
Abstract
Background
Myocardial infarction (MI) is a common cause of death worldwide. It is characterized by coronary artery occlusion that causes ischemia and hypoxia of myocardial cells, leading to irreversible myocardial damage.
Materials and Methods
To explore potential targets for treatment of MI, we reorganized and analyzed two microarray datasets (GSE4648 and GSE775). The GEO2R tool was used to screen for differentially expressed genes (DEGs) between infarcted and normal myocardium. We used the Database for Annotation, Visualization and Integrated Discovery (DAVID) to perform Gene Ontology functional annotation analysis (GO analysis) and the Kyoto Encyclopedia of Genes and Genomes for pathway enrichment analysis (KEGG analysis). We examined protein–protein interactions to characterize the relationship between differentially expressed genes, and we screened potential hub genes according to the degree of connection. PCR and Western blotting were used to identify the core genes.
Results
At different times of infarction, a total of 35 genes showed upregulation at all times; however, none of the genes showed downregulation at all 3 times. Similarly, 10 hub genes with high degrees of connectivity were identified. In vivo and in vitro experiments suggested that expression levels of MMP-9 increased at various times after myocardial infarction and that expression increased in a variety of cells simultaneously.
Conclusion
Expression levels of MMP-9 increase throughout the course of acute myocardial infarction, and this expression has both positive and negative sides. Further studies are needed to explore the role of MMP-9 in MI treatment. The potential values of Il6, Spp1, Ptgs2, Serpine1, Plaur, Cxcl5, Lgals3, Serpinb2, and Cd14 are also worth exploring.
Introduction
Ischemic heart disease is the main cause of death all over the world.Citation1,Citation2 Acute myocardial infarction (AMI) can be thought of as a severe, even fatal type of ischemic cardiomyopathy.Citation3,Citation4 Coronary artery acute occlusion leads to myocardial ischemia, hypoxia, and impaired energy supply,Citation5 leading acute imbalance of hemodynamics and cardiomyocyte irreversible damage.Citation6 The current treatments of acute myocardial infarction include thrombolysis and surgery. However, recanalization of the blood vessels causes reperfusion injury, which aggravates myocardial damage and microcirculation disturbances, further damaging cardiac function.Citation7,Citation8 Myocardiocytes are non-renewable cells,Citation9 and remaining viable myocardium is insufficient to fully compensate for cardiac function. There is currently no treatment that can solve this problem completely. Therefore, it is necessary to find new and better ways to augment current treatment.
In the present study, we explored changes in acute myocardial infarction in an animal model and attempted to identify possible therapeutic targets. In order to identify differentially expressed genes (DEGs) between AMI and sham groups in myocardial tissue originated from mice, we use bioinformatics methods to analyze the gene expression profiling data, downloaded from the Gene Expression Omnibus (GEO) database. Functional enrichment analysis was performed of the screened DEGs using Gene Ontology (GO) functional annotation and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway. Protein–protein interaction network analyses of the DEGs were also performed to help us explore the potential hub genes and the underlying molecular mechanisms of AMI.
Materials and Methods
Study Design
Gene expression data were obtained from GEO database, and further screening was carried out according to the conditions of a myocardial infarction in vivo model. We used the GEO2R online analysis tool to screen out the differentially expressed genes between the infarct and the sham groups at various time points, and compare the differentially expressed genes at multiple time points. Using Venn diagrams, we identified genes that were all upregulated or all downregulated at various time points. Then, we perform GO and KEGG pathway analysis and protein–protein interaction network analysis for this segment of DEGs. By evaluating the degree of each protein node in the protein–protein interaction network, the 10 most important core genes were identified. From this core gene screen, meaningful genes were verified using in vivo animal models and several in vitro cell models.
Data Sources
The gene expression datasets used in our research were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/), which were created by NCBI, containing high-throughput gene expression data from research institutes worldwide. After careful comparison, two microarray datasets (GSE775 and GSE4648) were downloaded from GEO. GSE775 and GSE4648 were based on platform GPL 81 ([MG_U74Av2] Affymetrix Murine Genome U74A Version 2 Array). We further screened according to animal modeling conditions and obtained results (). All data in this study are available free of charge online.
Table 1 Statistics and Further Screened of the Two Microarray Databases Derived from the GEO Database
Data Processing of DEGs
The GEO2R online analysis tool (https://www.ncbi.nlm.nih.gov/geo/geo2r/) was used to identify the DEGs between AMI and sham operation samples from mice. We defined the adjusted P-value < 0.05 and |log FC| ≥ 2.0 as the cutoff criteria in advance. Genes that met this criteria were considered DEGs and genes that did not meet the criteria were temporarily discarded. Statistical analysis was carried out for each dataset, and the intersections were identified using the Venn diagram web tool (http://bioinformatics.psb.ugent.be/webtools/Venn/) to screen out genes that showed the same changes at three different time points.
GO and KEGG Pathway Analysis of DEGs
GO analysis is a more common method for large-scale gene function enrichment research. Gene function can be divided into the following three categories: BP (biological process), MF (molecular function), and CC (cellular component). KEGG is a useful database that stores large amounts of data on genomics, biological pathways, signaling pathways, diseases, drugs, and chemicals. We used Database for Annotation, Visualization and Integrated Discovery (DAVID) tools (https://david.ncifcrf.gov/) to explore GO annotation analysis and KEGG pathway enrichment analysis of DEGs. P < 0.05 and counts ≥ 2.0 as the cutoff criteria were considered to be statistically significant.
Protein–Protein Interaction Network Construction
The Search Tool for the Retrieval of Interacting Genes (STRING) database (http://string-db.org/) is designed to accomplish the protein–protein interaction network analyses. To explore possible protein–protein interaction network interactions, the DEGs screened previously were mapped to the STRING database. The protein–protein interaction pairs were extracted with a minimum required interaction score > 0.4.
Potential Hub Gene Identification
We used Cytoscape software (www.cytoscape.org/) and a plugin called CytoHubba to evaluate the degree of each protein node. Nodes with higher connectivity degree are often more important in maintaining the stability of the entire network, and nodes with lower connectivity degree are relatively unimportant. In our research, the 10 most connected protein nodes were considered to be our potential hub genes.
Animals and Myocardial Infarction Model
All related operations involving animals followed the “Guidelines for Care and Use of Laboratory Animals” (NIH Publication No. 85–23, revised 1996) published by the US National Institutes of Health (Bethesda, MD, USA). Procedures were approved by the Animal Care and Use Committee at the Wenzhou Medical University. All male C57/BL6 mice (6–7 weeks old and 20–25 g in weight) were purchased from the SLAC Laboratory Animal Centre of Shanghai (Shanghai, China). The myocardial infarction model (MI) was implemented according to our previous study.Citation10 In brief, the mice were first anesthetized with isoflurane and were intubated. Then, a thoracotomy was performed to expose the heart at the third or fourth rib. The left anterior descending coronary artery was ligated using 7–0 silk thread to induce myocardial ischemia. Mice in the sham group underwent the same operation except that the arteries were not ligated.
Cell Culture and Glucose and Oxygen Deprivation Model (OGD)
Mouse cardiomyocytes and fibroblasts were extracted from newborn C57/B6J mice. The method was previously described by our group.Citation11 The macrophages were RAW264.7. Mouse coronary endothelial cells were purchased from ATCC. Neutrophils were obtained from peripheral blood according to manufacturer’s instructions (Solarbio; Beijing, China, P9201). Cells were maintained in a hypoxic incubator (N2 94%, O2 1% and CO2 5%) for 12 hours in medium deprived of serum and glucose.
Real-Time Quantitative PCR
Total RNA was extracted from the treated cells using TRIzol reagent (Ambion) and was further purified. A one-step reverse transcription amplification was performed using a cDNA reverse transcription kit (TaKaRa), and the corresponding primers (ShengGong, Shanghai, China) were used for quantitative analysis.
Western Blot Analysis
Myocardial tissue/cell total protein preparation and concentration determination kits were from Beyotime (Shanghai, China). Proteins of different molecular weights were separated on SDS-PAGE gels, transferred to 0.22-μm PVDF membranes, blocked with skimmed milk powder, and then incubated with primary antibodies as follows: MMP-9 (Abcam), GAPDH (CST) at 4°C overnight. After incubating with the corresponding secondary antibody the next day, protein bands were scanned by a ChemiDoc MP device (Bio-Rad, Hercules, CA, USA). Finally, ImageJ software (NIH, USA) was used for analysis.
Statistical Analysis
Data were presented as mean ± SD and were analyzed using SPSS version 21.0 (IBM, Armonk, NY, USA). All data we used were normally distributed. A two-tailed unpaired Student’s t-test was conducted to compare two experimental groups. One way analysis of variance (ANOVA) followed by Duncan’s T3 multiple-range test was used for comparisons of more than two groups. P-values less than 0.05 were considered statistically significant.
Results
Identification of DEGs
Two gene expression profiles (GSE4648 and GSE775) were selected (). GSE4648 included two AMI samples and two normal samples each at 1 and 2 days after myocardial infarction, and GSE775 at 2 days and 7 days contained three AMI specimens and one normal specimen, respectively. Then, we sorted again according to the infarct time ().
Table 2 Reclassifying Two Gene Expression Profiles Based on Infarct Time
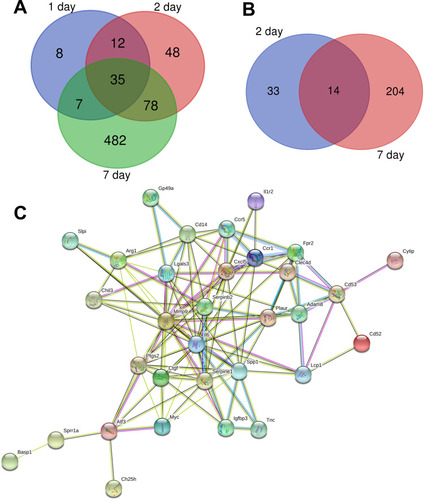
According to the standard of P < 0.05 and |log FC| ≥ 2.0, a total of 62 DEGs were identified from 1 day, including 62 upregulated genes and 0 downregulated gene. In another 2-day infarct group, 220 DEGs were identified, 173 genes were upregulated and 47 genes were downregulated. In the 7-day infarction group, there were 820 DEGs, containing 602 upregulated genes and 218 downregulated genes (). All DEGs were compared between AMI samples and normal samples. The differentially expressed genes that intersected with one another were then obtained using Venn analysis (). Finally, we obtained 35 upregulated DEGs with significantly differential expression in all three infarct groups; however, there were no differential genes that were downregulated.
Table 3 Number of Differential Genes in Different Infarct Groups
Figure 1 Venn diagram of DEGs and protein–protein interaction network. (A) Upregulated genes of DEGs common to all three infarct groups. (B) Downregulated genes of DEGs common to all three infarct groups. (C) Protein–protein interaction network constituted with the DEGs.
Functional Enrichment Analyses of DEGs
We used DAVID to work on our DEGs to perform GO annotation analysis and KEGG pathway enrichment analysis ().
Table 4 GO and KEGG Pathway Enrichment Analysis of DEGs
The GO analysis results were composed of three parts: biological process, cell component, and molecular function. The biological process (BP) showed significantly altered inflammatory responses, positive regulation of gene expression, positive regulation of cell proliferation, negative regulation of apoptotic process, regulation of cell proliferation, immune response, wound healing, chemotaxis, response to lipopolysaccharide, cellular response to lipopolysaccharide, angiogenesis and negative regulation of gene expression. The DEGs were enriched in cell component (CC), including extracellular region, extracellular space, extracellular exosome, extracellular matrix, and external side of plasma membrane. For the molecular function (MF), the DEGs were enriched in fibronectin binding and peptidase inhibitor activity. Otherwise, the enriched KEGG pathway included transcriptional misregulation in cancer, amoebiasis and cytokine–cytokine receptor interaction.
Protein–Protein Interaction Network Construction and Hub Gene Identification
The protein–protein interactions of the DEGs were predicted using STRING tools. A total of 32 nodes and 117 edges were related in the protein–protein interaction network (). The top 10 core genes were selected using connectivity degrees in the protein–protein interaction network. Their names and functions are display in . Interleukin 6 (Il6) was the most obvious gene with connectivity degree = 22; the next one was matrix metallopeptidase 9 (MMP-9; degree = 20), followed by secreted phosphoprotein 1 (Spp1; degree = 12), prostaglandin-endoperoxide synthase 2 (Ptgs2; degree = 12), serine (or cysteine) peptidase inhibitor, clade E, member 1 (Serpine1; degree = 11), plasminogen activator, urokinase receptor (Plaur; degree = 10), chemokine (C-X-C motif) ligand 5 (Cxcl5; degree = 10), lectin, galactose binding, soluble 3 (Lgals3; degree = 10), serine (or cysteine) peptidase inhibitor, clade B, member 2 (Serpinb2; degree = 9), and CD14 antigen (Cd14; degree = 9). All of the above core genes belong to the upregulation group.
Table 5 Top 10 Core Genes with Higher Degrees of Connectivity
In vivo and vitro Experimental Verification
Considering that there have been many reports related to IL-6 and myocardial infarction,Citation12 and connectivity degrees, we focused on measuring changes in MMP-9 expression levels. First, we measured changes in MMP-9 mRNA levels in myocardial infarction samples. Consistent with the microarray results, there were increases in expression levels at 1, 2, and 7 days after infarction (). Because each microarray result is a comparison between the infarct group and the sham group, we also focused on the comparison in mRNA levels between the infarct group and the sham group. However, we also saw that mRNA levels at 2 days of infarction were significantly higher than those of the previous day, and that changes in MMP-9 mRNA levels between different infarction groups were also worthy of follow-up study. We also measured changes in individual cells, including cardiomyocytes, fibroblasts, endothelial cells, macrophages and neutrophils. We found that there were different degrees of elevation in different cells (). Among them, the changes in macrophages and neutrophils were the most obvious, which preliminarily suggests that these two kinds of cells play a substantial role in the increase of MMP-9 expression during myocardial infarction.
Figure 2 mRNA and protein expression levels in vivo and in vitro models. (A) The mRNA expression of MMP-9 within the myocardial infarction tissue, (n=3). (B–F) The mRNA expression of MMP-9 within the cardiomyocytes, fibroblasts, endothelial cells, macrophage and neutrophils, (n=3). (G, H) The protein expression of MMP-9 within the myocardial infarction tissue, (n=4). (I, J) The protein expression of MMP-9 within the cardiomyocytes, (n=3). (K, L) The protein expression of MMP-9 within the fibroblasts, (n=3). (M, N) The protein expression of MMP-9 within the endothelial cells, (n=3). (O, P) The protein expression of MMP-9 within the macrophage, (n=3). Data are means ± SDs, *P < 0.05; **P < 0.01.
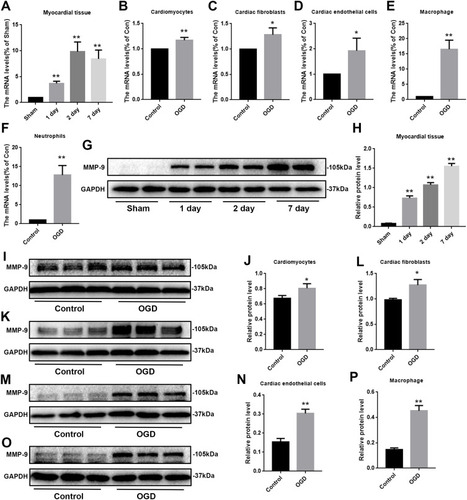
We also needed to detect changes in protein levels. Looking at the in vivo model first, we found that expression levels of MMP-9 increased sequentially over time (), which was not consistent with the results of mRNA measurements. The mRNA results showed that the 7th day was slightly lower than the 2nd day; however, protein levels on the 7th day higher than those of the 2nd day. We speculated that the change in MMP-9 mRNA expression may occur earlier than the protein level changes. We monitored expression levels of MMP-9 in in vitro models, including cardiomyocytes, fibroblasts, endothelial cells, and macrophages, and found that MMP-9 expression all increased after 12 hours of OGD treatment (). The difference was that the increase in MMP-9 was not consistent in various cell types.
Discussion
Although some progress has been made in terms of revascularization, surgical treatment, and drug treatment, cardiovascular diseases, especially acute myocardial infarction (AMI), remain the leading cause of death in developed countries.Citation1,Citation2 Cardiomyocytes are non-renewable cells,Citation9 that will cause under-compensation for residual myocardium in the context of AMI. To better study the pathophysiological changes after the onset of acute myocardial infarction, the greatest limitation is that it is difficult to obtain myocardial tissue samples after the event.Citation13,Citation14 Therefore, in the present study, we screened microarray datasets after simulated acute myocardial infarction in C57/BL6 mice. To better simulate acute myocardial infarction, we set the infarct time between 1 and 7 days.Citation15 The expectation was that we would identify new targets for the treatment of acute myocardial infarction or that we would identify pathways to advance relevant research.
In the present study, differentially expressed genes, protein–protein interactions, and biological functional enrichment were used based on publicly available databases to identify potential core genes associated with acute myocardial infarction. DEGs between AMI and healthy heart were screened out based on the GEO database. We identified 35 upregulated DEGs. These DEGs were associated with GO terms such as extracellular region, extracellular space, extracellular exosome and inflammatory response, and were significantly enriched in the KEGG terms transcriptional misregulation in cancer, amoebiasis and cytokine–cytokine receptor interaction. A protein–protein interaction network was built to investigate the interrelationship of the DEGs, and 10 core genes were identified, including Il6, MMP-9, Spp1, Ptgs2, Serpine1, Plaur, Cxcl5, Lgals3, Serpinb2, and Cd14. All the core genes were in the upregulation group.
Our GO analysis found that the main differences were concentrated in the cell component (CC) part, especially extracellular. Among the core genes we screened from the protein–protein interaction network, IL6 and MMP-9 were the most significant. IL6 is often used as an evaluative indicator,Citation16 and there have been many studies of it related to MI;Citation12 therefore, here, we discussed changes in MMP-9 expression.
MMP-9, also called matrix metalloproteinase 9, is encoded by the MMP-9 gene and is part of the zinc-dependent endopeptidase family.Citation17 MMP-9 is secreted by a variety of cells, including endogenous cardiomyocytes, fibroblasts, and endothelial cells, as well as leukocytes that temporarily infiltrate by infarcted heart muscle.Citation18–Citation20 Studies have reported that, in human and animal models, infiltrating neutrophils are the main source of MMP-9 after myocardial infarction.Citation21–Citation24 Of course, macrophages are also a major source of MMP-9,Citation25,Citation26 and all this is in line with our experimental results. Authors believe that MMP-9 released by neutrophils is highest on the first day of acute myocardial infarction, and it is released on day 5 by macrophages.Citation22,Citation27 However, although our results also show that neutrophils and macrophages secrete MMP-9 much higher than other cells, our in vitro model adopted a single oxygen deprivation condition that could not fully simulate various time points after myocardial infarction. It could only show that the expression of MMP-9 in the in vivo model continued to increase over time, and was accomplished by a variety of cells. We could no measure specific changes in certain cells over time, and this was a limitation. In addition, our verification results displayed an unusual finding: We found that protein levels of MMP-9 were higher on day 7 than on day 2; however, mRNA levels of MMP-9 on day 7 appeared to be slightly lower than those of day 2. We believe this is reasonable because the translation of mRNA into protein requires some time, which means that it takes a while to complete from the change in mRNA to the subsequent change at the protein level. We took samples of myocardial infarction tissue at the same times; nevertheless, there was a period of time between 2 and 7 days after myocardial infarction. During this time, the mRNA may have increased to a peak and began to decline, and the protein change may have just begun or even had not started to change.
MMP-9 has many roles in myocardial infarction. First, MMP-9 was involved in the degradation of extracellular matrix (ECM),Citation28 and the degradation of ECM increased leukocyte infiltration at the expense of increased microvascular permeability.Citation22,Citation29 MMP-9 also regulated left ventricular remodeling after myocardial infarction.Citation30,Citation31 Attenuating MMP-9 expression attenuated collagen degradation and reduce left ventricular rupture in mice.Citation32 It activates or inactivates chemokines and cytokines to exercise completely different functions.Citation30 Angiogenesis is an important compensatory factor after myocardial infarction that is also closely related to MMP-9. MMP-9 clears the ECM around the endothelial cells after myocardial infarction, thereby promoting proliferation and migration.Citation33 MMP-9 also enhances the interactions between VEGF and its receptor, opening the switch of angiogenesis.Citation33,Citation34 MMP-9 is directly required for angiogenesis and vascular remodeling. Indirectly, MMP-9 cleaves type 4 collagen in the basement membrane to induce endothelial growth and migration.Citation33,Citation35 In this way, MMP-9 is helpful for angiogenesis. Paradoxically, however, studies have reported that the loss of MMP-9 promoted the formation of blood vessels.Citation27,Citation36 It is worthwhile speculating whether MMP-9 promotes or inhibits the formation of blood vessels in different ways. We speculate that MMP-9 promotes blood vessel formation, at least for certain periods of time. Nevertheless, there are stages in which MMP-9 inhibits angiogenesis or regulatory functions. Regarding the lack of MMP-9 promoting angiogenesis, it can be suggested that MMP-9 plays a more important role in mediating damage of the entire infarct. We speculate that this occurs at some specific times or for some specific cells. The protection provided by MMP-9 is indispensable. Similarly, the ability of MMP-9 to degrade some proteins can help clear necrotic cells after ischemia, thereby reducing secondary damage.Citation37
Taken together, the data suggest that MMP-9 has both a good side and a bad side after myocardial infarction, especially with respect to angiogenesis. There may be conflicts in other areas; however, we have not mentioned them here and further studies are needed to explore them. Because MMP-9 has so many effects in myocardial infarction, it is clear that targeted regulation of MMP-9 can improve myocardial ischemic injury to some extent. Unfortunately, the only currently approved MMP inhibitor is doxycycline, and unfortunately, this is an antibacterial drug, although studies have reported that it can weaken the damage of myocardial infarction;Citation38,Citation39 nevertheless, we also need to consider what side effects can be caused by direct application. Fortunately, specific inhibitors of MMP-9 have been tested in some disease models; these inhibitors include SB-3CT,Citation40 JNJ0966,Citation41 and salvianolic acid A.Citation42
However, as mentioned above, MMP-9 is produced by different cells at various times after the occurrence of myocardial infarction, or multiple cells are produced at the same time that also produce the same or opposite effects through different signaling pathways. Therefore, MMP-9 inhibitors cannot be simply used, and it is necessary to consider different periods and duration of action. It is compelling to consider the use of MMP-9 inhibitors to inhibit all the functions of MMP-9 functions while using some means to maintain the beneficial properties of MMP-9 regulation.
In addition to MMP-9, we identified nine other core genes associated with acute myocardial infarction, including Il6, Spp1, Ptgs2, Serpine1, Plaur, Cxcl5, Lgals3, Serpinb2, and Cd14. Many of these genes are also closely related to myocardial infarction. Il6 is a well-known inflammation-related gene, and many studies regard it as an indicator of inflammation.Citation43,Citation44 We did not discuss it in depth. Ptgs2, also called COX-2, promotes cell proliferation in tumors, accelerates angiogenesis,Citation45 and reduces inflammation in myocardial infarction.Citation46 Spp1 is involved in the repair of osteopontin in myocardial infarction.Citation47 Mutations in the Serpine1 gene affect the risk of myocardial infarction.Citation48 Plaur, which codes for urokinase-plasminogen activator receptor (Upar), may affect the susceptibility to MI,Citation49 Cxcl5 affects the formation of new blood vessels in myocardial infarction.Citation50 Regarding Lgals3 and Serpinb2 in myocardial infarction, there are not many studies. Cd14 is used as a cell marker.Citation51 Its specific function is not very clear, and further research is needed. Nevertheless, these genes all have promise as potential therapeutic targets.
Conclusion
Our bioinformatics analysis was based on the gene expression dataset obtained from the GEO database and identified 10 hub genes that are differentially expressed between normal myocardium and infarcted myocardium: MMP-9, Il6, Spp1, Ptgs2, Serpine1, Plaur, Cxcl5, Lgals3, Serpinb2, and Cd14. All these genes are upregulated when myocardial infarction occurs, and are likely to be related to the progression of myocardial infarction. In AMI patients, we believe MMP-9 is likely to be an important potential target. Our research results need further research. In short, MMP-9 may play an important role in AMI.
Data Sharing Statement
The data used to support the findings of this study are available from the corresponding author upon request.
Ethics Approval
All our operations have been approved by the Animal Care and Use Committee at the wenzhou medical university.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Acknowledgments
The authors thank Institute of Cardiovascular Molecular Pharmacology, Second Affiliated Hospital of Wenzhou Medical University.
Disclosure
The authors report no conflicts of interest for this work.
References
- Lopez A, Mathers C, Ezzati M, Jamison D, Murray C. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet. 2006;367(9524):1747–1757. doi:10.1016/S0140-6736(06)68770-916731270
- Mozaffarian D, Benjamin E, Go A, et al. Heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation. 2016;133(4):e38–360.26673558
- Ibanez B, James S, Agewall S, et al. 2017 ESC guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: the task force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J. 2018;39(2):119–177.28886621
- Rao S, Kaul P, Newby L, et al. Poverty, process of care, and outcome in acute coronary syndromes. J Am Coll Cardiol. 2003;41(11):1948–1954. doi:10.1016/S0735-1097(03)00402-912798563
- Xia J, Wu H, Lai B, et al. Gene delivery of hypoxia-inducible VEGF targeting collagen effectively improves cardiac function after myocardial infarction. Sci Rep. 2017;7(1):13273. doi:10.1038/s41598-017-13547-129038511
- Rieckmann M, Delgobo M, Gaal C, et al. Myocardial infarction triggers cardioprotective antigen-specific T helper cell responses. J Clin Invest. 2019;129(11):4922–4936. doi:10.1172/JCI12385931408441
- Galiuto L, DeMaria A, Iliceto S. Microvascular damage during myocardial ischemia-reperfusion: pathophysiology, clinical implications and potential therapeutic approach evaluated by myocardial contrast echocardiography. Ital Heart J. 2000;1(2):108–116.10730610
- Eefting F, Rensing B, Wigman J, et al. Role of apoptosis in reperfusion injury. Cardiovasc Res. 2004;61(3):414–426. doi:10.1016/j.cardiores.2003.12.02314962473
- Mathur A, Martin J. Stem cells and repair of the heart. Lancet. 2004;364(9429):183–192. doi:10.1016/S0140-6736(04)16632-415246732
- Huang K, Wang J, Zhou Y, et al. Antithrombin III alleviates myocardial ischemia/reperfusion injury by inhibiting excessive autophagy in a phosphoinositide 3-Kinase/Akt-dependent manner. Front Pharmacol. 2019;10:516. doi:10.3389/fphar.2019.0051631133861
- Zeng J, Jin Q, Ruan Y, et al. Inhibition of TGFβ-activated protein kinase 1 ameliorates myocardial ischaemia/reperfusion injury via endoplasmic reticulum stress suppression. J Cell Mol Med. 2020;24(12):6846–6859. doi:10.1111/jcmm.1534032378287
- Podgoreanu M, White W, Morris R, et al. Inflammatory gene polymorphisms and risk of postoperative myocardial infarction after cardiac surgery. Circulation. 2006;114:I275–1281. doi:10.1161/CIRCULATIONAHA.105.00103216820586
- Devaux Y, Bousquenaud M, Rodius S, et al. Transforming growth factor β receptor 1 is a new candidate prognostic biomarker after acute myocardial infarction. BMC Med Genomics. 2011;4(1):83. doi:10.1186/1755-8794-4-8322136666
- Kim J, Ghasemzadeh N, Eapen D, et al. Gene expression profiles associated with acute myocardial infarction and risk of cardiovascular death. Genome Med. 2014;6(5):40. doi:10.1186/gm56024971157
- Lyu J, Wang M, Kang X, et al. Macrophage-mediated regulation of catecholamines in sympathetic neural remodeling after myocardial infarction. Basic Res Cardiol. 2020;115(5):56. doi:10.1007/s00395-020-0813-332767120
- Hunter C, Jones S. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015;16(5):448–457. doi:10.1038/ni.315325898198
- Lyngbakken M, Myhre P, Røsjø H, Omland T. Novel biomarkers of cardiovascular disease: applications in clinical practice. Crit Rev Clin Lab Sci. 2019;56(1):33–60.30457415
- Halade G, Jin Y, Lindsey M. Matrix metalloproteinase (MMP)-9: a proximal biomarker for cardiac remodeling and a distal biomarker for inflammation. Pharmacol Ther. 2013;139(1):32–40. doi:10.1016/j.pharmthera.2013.03.00923562601
- Turner N, Porter K. Regulation of myocardial matrix metalloproteinase expression and activity by cardiac fibroblasts. IUBMB Life. 2012;64(2):143–150. doi:10.1002/iub.59422215527
- Xie Z, Singh M, Singh K. Differential regulation of matrix metalloproteinase-2 and −9 expression and activity in adult rat cardiac fibroblasts in response to interleukin-1beta. J Biol Chem. 2004;279(38):39513–39519. doi:10.1074/jbc.M40584420015269222
- Ardi V, Kupriyanova T, Deryugina E, Quigley J. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2007;104(51):20262–20267. doi:10.1073/pnas.070643810418077379
- Lindsey M, Wedin K, Brown M, et al. Matrix-dependent mechanism of neutrophil-mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation. 2001;103(17):2181–2187. doi:10.1161/01.CIR.103.17.218111331260
- Romanic A, Harrison S, Bao W, et al. Myocardial protection from ischemia/reperfusion injury by targeted deletion of matrix metalloproteinase-9. Cardiovasc Res. 2002;54(3):549–558. doi:10.1016/S0008-6363(02)00254-712031700
- Kelly D, Cockerill G, Ng L, et al. Plasma matrix metalloproteinase-9 and left ventricular remodelling after acute myocardial infarction in man: a prospective cohort study. Eur Heart J. 2007;28(6):711–718. doi:10.1093/eurheartj/ehm00317339265
- Creemers E, Cleutjens J, Smits J, Daemen M. Matrix metalloproteinase inhibition after myocardial infarction: a new approach to prevent heart failure? Circ Res. 2001;89(3):201–210. doi:10.1161/hh1501.09439611485970
- Mauviel A. Cytokine regulation of metalloproteinase gene expression. J Cell Biochem. 1993;53(4):288–295. doi:10.1002/jcb.2405304048300745
- Lindsey M, Escobar G, Dobrucki L, et al. Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;290(1):H232–H239. doi:10.1152/ajpheart.00457.200516126817
- Page-McCaw A, Ewald A, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8(3):221–233. doi:10.1038/nrm212517318226
- Stefanidakis M, Ruohtula T, Borregaard N, Gahmberg C, Koivunen E. Intracellular and cell surface localization of a complex between alphaMbeta2 integrin and promatrix metalloproteinase-9 progelatinase in neutrophils. J Immunol. 2004;172(11):7060–7068. doi:10.4049/jimmunol.172.11.706015153528
- Deleon-Pennell K, Altara R, Yabluchanskiy A, Modesti A, Lindsey M. The circular relationship between matrix metalloproteinase-9 and inflammation following myocardial infarction. IUBMB Life. 2015;67(8):611–618. doi:10.1002/iub.140826269290
- Wagner D, Delagardelle C, Ernens I, Rouy D, Vaillant M, Beissel J. Matrix metalloproteinase-9 is a marker of heart failure after acute myocardial infarction. J Card Fail. 2006;12(1):66–72. doi:10.1016/j.cardfail.2005.08.00216500583
- Parajuli N, Yuan Y, Zheng X, Bedja D, Cai Z. Phosphatase PTEN is critically involved in post-myocardial infarction remodeling through the Akt/interleukin-10 signaling pathway. Basic Res Cardiol. 2012;107(2):248. doi:10.1007/s00395-012-0248-622298084
- Bendeck M. Macrophage matrix metalloproteinase-9 regulates angiogenesis in ischemic muscle. Circ Res. 2004;94(2):138–139. doi:10.1161/01.RES.0000117525.23089.1A14764648
- Xu J, Rodriguez D, Petitclerc E, et al. Proteolytic exposure of a cryptic site within collagen type IV is required for angiogenesis and tumor growth in vivo. J Cell Biol. 2001;154(5):1069–1079. doi:10.1083/jcb.20010311111535623
- Johnson C, Sung H, Lessner S, Fini M, Galis Z. Matrix metalloproteinase-9 is required for adequate angiogenic revascularization of ischemic tissues: potential role in capillary branching. Circ Res. 2004;94(2):262–268. doi:10.1161/01.RES.0000111527.42357.6214670843
- DeLeon-Pennell K, de Castro Brás L, Iyer R, et al. P. gingivalis lipopolysaccharide intensifies inflammation post-myocardial infarction through matrix metalloproteinase-9. J Mol Cell Cardiol. 2014;76:218–226. doi:10.1016/j.yjmcc.2014.09.00725240641
- Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–175.23435331
- Hori Y, Kunihiro S, Sato S, et al. Doxycycline attenuates isoproterenol-induced myocardial fibrosis and matrix metalloproteinase activity in rats. Biol Pharm Bull. 2009;32(10):1678–1682. doi:10.1248/bpb.32.167819801827
- Fana X, Zhu H, Wu X, Yan J, Xu J, Wang D. Effects of doxycycline on cx43 distribution and cardiac arrhythmia susceptibility of rats after myocardial infarction. Iran J Pharm Res. 2014;13(2):613–621.25237357
- Albrengues J, Shields M, Ng D, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science (New York, NY). 2018;361(6409):eaao4227. doi:10.1126/science.aao4227
- Herich S, Schneider-Hohendorf T, Rohlmann A, et al. Human CCR5high effector memory cells perform CNS parenchymal immune surveillance via GZMK-mediated transendothelial diapedesis. Brain. 2019;142(11):3411–3427. doi:10.1093/brain/awz30131563951
- Zhang W, Song J, Zhang X, et al. Salvianolic acid A attenuates ischemia reperfusion induced rat brain damage by protecting the blood brain barrier through MMP-9 inhibition and anti-inflammation. Chin J Nat Med. 2018;16(3):184–193. doi:10.1016/S1875-5364(18)30046-329576054
- Eghbalzadeh K, Georgi L, Louis T, et al. Compromised anti-inflammatory action of neutrophil extracellular traps in PAD4-deficient mice contributes to aggravated acute inflammation after myocardial infarction. Front Immunol. 2019;10:2313. doi:10.3389/fimmu.2019.0231331632398
- Huang P, Wang L, Li Q, et al. Atorvastatin enhances the therapeutic efficacy of mesenchymal stem cells-derived exosomes in acute myocardial infarction via up-regulating long non-coding RNA H19. Cardiovasc Res. 2020;116(2):353–367.31119268
- Gray R, Cantwell M, Coleman H, et al. Evaluation of PTGS2 expression, PIK3CA mutation, aspirin use and colon cancer survival in a Population-Based Cohort Study. Clin Transl Gastroenterol. 2017;8(4):e91. doi:10.1038/ctg.2017.1828448072
- Ge Z, Zhu X, Wang B, et al. MicroRNA-26b relieves inflammatory response and myocardial remodeling of mice with myocardial infarction by suppression of MAPK pathway through binding to PTGS2. Int J Cardiol. 2019;280:152–159. doi:10.1016/j.ijcard.2018.12.07730679074
- Shirakawa K, Endo J, Kataoka M, et al. IL (interleukin)-10-STAT3-galectin-3 axis is essential for osteopontin-producing reparative macrophage polarization after myocardial infarction. Circulation. 2018;138(18):2021–2035. doi:10.1161/CIRCULATIONAHA.118.03504729967195
- Morange P, Saut N, Alessi M, et al. Association of plasminogen activator inhibitor (PAI)-1 (SERPINE1) SNPs with myocardial infarction, plasma PAI-1, and metabolic parameters: the HIFMECH study. Arterioscler Thromb Vasc Biol. 2007;27(10):2250–2257. doi:10.1161/ATVBAHA.107.14946817656673
- Xu J, Li W, Bao X, et al. Association of putative functional variants in the PLAU gene and the PLAUR gene with myocardial infarction. Clin Sci. 2010;119(8):353–359. doi:10.1042/CS20100151
- Wang N, Liu C, Wang X, et al. Hyaluronic acid oligosaccharides improve myocardial function reconstruction and angiogenesis against myocardial infarction by regulation of macrophages. Theranostics. 2019;9(7):1980–1992. doi:10.7150/thno.3107331037151
- Gupta R, Liu L, Zhang X, et al. IL-10 provides cardioprotection in diabetic myocardial infarction via upregulation of Heme clearance pathways. JCI Insight. 2020;5(17). doi:10.1172/jci.insight.133050