IL-5, and IL-13, mediating B-cell-associated humoral immunity, and TH17 cells produce IL-17, IL-6, and IL-23, regulating chronic inflammation via regulatory T-cells (Treg).Citation12
The extent of cytokine release by innate, TH, and Treg cells and the cytokine composition is crucial for the degree and duration of inflammation.Citation13
The role of inflammation in cancer development and maintenance
Genetic instability and cell death resistance
In more than 15% of cancer patients worldwide, the cancer is believed to be caused by infection. Persistent infections, like some other causes, can be a reason for chronic inflammation.Citation14,Citation15 At sites of acute and chronic inflammation, cells are exposed to oxidative cell stress as well as to aggressive oxygen and nitrogen radicals from mononuclear phagocytes and leukocytes. Release of reactive oxygen and nitrogen species is enhanced by proinflammatory cytokines such as TNF, IL-1β, and IFN-α. Particularly in proliferating cells, these radicals can cause DNA damage by nitration and strand breakage.Citation16 Thus, chronic inflammation creates an environment of genomic instability.
Cells can accumulate several mutations as DNA control and repair mechanisms are suppressed in inflammatory tissue. Nitric oxide itself can inhibit DNA mismatch repair (MMR) directly at the protein level and also by induction of hypermethylation.Citation17,Citation18 In this fashion, it inactivates the promoter of the MMR protein hMLH1.Citation19 Another mechanism to alter MMR activity is the induction of hypoxia-inducible factor (HIF)-1α by TNF, IL-1β, and prostaglandin E2. Furthermore, HIF-1α removes c-MYC from the promoter of the MMR genes MSH2 and MSH6.Citation20,Citation21
Besides downregulation of MMR activity, it has been shown that DNA damage-induced apoptosis via the p53 pathway is impaired in chronic inflammation. TNF-α and the TH2 cytokines IL-4 and IL-13, for example, can lead to mutations in the TP53 gene by amplification of activation-induced cytidine deaminase expression.Citation22 Furthermore, migration inhibitory factor (MIF) from macrophages can repress p53 function.Citation23 The dimension of genomic instability together with impaired MMR and cell cycle control in chronic inflammation is emphasized by a high frequency of p53 mutations in chronic inflammatory diseases such as rheumatoid arthritis.Citation24
In addition to p53, inflammation alters a wide range of other cell-cycle control mechanisms. TNF induces fibroblast proliferation (compare page 2), and IL-1 advances the growth of gastric carcinoma.Citation25 Proliferation is stimulated by the activation of toll-like receptor (TLR)-4 by bacteria. This results in increased prostaglandin E2 production by induction of cyclooxygenase (COX)-2 and activation of epidermal growth factor receptor (EGFR) signaling.Citation26
Another mechanism leading to activation of signal transducer and activator of transcription (STAT)-3 is the secretion of IL-6 from cells of the innate immune system and IL-22 from TH17 cells. Apart from various other genes, STAT3 is involved in the transcriptional regulation of cyclin B, cyclin D1, and cyclin D2, which play important roles in cell-cycle control and proliferation.Citation27 Similarly, upregulation of the expression of BCL2 and BCL2-like1 genes by STAT3 results in prolonged cell survival by avoiding apoptosis.Citation28,Citation29
In summary, inflammation leads to DNA damage by oxygen and nitrogen radicals, especially in cells that are prone because of diminished capacity for DNA repair, apoptosis, and/or cell-cycle control.
Persistent proliferation and perpetual tumorigenic signaling in chronic inflammation
Persistence of this mutagenic environment in chronic inflammation enables cells to accumulate several mutations that are required for neoplastic transformation. For example, Kras and p53 gene mutations are very common in patients with GI malignancies. Tumor suppressor p53 is often designated as ‘guardian of the genome’ as it prompts cell-cycle arrest and apoptosis in defective cells via Mdm2, p21, mammalian target of rapamycin (mTOR), and PUMA signaling. In contrast, Kras is an oncogene, which is activated in many different tumors. It belongs to the Ras family of guanosine triphosphates (GTPases) and is involved in many cellular processes, such as cell proliferation, differentiation, apoptosis, and senescence. Phosphatidylinositol 3-kinase (PI-3-K), Raf, Rac, Rho, and TGF-β belong to its key downstream effector pathway.Citation30–Citation32
IL-6/STAT3 signaling can maintain a mutagenic state. Upregulation of sphingosine-1-phosphate (S1P) activates nuclear factor (NF)-κB, which in turn induces IL-6 transcription.Citation33 NF-κB can also be activated by TNF and IL-1.Citation34 A self-augmenting loop maintaining chronic inflammation is described after the activation of the proto-oncogene SRC. SRC activation as the triggering event initiates NF-κB/STAT3-mediated cell growth and malignant transformation. Concurrently, NF-κB rapidly reduces let7-microRNA via Lin28 activation, which acts as a direct inhibitor of IL-6. High IL-6 concentrations in return activate NF-κB signaling and by these means start a positive feedback loop and link signaling in chronic inflammation to malignant transformation.Citation35 NF-κB directly, but also via WNT-β-catenin activation, has been shown to provoke dedifferentiation, proliferation, and resistance to cell death in many tumors.Citation36,Citation37 NF-κB as the signal pathway relating carcinogenesis to inflammation was first described by Greten et alCitation38 in colorectal cancer (CRC). In fact, there is evidence for a role of NF-κB in the carcinogenesis of various GI tumors. NF-κB can be activated by TNF and is involved in the transcriptional regulation of many cellular functions. Amongst these are a plethora of tumorigenic functions such as proliferation, invasion, and suppression of apoptosis. Beyond that, NF-κB influences the inflammatory response by controlling the secretion of cytokines such as TNF-α and IL-1.Citation39
Angiogenesis, invasion, and migration
Genomic instability and evasion of cell death are considered to be key steps in carcinogenesis and metastasis formation. But inflammatory cytokines are involved in several of these processes.Citation40 For example, TNF, IL-1, and IL-6 from neutrophils, mast cells, and macrophages increase transcription of vascular endothelial growth factor (VEGF) – an important stimulating factor for angiogenesis.Citation41,Citation42 Another example is the pro-angiogenic effect of IL-8. IL-8 can also be induced by TNF and IL-1.Citation43,Citation44
In accordance with Hanahan and Weinberg’s Hallmarks of Cancer,Citation40 infiltration and migration are crucial features of malignant cells. Altered endothelial permeability by TNF and degradation of extracellular matrix by MMP and cathepsins not only fosters leukocyte infiltration during inflammation, but also paves the way for invasive growth, migration, and intravasation of tumor cells.Citation45,Citation46 Inhibition of IL-1 in a metastatic mouse model actually revealed a distinct decrease in the rate of metastasis.Citation47 To detach epithelial cells and enable these cells to migrate, an epithelial mesenchymal transition (EMT) is assumed to be decisive. STAT3 transcriptional regulation also includes transcription of E-cadherin, which is characteristic for epithelial cells and transcription of MMP. TNF, IL-1, and IL-6-associated NF-κB and STAT3 activation leads to downregulation of E-cadherin and increased MMP transcription, constituting a mesenchymal cell phenotype. A switch from E-cadherin to N-cadherin is typical of EMT. As the switch from E- to N-cadherin depends on the transcription factor Snail, Snail is an important regulator of EMT.Citation48,Citation49 In CRC, active NF-κB has been shown to inhibit Snail ubiquitination and degradation downstream, thereby highlighting the significance of inflammatory cytokine signaling for metastasis.Citation50–Citation52 Through Wnt and Ras signaling, TGF-β can also induce EMT.Citation53
Through these mechanisms, chronic inflammation is a decisive player in early carcinogenesis. It generates a mutagenic environment and may enable susceptible cells with inherited or spontaneously altered genes to accumulate enough mutations for malignant transformation. This is supported by studies that show that treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) consistently decreases the risk of cancer, most likely by direct inhibition of the NF-κB/STAT3 signaling pathway. NF-κB/STAT3 signaling increases expression of COX-2 and in turn is activated itself by COX-2. COX-2 is significantly overexpressed in malignancies, and NSAIDs also increase cell death in existing tumors.Citation27,Citation54–Citation58 The anti-neoplastic activity of NSAIDs seems to be mediated by several effectors. NSAIDs without COX inhibitory activity, for example, have similar anti-tumorigenic efficacy. Among others, induction of apoptosis as well as suppression of cyclic guanosine monophosphate (cGMP)- and β-catenin-dependent transcription were identified as alternative NSAID properties. This leads to the assumption that inflammatory cytokines have an influence on tumor progression.Citation59,Citation60
Inflammation in tumor progression
Tumor interactions with the immune system have recently been the focus of extensive research, and a comprehensive description would be beyond the scope of this article. However, as tumors induce a peritumoral chronic state of inflammation, major aspects of tumor-immune cell interactions will be illustrated in this article. To maintain carcinogenic signaling from immune cells, tumors actively attract macrophages and T-cells by chemokines such as IL-4 and TGF-β, bridging the gap to Virchow’s description of leukocyte infiltration of tumors. Cytokines, MMPs, and cathepsins from the so-called tumor-associated macrophages (TAMs) are pivotal for further growth, angiogenesis, and invasion. In addition, macrophage-derived EGF forms a paracrine signaling loop, with tumor-derived colony-stimulating factor (CSF)-1 driving neoplastic invasion.Citation61,Citation62 These experimental findings have been confirmed by demonstrating an association of macrophage infiltration with a poor prognosis in human cancer patients.Citation63 As necrotic tissue fosters macrophage invasion, tumors might even benefit from limited intra-tumoral necrosis.Citation62 In contrast, the immune system poses a threat to malignant cells and it is essential for tumors to escape a cytotoxic immune response. Gabrilovich et al.Citation64 discovered tumor-derived VEGF-A, which has the potential to suppress dendritic cell antigen presentation. APC function is also directly impaired by TGF-β and IL-10 secretion of tumor cells.Citation62,Citation65 Another mechanism to avoid natural killer cells is to attract myeloid-derived suppressor cells (MDSCs). Amongst others, the release of chemokines such as CC-chemokine ligands (CCL) -2, -5, and -12, prostaglandins, and granulocyte colony-stimulating factor (G-CSF) can evoke MDSC accumulation in tumor tissue. As a consequence, MDSCs suppress T-cell function by production of IL-10 and TGF-β.Citation42,Citation62,Citation66,Citation67 Only the immune-suppressive function of Treg cells is maintained by TGF-β, and elevated levels of MDSC and Treg correlate with tumor burden.Citation68
In conclusion, inflammatory signaling can both support and suppress the growth of established tumors. By partial immune suppression with concomitant selective activation of myeloid cells, tumors influence the inflammatory response to their favor.
Specific role of chronic inflammation in gastrointestinal cancer
Esophagus
Barrett’s esophagus and esophageal adenocarcinoma
Gastroesophageal reflux disease (GERD), smoking, body mass index, and a low fruit consumption are well established risk factors for EAC and account for the majority of cases. Although the interaction of several of these risk factors is crucial for the development of this cancer, GERD appears to be the most important individual risk factor.Citation69 The reflux of gastric acid causes mucosal damage of the distal esophagus, leading to chronic esophagitis. In some patients, metaplasia from a squamous to a columnar epithelium occurs as a consequence of chronic inflammation during mucosal regeneration – the so-called Barrett’s mucosa. Whether Barrett’s mucosa is caused by a translocation or migration of gastric cells, by bone marrow stem cells, or by true metaplasia of esophageal cells is controversial. However, proinflammatory cytokines such as IL-6, IL-8, and IL-1β appear to be involved in the development of Barrett’s mucosa.Citation70–Citation73 Consistently, inhibition of COX-2 inhibits proliferation of Barrett’s mucosal cells.Citation74 As a result, the incidence of EAC is 30 times higher in people with Barrett’s esophagus than in the general population.Citation74 Nevertheless, the annual risk of patients with Barrett’s mucosa for developing EAC is 0.12%–0.4% and is relatively low overall.Citation75,Citation76
Squamous cell carcinoma
Besides the major risk factors of alcohol, smoking, and genetic polymorphisms, a carcinogenesis-promoting role of chronic inflammation in squamous cell carcinoma is assumed. Hot beverages and fungal invasion are suspected to cause chronic irritation and inflammation of the squamous cell mucosa. The influence of human papilloma virus is still under debate, but its pathogenetic influence on carcinogenesis might be by way of genomic integration rather than inflammation.Citation77–Citation79
Stomach
Carcinomas other than adenocarcinomas are very rare in the stomach. Gastric adenocarcinomas are classified into an intestinal and a diffuse type. A total of 80% of gastric cancer cases are associated with H. pylori infections, and H. pylori-associated gastric cancer is one of the best elucidated relationships between chronic inflammation and cancer overall. While being protected by antioxidants itself, H. pylori can expose host cells to oxidative stress, impair MMR, and alter DNA methylation.Citation80,Citation81 In response to H. pylori, the gastric mucosa releases the inflammatory cytokine IL-8, an overexpression of which correlates with a poor prognosis.Citation82 Virulence factor cytotoxin-associated gene A (cagA)-positive H. pylori strains were identified as particularly carcinogenic. After delivering cagA into gastric epithelial cells, H. pylori can initiate signaling that activates growth factor receptors that increase proliferation, invasion, and angiogenesis and also inhibit apoptosis.Citation83 Additionally, chronic inflammation caused by infection with H. pylori contributes to neoplastic transformation by establishing a positive feedback loop via STAT3-dependent COX-2 induction, which in turn influences STAT3 regulation via IL-6.Citation84
However, gastric carcinogenesis is considered a multistep process, and other inflammatory risk factors have been identified that either act independently of H. pylori infections or further enhance their effects. For example, chronic gastritis caused by bile reflux can cause intestinal metaplasia as a neoplastic precursor lesion. Also, T-cell-mediated autoimmune gastritis fosters the development of intestinal-type gastric cancer.Citation85,Citation86
All of these risk factors lead to a state of chronic inflammation. Activation of signaling mediators that are typical of chronic inflammation such as TNF, IL-6, and prostaglandin E2 has been identified as a promoter of gastric tumorigenesis. For example, TNF-α fosters carcinogenesis by upregulating nicotinamide adenine dinucleotide phosphate (NADPH) oxidase organizer 1 (Noxo1) and Gna14, resulting in dedifferentiation of gastric epithelial cells.Citation87–Citation89 Furthermore, tumor suppressor microRNA miR-7, which inhibits proliferation in vitro, is downregulated in gastric tumors in an inflammation-dependent manner.Citation90
Colorectal cancer
Inflammatory bowel disease (IBD) has long been identified as risk factor for CRC. As CRC in IBD arises from flat dysplastic tissue or dysplasia-associated lesions or masses, carcinogenesis seems to differ from the well described adenoma–carcinoma sequence. Concomitantly, IBD-associated CRC has a worse prognosis. The risk of CRC in patients with colonic manifestation of Crohn’s disease and in patients with ulcerative colitis is similar. Recent population-based studies have shown an overall standardized incidence ratio of CRC in patients with IBD of 1.7, with a significantly lower age of CRC manifestation (7–12 years earlier). However, this ratio is lower than those reported for years before 2000, which might be a result of aging cohorts.Citation91,Citation92 The risk of malignancy correlates with the duration of IBD (10 years after IBD diagnosis: increase of approximately 1% per year). Several clinical symptoms of IBD, such as anatomic extent of disease, disease activity, and primary sclerosing cholangitis (PSC) characterize patients with a comparatively higher risk for malignancy.Citation93,Citation94 Inflammation-driven carcinogenesis in patients with CRC is based on the above-illustrated conditions in chronic inflammation, with oxidative stress and impaired MMR combined with proliferation, invasion, and angiogenesis promoting signaling. In contrast with sporadic CRC, p53 mutations occur rather early in disease development, and APC mutations rather late.Citation95 Downregulation of tumor-suppressor genes by hypermethylation was often found at sites of chronic inflammation, and recently the role of intestinal pathogens in carcinogenesis has attracted increasing attention. The DNA repair gene MUTYH is a homologue to the Escherichia coli gene mutY, and colonization by exclusively intracellular E. coli was detected at tumor sites.Citation95,Citation96 Pathogen antigen binding to TLRs, such as TLR9, can initiate inflammation and could provide a link between pathogens such as E. coli and neoplastic transformation.Citation97 As Treg concentration in CRC correlates with morbidity, activation of Tregs in IBD might also contribute to cancer initiation and progression.Citation98
Pancreas
Chronic pancreatitis is a well established risk factor for the development of PDAC, and the risk correlates with the duration of chronic pancreatitis. As mutations found in hereditary pancreatitis differ from those found in tumors, this epidemiologic observation has to be based on enhanced carcinogenesis due to inflammation.Citation99,Citation100 In fact, chemokine receptor 2 (CXCR2) ligands such as IL-6 and IL-8 have been identified as autocrine promoters of tumor growth in pancreatic cancer.Citation101 Another inflammatory chemokine, the so-called macrophage pro-inflammatory chemokine-3a (MIP-3a/CCL20), is overexpressed in pancreatic cancer cells and TAMs. It stimulates growth and migration of neoplastic cells and activates further macrophages.Citation102 Furthermore, patients with homozygous allele 2 of IL-1β, which is associated with higher IL-1β concentrations in pancreatic cancer, had a significantly shorter survival, which once again points to the importance of crosstalk of pancreatic cancer and immune cells.Citation103 TGF-β signaling is enhanced in chronic inflammation and plays a prominent role in its regulation (compare pages 2 and 4). Concomitantly, inhibition of TGF-β signaling in pancreatic cancer resulted in increased survival.Citation104 In 90% of pancreatic cancers, Kras-activating mutations are responsible for continuous proliferative and anti-apoptotic signaling. Interestingly, activation of NF-κB signaling in chronic pancreatitis can ultimately lead to Kras activation and neoplastic transformation. Also, permanently mutated Kras has been detected in chronic pancreatitis.Citation105–Citation108 Only recently has a pivotal role of the IL-6/STAT3 pathway in pancreatic cancer been described. Oncogenic Ras (KrasG12D) induces the secretion of IL-6 from different cell types, and genetic inactivation of IL-6 impedes Ras-driven tumorigenesis.Citation109 The activation of mutant Kras (KrasG12D) in pancreatic ductal epithelial cells reprograms the tumor microenvironment and is associated with a robust inflammatory response characterized by release of inflammatory cytokines, such as granulocyte macrophage (GM)-CSF. KrasG12D-dependent GM-CSF production promotes infiltration of myeloid cells into the surrounding stroma.Citation110,Citation111 TAMs secrete IL-6 that directly activates the Janus kinases (JAKs), thus influencing pancreatic intraepithelial neoplasia (PanIN) progression and pancreatic cancer development.Citation112,Citation113 The activated JAK in turn phosphorylates several downstream targets, including cytoplasmic STAT3, which after dimerization rapidly translocates into the nucleus and promotes PanIN progression through transcriptional regulation of anti-apoptotic and pro-proliferative genes. Pancreatic epithelial STAT3 deletion and inactivation of IL-6 trans-signaling impairs PanIN formation and inhibits PDAC development.Citation113 These studies, together with numerous other observations, suggest that Kras serves as a link between chronic inflammation and carcinogenesis. In combination with other risk factors, H. pylori infection and periodontal disease also appear to contribute to carcinogenesis in the pancreas.Citation114
Hepatobiliary
Hepatocellular carcinoma
In recent decades, incidence of HCC has been increasing in the USA, and a growing number of hepatitis C virus (HCV) and hepatitis B virus (HBV) infections have been identified as the underlying cause. Other risk factors for HCC have not increased in incidence during this time.Citation115 HBV is a DNA-integrating virus. Accordingly, activation or suppression of oncogenes and tumor suppressor genes by random integration of viral DNA is suspected to be the key effect in HBV-associated carcinogenesis. However, HBV infection-induced inflammation with activation of NF-κB signaling and DNA damage by oxidative stress appear to be important pathogenetic mechanisms.Citation116 In contrast, no specific virally induced oncogenes have been identified in HCV infection. Carcinogenesis in patients with HCV (an RNA virus) infection is considered to be an inflammation-mediated process (eg, TGF-β signaling) with a high cell turnover, fibrosis, and cirrhosis. This hypothesis is supported by the fact that HCV-related HCC correlates with the degree of inflammation, whereas HCC can emerge in HBV-infected patients even without evidence of cirrhosis.Citation117–Citation119 This hypothesis was further supported by the finding that antiviral therapy was beneficial in select patients with HBV/HCV-related HCC.Citation120
However, as these lesions develop secondary to hepatic cirrhosis in most patients with HCC, this is considered to be the major risk factor. While hepatocytes are more susceptible to mutagenesis because of the high cell turnover during regeneration, recurrent necrosis of liver cells induces chronic inflammation. This process leads to hepatic fibrosis, which ultimately culminates in cirrhosis.Citation121–Citation123 Hepatocellular necrosis can be caused by toxins such as alcohol, other environmental factors such as diet, or by chronic inflammation itself. Accordingly, non-alcoholic fatty liver disease and primary biliary cirrhosis have also been identified as risk factors of HCC that are based on chronic inflammation.Citation124–Citation126
During inflammation, macrophages (ie, Kupffer cells) activate fibroblasts and stellate cells by release of TNF and PDGF. Activated fibroblasts produce numerous cytokines, including EGF, hepatocyte growth factor, fibroblast growth factor, IL-6, and MMP-9. This combined signaling causes fibroblastic differentiation of stellate cells, which plays a central role in the pathogenesis of fibrosis. After initiation of the tumor, infiltrating Treg cells that are attracted by Kupffer cells and TAMs modify the immune response and stimulate angiogenesis. Taken together, accumulation of mutations during inflammation-driven fibrosis or cirrhosis is a prerequisite for HCC development.Citation127,Citation128
Cholangiocarcinoma
The risk factor that accounts for the most cases of cholangiocarcinoma worldwide is infection with parasites such as Opisthorchis viverrini, Clonorchis sinensis, or Ascaris lumbricoides. These parasites are endemic in several Asian regions, cause chronic inflammation of the biliary tree, and expose the biliary epithelium to reactive oxygen and nitrogen species.Citation129,Citation130
In Western nations, almost one-third of the patients with cholangiocarcinoma suffer from PSC and, on average, develop the malignancy at a significantly lower age.Citation131 Interestingly, the duration of PSC does not correlate with the incidence of cholangiocarcinoma. DNA damage and proliferative and mutagenic signaling in chronic inflammation of PSC are considered key steps in the development of cholangiocarcinoma.Citation132
Similar to HCC, chronic infection with HBV or HCV, hepatic cirrhosis, and non-alcoholic fatty liver disease also increase the risk of cholangiocarcinoma.Citation133,Citation134
Recurrent bacterial infections due to cholestasis by biliary stones, congenital abnormalities such as fibropolycystic liver disease, or due to choledocho-enteric anastomoses are also associated with a higher incidence of cholangiocarcinoma. Chronic infections of the biliary tree are presumably not solitary triggers of carcinogenesis and can provide the necessary second hit for malignant transformation in genetically predisposed patients.Citation135
Several causative signaling pathways linking inflammation to carcinogenesis in biliary disease have been identified. Human immune response to parasitic biliary infections is mediated by metastasis-associated protein 1 (MTA1) with its adjunctive cytokeratins K-18 and 19. Besides regulating TH1 and TH2 immune responses, MTA1 has a central role in carcinogenesis and is overexpressed in malignant cells.Citation136 Auto-and paracrine stimulation of proliferation mediated by IL-6, IL-8, TGF-β, and TNF-α can be found in cholangiocytes. This stimulation is mediated by IL-6-related activation of p38 mitogen-activated protein kinase and overexpression of the anti-apoptotic myeloid cell leukemia (Mcl)-1.Citation137–Citation139
A similar correlation of recurrent cholecystitis and chronic inflammation of the bile ducts with malignancy is evident for the gallbladder.Citation140
Mucosa-associated lymphoid tissue lymphoma
MALT lymphoma is an extra-nodal B-cell non-Hodgkin lymphoma that can manifest itself in epithelial tissues throughout the body. Typical GI organs of manifestation are the stomach and the small intestine. Although several chromosomal translocations have been identified in MALT lymphomas, recent research provides augmenting evidence for an inflammatory genesis. Chronic inflammation by infections with Campylobacter jejuni, HCV, or, predominantly, H. pylori precede many cases of MALT lymphoma and an association of extra-intestinal MALT and autoimmune disease has been described.Citation141–Citation143 The release of a proliferation-inducing ligand (APRIL), which belongs to the TNF family, by macrophages as response to infection is a potential link between inflammation and neoplastic transformation. APRIL is important for B-cell maturation and survival. Also, NF-κB is frequently activated in MALT lymphoma. This signaling pathway can be induced in inflammation or might be permanently mutated by oxygen radicals during chronic inflammation.Citation141,Citation144 H. pylori virulence factor cagA is found in approximately 90% of patients with gastric MALT lymphoma. Also, eradication of H. pylori leads to regression of GI MALT lymphomas in about 90% of the cases, and H. pylori eradication is the standard therapy for patients affected.Citation145,Citation146
Clinical perspective
There is accumulating evidence for the involvement of chronic inflammation in the carcinogenesis of most GI malignancies, and the underlying cellular and molecular processes are increasingly understood. Chronic inflammation as a potent player in tumor initiation and progression has extended our concept of the multivariate process of neoplastic transformation (). This knowledge has already been implemented in clinical routine. For example, in patients with Barrett’s esophagus, H. pylori is eradicated to prevent esophageal or gastric cancer. In patients with MALT lymphoma, eradication of H. pylori is used to induce remission in these patients. In many inflammatory diseases such as IBD, anti-inflammatory drugs are the mainstay of therapy. However, while mucosal healing in IBD patients results in a decreasing rate of dysplasia, a similar effect could not be proven for single agents such as 5-aminosalicylates or thiopurines.Citation147,Citation148 In contrast, many studies showed a chemoprophylactic property of NSAIDs in other inflammation-associated malignancies, such as gastric carcinoma and CRC. Nevertheless, unselective COX inhibitors carry a notable risk of GI bleeding, and selective COX-2 inhibitors also carry a risk of cardiovascular complications that outweigh the anti-neoplastic benefits.Citation149 Thus, the identification of COX-independent anti-neoplastic activity of NSAIDs and the development of NSAIDs that are more selective might offer future treatment options for defined patient populations that carry a high risk of cancer, such as patients with a history of familial adenomatous polyposis.
Figure 1 Chronic inflammation in carcinogenesis.
Nowadays, immune-suppressive drugs not only restrict chronic inflammation but are also associated with an increased risk of developing malignancies. More specific agents that interfere with selected immune functions might actually be equally effective as an anti-inflammatory and as an anti-neoplastic medication. Inhibition of IL-6 signaling via IL-6 receptor antibodies (Tocilizumab®; Hoffmann-La Roche AG, Basel, Swiss) represents an effective treatment for patients with rheumatoid arthritis, but also showed promising results in a mouse model of Crohn’s disease.Citation150
However, similar to TNF, with its pro- and anti-angiogenic effects, many mediators of the immune response have a dual role in carcinogenesis; further research is required for clarification.
Also complicating is the fact that many cytokines and signaling pathways have multiple functions throughout the human body. For example, Tocilizumab-induced inhibition of IL-6 – a decisive cytokine linking inflammation and carcinogenesis – is associated with severe side effects such as gastric perforation.Citation151
More importantly, it is crucial to obtain further insight into the molecular pathways that drive carcinogenesis in inflammation. Identification of such signaling pathways is therefore of utmost importance. For example, blocking the soluble IL-6 receptor that is predominantly activated during stress situations such as chronic inflammation and cancer might evolve into a future treatment option.Citation152 Another potentially interesting target is the JAK2/STAT3 pathway. In fact, several studies are currently underway that address the pharmacological inhibition of the IL-6-dependent JAK2/STAT3 pathway in patients with various solid GI tumors.
Furthermore, there is already evidence of NK cells being capable of restoring the antigen-presenting function of dendritic cells and MDSC.Citation68 Detailed understanding of the tumor–immune system interaction would provide the possibility to redirect the immune response against neoplastic cells. Such immune-modulatory drugs could be beneficial for cancer prevention as well as for limiting cancer progression or improving response to existing therapies.
Conclusion
Chronic inflammation is a major risk factor for the development of GI malignancies. As ‘wounds that do not heal’, tumors benefit from inflammatory signaling while escaping damage through the immune system by modifying the immune response in their favor. Further insight into the function of the mediators and the regulation of the immune response could help to identify more specific future treatment targets. In our opinion, understanding of the tumor microenvironment and tumor–immune system interactions will be crucial, as these aspects have not yet been implemented in therapeutic regimens. Altering a tumor-susceptible micro environment, or even avoiding formation of the so-called metastatic niche, might reduce metastasis. In addition, re-establishing an immune response against neoplastic cells might improve the efficacy of existing therapies. This estimation is supported by the finding that assessment of the immunologic tumor environment allows better conclusions regarding prognosis and response to therapy than the traditional anatomic International Union Against Cancer ‘Tumor/Node/Metastasis’ (UICC-TNM) classification system for tumor staging.Citation153 As assay availability and protocols vary among laboratories, an international task force has proposed the ‘Immunoscore’ as a standardized prognostic tool.Citation154 The ongoing clinical evaluation of this score will reveal its relevance for clinical practice.
Disclosure
The authors do not have any conflicts of interest to disclose.
References
- FerlayJShinHRBrayFFormanDMathersCParkinDMEstimates of worldwide burden of cancer in 2008: GLOBOCAN 2008Int J Cancer2010127122893291721351269
- CookMBChowWHDevesaSSOesophageal cancer incidence in the United States by race, sex, and histologic type, 1977–2005Br J Cancer2009101585585919672254
- LiuJWaalkesMPLiver is a target of arsenic carcinogenesisToxicol Sci20081051243218566022
- PeekRMJrCrabtreeJEHelicobacter infection and gastric neoplasiaJ Pathol2006208223324816362989
- Varela-ReyMWoodhooAMartinez-ChantarMLMatoJMLuSCAlcohol, DNA methylation, and cancerAlcohol Res2013351253524313162
- VirchowRAn address on the value of pathological experimentsBr Med J188121075198203
- SpitsHCupedoTInnate lymphoid cells: emerging insights in development, lineage relationships, and functionAnnu Rev Immunol20123064767522224763
- SpitsHArtisDColonnaMInnate lymphoid cells – a proposal for uniform nomenclatureNat Rev Immunol201313214514923348417
- LejeuneFJRüeggCLiénardDClinical applications of TNF-alpha in cancerCurr Opin Immunol19981055735809794839
- DongCDiversification of T-helper-cell lineages: finding the family root of IL-17-producing cellsNat Rev Immunol20066432933316557264
- RomagnaniSLymphokine production by human T cells in disease statesAnnu Rev Immunol1994122272578011282
- MiossecPIL-17 and Th17 cells in human inflammatory diseasesMicrobes Infect200911562563019371791
- GallinJISnydermanRFearonDTHaynesBFNathanCInflammation: Basic Principles and Clinical Correlates3rd edPhiladelphiaLippincott Williams & Wilkins1999
- KuperHAdamiHOTrichopoulosDInfections as a major preventable cause of human cancerJ Intern Med2000248317118310971784
- de MartelCFerlayJFranceschiSGlobal burden of cancers attributable to infections in 2008: a review and synthetic analysisLancet Oncol201213660761522575588
- MaedaHAkaikeTNitric oxide and oxygen radicals in infection, inflammation, and cancerBiochemistry (Mosc)19986378548659721338
- JaiswalMLaRussoNFBurgartLJGoresGJInflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanismCancer Res200060118419010646872
- ChangCLMarraGChauhanDPOxidative stress inactivates the human DNA mismatch repair systemAm J Physiol Cell Physiol20022831C148C15412055083
- FleisherASEstellerMHarpazNMicrosatellite instability in inflammatory bowel disease-associated neoplastic lesions is associated with hypermethylation and diminished expression of the DNA mismatch repair gene, hMLH1Cancer Res200060174864486810987299
- KoshijiMToKKHammerSHIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expressionMol Cell200517679380315780936
- JungYJIsaacsJSLeeSTrepelJNeckersLIL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesisFASEB J200317142115211712958148
- EndoYMarusawaHKouTActivation-induced cytidine deaminase links between inflammation and the development of colitis-associated colorectal cancersGastroenterology20081353889898898. e1e318691581
- HudsonJDShoaibiMAMaestroRCarneroAHannonGJBeachDHA proinflammatory cytokine inhibits p53 tumor suppressor activityJ Exp Med1999190101375138210562313
- YamanishiYBoyleDLRosengrenSGreenDRZvaiflerNJFiresteinGSRegional analysis of p53 mutations in rheumatoid arthritis synoviumProc Natl Acad Sci U S A20029915100251003012119414
- El-OmarEMCarringtonMChowWHInterleukin-1 polymorphisms associated with increased risk of gastric cancerNature2000404677639840210746728
- FukataMChenAVamadevanASToll-like receptor-4 promotes the development of colitis-associated colorectal tumorsGastroenterology200713361869188118054559
- YuHPardollDJoveRSTATs in cancer inflammation and immunity: a leading role for STAT3Nat Rev Cancer200991179880919851315
- BollrathJPhesseTJvon BurstinVAgp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesisCancer Cell20091529110219185844
- SonnenbergGFFouserLAArtisDBorder patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22Nat Immunol201112538339021502992
- CastellanoEDownwardJRAS interaction with PI3K: more than just another effector pathwayGenes Cancer20112326127421779497
- Pylayeva-GuptaYGrabockaEBar-SagiDRAS oncogenes: weaving a tumorigenic webNat Rev Cancer2011111176177421993244
- SuppiahAGreenmanJClinical utility of anti-p53 auto-antibody: systematic review and focus on colorectal cancerWorld J Gastroenterol201319294651467023922463
- LiangJNagahashiMKimEYSphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancerCancer Cell201323110712023273921
- FaustmanDDavisMTNF receptor 2 pathway: drug target for autoimmune diseasesNat Rev Drug Discov20109648249320489699
- IliopoulosDHirschHAStruhlKAn epigenetic switch involving NF-kappaB, Lin28, Let-7 microRNA, and IL6 links inflammation to cell transformationCell2009139469370619878981
- Ben-NeriahYKarinMInflammation meets cancer, with NF-kappaB as the matchmakerNat Immunol201112871572321772280
- SchwitallaSFingerleAACammareriPIntestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like propertiesCell20131521–2253823273993
- GretenFREckmannLGretenTFIKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancerCell2004118328529615294155
- KimHJHawkeNBaldwinASNF-kappaB and IKK as therapeutic targets in cancerCell Death Differ200613573874716485028
- HanahanDWeinbergRAHallmarks of cancer: the next generationCell2011144564667421376230
- FerraraNVascular endothelial growth factor: basic science and clinical progressEndocr Rev200425458161115294883
- MurdochCMuthanaMCoffeltSBLewisCEThe role of myeloid cells in the promotion of tumour angiogenesisNat Rev Cancer20088861863118633355
- KeaneMPStrieterRMThe role of CXC chemokines in the regulation of angiogenesisChem Immunol1999728610110550932
- TorisuHOnoMKiryuHMacrophage infiltration correlates with tumor stage and angiogenesis in human malignant melanoma: possible involvement of TNFalpha and IL-1alphaInt J Cancer200085218218810629075
- PeinadoHLavotshkinSLydenDThe secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughtsSemin Cancer Biol201121213914621251983
- QianBZPollardJWMacrophage diversity enhances tumor progression and metastasisCell20101411395120371344
- Vidal-VanaclochaFFantuzziGMendozaLIL-18 regulates IL-1beta-dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1Proc Natl Acad Sci U S A200097273473910639148
- KalluriRWeinbergRAThe basics of epithelial-mesenchymal transitionJ Clin Invest200911961420142819487818
- WuYZhouBPTNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasionBr J Cancer2010102463964420087353
- SullivanNJSasserAKAxelAEInterleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cellsOncogene200928332940294719581928
- WangHWangHSZhouBHEpithelial-mesenchymal transition (EMT) induced by TNF-α requires AKT/GSK-3β-mediated stabilization of snail in colorectal cancerPLoS One201382e5666423431386
- WuYDengJRychahouPGQiuSEversBMZhouBPStabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasionCancer Cell200915541642819411070
- FuxeJKarlssonMCTGF-β-induced epithelial-mesenchymal transition: a link between cancer and inflammationSemin Cancer Biol2012225–645546122627188
- ElderDJHaltonDEHagueAParaskevaCInduction of apoptotic cell death in human colorectal carcinoma cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug: independence from COX-2 protein expressionClin Cancer Res1997310167916839815550
- García-RodríguezLAHuerta-AlvarezCReduced risk of colorectal cancer among long-term users of aspirin and nonaspirin nonsteroidal antiinflammatory drugsEpidemiology2001121889311138826
- GuptaRADuboisRNColorectal cancer prevention and treatment by inhibition of cyclooxygenase-2Nat Rev Cancer200111112111900248
- YinMJYamamotoYGaynorRBThe anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-betaNature1998396670677809817203
- RoelofsHMTe MorscheRHvan HeumenBWNagengastFMPetersWHOver-expression of COX-2 mRNA in colorectal cancerBMC Gastroenterol201414124383454
- GurpinarEGrizzleWEPiazzaGACOX-independent mechanisms of cancer chemoprevention by anti-inflammatory drugsFront Oncol2013318123875171
- GurpinarEGrizzleWEPiazzaGANSAIDs inhibit tumorigenesis, but how?Clin Cancer Res20142051104111324311630
- QuailDFJoyceJAMicroenvironmental regulation of tumor progression and metastasisNat Med201319111423143724202395
- SmithHAKangYThe metastasis-promoting roles of tumor-associated immune cellsJ Mol Med (Berl)201391441142923515621
- LeekRDLandersRJHarrisALLewisCENecrosis correlates with high vascular density and focal macrophage infiltration in invasive carcinoma of the breastBr J Cancer1999795–699199510070902
- GabrilovichDIChenHLGirgisKRProduction of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cellsNat Med1996210109611038837607
- GeissmannFRevyPRegnaultATGF-beta 1 prevents the non-cognate maturation of human dendritic Langerhans cellsJ Immunol199916284567457510201996
- HuangBLeiZZhaoJCCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancersCancer Lett20072521869217257744
- GabrilovichDINagarajSMyeloid-derived suppressor cells as regulators of the immune systemNat Rev Immunol20099316217419197294
- LindauDGielenPKroesenMWesselingPAdemaGJThe immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cellsImmunology2013138210511523216602
- ZhaiRChenFLiuGInteractions among genetic variants in apoptosis pathway genes, reflux symptoms, body mass index, and smoking indicate two distinct etiologic patterns of esophageal adenocarcinomaJ Clin Oncol201028142445245120385987
- QuanteMBhagatGAbramsJABile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasiaCancer Cell2012211365122264787
- SarosiGBrownGJaiswalKBone marrow progenitor cells contribute to esophageal regeneration and metaplasia in a rat model of Barrett’s esophagusDis Esophagus2008211435018197938
- WangXOuyangHYamamotoYResidual embryonic cells as precursors of a Barrett’s-like metaplasiaCell201114571023103521703447
- FitzgeraldRCAbdallaSOnwuegbusiBAInflammatory gradient in Barrett’s oesophagus: implications for disease complicationsGut200251331632212171950
- Van der VeenAHDeesJBlankensteijnJDVan BlankensteinMAdenocarcinoma in Barrett’s oesophagus: an overrated riskGut198930114182920919
- de JongePJvan BlankensteinMLoomanCWCasparieMKMeijerGAKuipersEJRisk of malignant progression in patients with Barrett’s oesophagus: a Dutch nationwide cohort studyGut20105981030103620639249
- Hvid-JensenFPedersenLDrewesAMSørensenHTFunch-JensenPIncidence of adenocarcinoma among patients with Barrett’s esophagusN Engl J Med2011365151375138321995385
- StonerGDGuptaAEtiology and chemoprevention of esophageal squamous cell carcinomaCarcinogenesis200122111737174611698334
- SunLYuSMeta-analysis: non-steroidal anti-inflammatory drug use and the risk of esophageal squamous cell carcinomaDis Esophagus201124854454921539676
- ZhangYEpidemiology of esophageal cancerWorld J Gastroenterol201319345598560624039351
- KimSSRuizVECarrollJDMossSFHelicobacter pylori in the pathogenesis of gastric cancer and gastric lymphomaCancer Lett2011305222823820692762
- NaginiSCarcinoma of the stomach: a review of epidemiology, pathogenesis, molecular genetics and chemopreventionWorld J Gastrointest Oncol20124715616922844547
- LeeKEKhoiPNXiaYHelicobacter pylori and interleukin-8 in gastric cancerWorld J Gastroenterol201319458192820224363509
- HatakeyamaMOncogenic mechanisms of the Helicobacter pylori CagA proteinNat Rev Cancer20044968869415343275
- XiongHDuWSunTTA positive feedback loop between STAT3 and cyclooxygenase-2 gene may contribute to Helicobacter pylori-associated human gastric tumorigenesisInt J Cancer201413492030204024127267
- SobalaGMO’ConnorHJDewarEPKingRFAxonATDixonMFBile reflux and intestinal metaplasia in gastric mucosaJ Clin Pathol19934632352408463417
- NguyenTLKhuranaSSBelloneCJAutoimmune gastritis mediated by CD4+ T cells promotes the development of gastric cancerCancer Res20137372117212623378345
- OshimaHOshimaMThe role of PGE2-associated inflammatory responses in gastric cancer developmentSemin Immunopathol201335213915023053397
- OshimaHIshikawaTYoshidaGJTNF-α/TNFR1 signaling promotes gastric tumorigenesis through induction of Noxo1 and Gna14 in tumor cellsOncogene Epub8262013
- KinoshitaHHirataYNakagawaHInterleukin-6 mediates epithelial-stromal interactions and promotes gastric tumorigenesisPLoS One201384e6091423593346
- KongDPiaoYSYamashitaSInflammation-induced repression of tumor suppressor miR-7 in gastric tumor cellsOncogene201231353949396022139078
- LutgensMWvan OijenMGvan der HeijdenGJVleggaarFPSiersemaPDOldenburgBDeclining risk of colorectal cancer in inflammatory bowel disease: an updated meta-analysis of population-based cohort studiesInflammatory Bowel Diseases201319478979923448792
- Peyrin-BirouletLLepageCJoosteVGuéantJLFaivreJBouvierAMColorectal cancer in inflammatory bowel diseases: a population-based study (1976-2008)Inflamm Bowel Dis201218122247225122467511
- HerszenyiLMihellerPTulassayZCarcinogenesis in inflammatory bowel diseaseDig Dis200725326726917827953
- DysonJKRutterMDColorectal cancer in inflammatory bowel disease: what is the real magnitude of the risk?World J Gastroenterol201218293839384822876036
- RoglerGChronic ulcerative colitis and colorectal cancerCancer Lett2014345223524123941831
- KhanAACashPE. coli and colon cancer: is mutY a culprit?Cancer Lett2013341212713123933175
- FüriISiposFGermannTMEpithelial toll-like receptor 9 signaling in colorectal inflammation and cancer: clinico-pathogenic aspectsWorld J Gastroenterol201319264119412623864774
- MüzesGMolnárBSiposFRegulatory T cells in inflammatory bowel diseases and colorectal cancerWorld J Gastroenterol201218405688569423155308
- WhitcombDCInflammation and cancer V. Chronic pancreatitis and pancreatic cancerAm J Physiol Gastrointest Liver Physiol20042872G315G31915246966
- WörmannSMDiakopoulosKNLesinaMAlgülHThe immune network in pancreatic cancer development and progressionOncogene201433232956296723851493
- HertzerKMDonaldGWHinesOJCXCR2: a target for pancreatic cancer treatment?Expert Opin Ther Targets201317666768023425074
- KleeffJKusamaTRossiDLDetection and localization of Mip-3alpha/LARC/Exodus, a macrophage proinflammatory chemokine, and its CCR6 receptor in human pancreatic cancerInt J Cancer199981465065710225458
- BarberMDPowellJJLynchSFFearonKCRossJAA polymorphism of the interleukin-1 beta gene influences survival in pancreatic cancerBr J Cancer200083111443144711076651
- WörmannSMAlgülHRisk factors and therapeutic targets in pancreatic cancerFront Oncol2013328224303367
- KolodecikTShugrueCAshatMThrowerECRisk factors for pancreatic cancer: underlying mechanisms and potential targetsFront Physiol2013441524474939
- FarrowBEversBMInflammation and the development of pancreatic cancerSurg Oncol200210415316912020670
- RiveraJARallCJGraeme-CookFAnalysis of K-ras oncogene mutations in chronic pancreatitis with ductal hyperplasiaSurgery1997121142499001550
- HidalgoMPancreatic cancerN Engl J Med2010362171605161720427809
- AncrileBLimKHCounterCMOncogenic Ras-induced secretion of IL6 is required for tumorigenesisGenes Dev200721141714171917639077
- BayneLJBeattyGLJhalaNTumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancerCancer Cell201221682283522698406
- Pylayeva-GuptaYLeeKEHajduCHMillerGBar-SagiDOncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasiaCancer Cell201221683684722698407
- BeattyGLChioreanEGFishmanMPCD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humansScience201133160241612161621436454
- LesinaMKurkowskiMULudesKStat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancerCancer Cell201119445646921481788
- MichaudDSRole of bacterial infections in pancreatic cancerCarcinogenesis201334102193219723843038
- DavilaJAMorganROShaibYMcGlynnKAEl-SeragHBHepatitis C infection and the increasing incidence of hepatocellular carcinoma: a population-based studyGastroenterology200412751372138015521006
- AyubAAshfaqUAHaqueAHBV induced HCC: major risk factors from genetic to molecular levelBiomed Res Int2013201381046123991421
- BudhuAWangXWThe role of cytokines in hepatocellular carcinomaJ Leukoc Biol20068061197121316946019
- Kim doYHanKHEpidemiology and surveillance of hepatocellular carcinomaLiver Cancer20121121424159567
- MatsuzakiKMurataMYoshidaKChronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinomaHepatology2007461485717596875
- LokASDoes antiviral therapy for hepatitis B and C prevent hepatocellular carcinoma?J Gastroenterol Hepatol201126222122721070361
- AravalliRNCressmanENSteerCJCellular and molecular mechanisms of hepatocellular carcinoma: an updateArch Toxicol201387222724723007558
- SeveriTvan MalensteinHVerslypeCvan PeltJFTumor initiation and progression in hepatocellular carcinoma: risk factors, classification, and therapeutic targetsActa Pharmacol Sin201031111409142020953207
- RamakrishnaGRastogiATrehanpatiNSenBKhoslaRSarinSKFrom cirrhosis to hepatocellular carcinoma: new molecular insights on inflammation and cellular senescenceLiver Cancer201323–436738324400224
- WhiteDLKanwalFEl-SeragHBAssociation between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic reviewClin Gastroenterol Hepatol2012101213421359. e223041539
- TackeFYoneyamaHFrom NAFLD to NASH to fibrosis to HCC: role of dendritic cell populations in the liverHepatology201358249449623519833
- LiangYYangZZhongRPrimary biliary cirrhosis and cancer risk: a systematic review and meta-analysisHepatology20125641409141722504852
- AravalliRNRole of innate immunity in the development of hepatocellular carcinomaWorld J Gastroenterol201319437500751424282342
- YangJDNakamuraIRobertsLRThe tumor microenvironment in hepatocellular carcinoma: current status and therapeutic targetsSemin Cancer Biol2011211354320946957
- PinlaorSMaNHirakuYRepeated infection with Opisthorchis viverrini induces accumulation of 8-nitroguanine and 8-oxo-7,8-dihydro-2′-deoxyguanine in the bile duct of hamsters via inducible nitric oxide synthaseCarcinogenesis20042581535154215059927
- ShinHROhJKMasuyerEEpidemiology of cholangiocarcinoma: an update focusing on risk factorsCancer Sci2010101357958520085587
- ClaessenMMHVleggaarFPTytgatKMSiersemaPDvan BuurenHRHigh lifetime risk of cancer in primary sclerosing cholangitisJ Hepatol200950115816419012991
- EhlkenHSchrammCPrimary sclerosing cholangitis and cholangiocarcinoma: pathogenesis and modes of diagnosticsDig Dis201331111812523797133
- Al-BahraniRAbuetabhYZeitouniNSergiCCholangiocarcinoma: risk factors, environmental influences and oncogenesisAnn Clin Lab Sci201343219521023694797
- ChangJSTsaiCRChenLTMedical risk factors associated with cholangiocarcinoma in taiwan: a population-based case-control studyPLoS One201387e6998123894567
- KhanSAThomasHCDavidsonBRTaylor-RobinsonSDCholangiocarcinomaLancet200536694931303131416214602
- NairSSBommanaAPakalaSBInflammatory response to liver fluke Opisthorchis viverrini depends on host master coregulator MTA1, a marker for parasite-induced cholangiocarcinoma in humansHepatology20115441388139721725997
- BerthiaumeEPWandsJThe molecular pathogenesis of cholangiocarcinomaSemin Liver Dis200424212713715192786
- ParkJTadlockLGoresGJPatelTInhibition of interleukin 6- mediated mitogen-activated protein kinase activation attenuates growth of a cholangiocarcinoma cell lineHepatology19993051128113310534331
- IsomotoHKobayashiSWerneburgNWInterleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cellsHepatology20054261329133816317687
- KumarSKumarSKumarSInfection as a risk factor for gallbladder cancerJ Surg Oncol200693863363916724347
- WitkowskaMSmolewskiPHelicobacter pylori infection, chronic inflammation, and genomic transformations in gastric MALT lymphomaMediators Inflamm2013201352317023606792
- LibraMGloghiniAMalaponteGAssociation of t(14;18) translocation with HCV infection in gastrointestinal MALT lymphomasJ Hepatol200849217017418538438
- LecuitMAbachinEMartinAImmunoproliferative small intestinal disease associated with Campylobacter jejuniN Engl J Med2004350323924814724303
- ThieblemontCBertoniFCopie-BergmanCFerreriAJPonzoniMChronic inflammation and extra-nodal marginal-zone lymphomas of MALT-typeSemin Cancer Biol201424334224333758
- WangHPZhuYLShaoWRole of Helicobacter pylori virulence factor cytotoxin-associated gene A in gastric mucosa-associated lymphoid tissue lymphomaWorld J Gastroenterol201319458219822624363512
- ZulloAHassanCCristofariFEffects of Helicobacter pylori eradication on early stage gastric mucosa-associated lymphoid tissue lymphomaClin Gastroenterol Hepatol 2010201082105110
- LichtensteinGRRutgeertsPImportance of mucosal healing in ulcerative colitisInflamm Bowel Dis201016233834619637362
- NguyenGCGulamhuseinABernsteinCN5-aminosalicylic acid is not protective against colorectal cancer in inflammatory bowel disease: a meta-analysis of non-referral populationsAm J Gastroenterol2012107912981304 quiz 1297, 130522751467
- SahinIHHassanMMGarrettCRImpact of non-steroidal anti-inflammatory drugs on gastrointestinal cancers: current state-of-the scienceCancer Lett2014345224925724021750
- YaoXHuangJZhongHTargeting interleukin-6 in inflammatory autoimmune diseases and cancersPharmacol Ther2014141212513924076269
- RudermanEMOverview of safety of non-biologic and biologic DMARDsRheumatology (Oxford)201251Suppl 6vi37vi4323221586
- Rose-JohnSWaetzigGHSchellerJGrötzingerJSeegertDThe IL-6/sIL-6R complex as a novel target for therapeutic approachesExpert Opin Ther Targets200711561362417465721
- GalonJCostesASanchez-CaboFType, density, and location of immune cells within human colorectal tumors predict clinical outcomeScience200631357951960196417008531
- AngellHGalonJFrom the immune contexture to the Immunoscore: the role of prognostic and predictive immune markers in cancerCurr Opin Immunol201325226126723579076