
Abstract
Aducanumab is a monoclonal antibody selective for amyloid β (Aβ) aggregates. In June 2021, aducanumab became the first drug underlying the pathophysiology of Alzheimer’s disease (AD) approved by the US Food and Drug Administration (FDA), under the accelerated approval pathway. The decision was based on the ability of aducanumab to remove Aβ plaques, without any evidence that the Aβ clearance is correlated with less cognitive or functional decline. This decision has generated a considerable debate in the scientific community, especially because the results from the two Phase 3 trials, EMERGE and ENGAGE, were divergent and, even after the post hoc analysis, the data were insufficient to prove aducanumab efficacy. Moreover, some researchers think that this approval will be an obstacle to the progress and also demonstrated concerns about aducanumab cost and its safety profile. The European Medicines Agency’s rejection of aducanumab in December 2021 just brought more controversy over FDA’s decision. Now, Biogen is designing the FDA’s required confirmatory study, named ENVISION, which should be complete in 2026. Despite the controversy, the aducanumab showed to affect downstream tau pathology, which could open doors for a combination therapy approach for AD (anti-tau and anti-amyloid drug). This review summarizes the clinical development of aducanumab until regulatory agencies’ decisions, the available trials data and the controversy over aducanumab approval for AD.
Introduction
Alzheimer´s Disease (AD) is a neurodegenerative disorder characterized by progressive loss of memory and cognitive impairment, usually followed by behavioral changes and loss of functional abilities. The World Health Organization recognizes AD as a growing global health concern with a significant impact on individuals, caregivers, and society. By 2060, the number of AD patients is expected to reach 13.8 million, just in the United States (US).Citation1,Citation2
Until recently, only four drugs had been approved for AD treatment (donepezil, galantamine, rivastigmine, and memantine).Citation1 These drugs temporary attenuate symptoms but do not target AD’s two main recognized pathological features: extracellular deposits of amyloid β (Aβ) in plaques and intracellular neurofibrillary tangles (comprised of abnormal tau protein).Citation3,Citation4
On June 7, 2021, aducanumab (Aduhelm™, Biogen) became the first disease-modifying therapy approved by the US Food and Drug Administration (FDA) and the first AD drug approved since 2003 (memantine).Citation5 Aducanumab is a monoclonal antibody selective for aggregated forms of Aβ, with demonstrated efficacy in the clearance of brain Aβ plaques.Citation6 According to the amyloid cascade hypothesis, the extracellular accumulation of Aβ aggregates is the leading cause of synapses dysfunction, neuroinflammation, neuronal loss and is also the trigger of tau pathology.Citation7 Thus, researchers believe that Aβ clearance by aducanumab is a rational mechanism to slow cognitive decline in AD. However, there is a lack of correlation between the reduction of Aβ plaques and clinical improvements in trials to date.Citation8–Citation10 The known controversy over the FDA aducanumab approval decision begins here.Citation11–Citation13
Aducanumab was granted accelerated approval by the FDA, a provisional approval for drugs targeting serious diseases that appear to have considerable advantages over current treatment. This decision was based on the surrogate endpoint of Aβ plaques reduction, which the FDA believes to be a biomarker reasonably likely to predict clinical benefit.Citation5,Citation14 The scientific community has been divided and confounded with this decision once it was based on an unproven biomarker and without data showing clear evidence of clinical efficacy.Citation11,Citation12,Citation14–Citation17 Furthermore, the approval was taken after the FDA’s independent advisory committee recommended against it.Citation11,Citation18–Citation20 Although the FDA’s accelerated approval requires a confirmatory trial to verify clinical efficacy, until 2030, several experts are worried that aducanumab’s approval could set a dangerous precedent and lead companies to abandon other targets associated with AD (such as tau protein).Citation11,Citation13
In addition, there are also considerable concerns about the cost and the safety of this new drug. Firstly, Biogen set the price of aducanumab at US$ 56,000 per person annually. Secondly, aducanumab had a significantly higher incidence of brain swelling and intracerebral hemorrhages (amyloid-related imaging abnormalities, ARIA) in patients treated with aducanumab.Citation21 Bearing this last point in mind and the divergent outcomes of clinical trials, the European Medicines Agency (EMA) rejected the marketing authorization for aducanumab on December 17, 2021.Citation22 Similarly, on December 22, 2021, the Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) declined the Aduhelm™ approval.Citation23,Citation24
In this review, we first summarize the aducanumab path until drug agencies’ conclusions. Secondly, we analyze the trials data available in detail and, finally, explore the controversy and the challenges surrounding its approval.
Aducanumab: Selective Anti-Aβ Antibody
Over the last 25 years, several drugs targeting Aβ have failed to show clinical efficacy in trials, including five anti-Aβ antibodies: bapineuzumab, solanezumab, crenezumab, ponezumab, and gantenerumab.Citation25,Citation26 Thus, supporters of the amyloid hypothesis placed high hopes on aducanumab.
Aducanumab is part of a new generation of monoclonal anti-Aβ antibodies that specifically target Aβ aggregates. These aggregates, in particular the soluble oligomers, appear to be the most neurotoxic forms of Aβ, whereas the monomeric Aβ possibly have a neuroprotective role in the brain.Citation10,Citation27–Citation29 Hence, the high selectivity of aducanumab for Aβ aggregates, including soluble oligomers and insoluble fibrils, constitutes a major advantage over previous anti-Aβ antibodies.Citation30 Studies demonstrated that aducanumab’s binding promotes the clearance of Aβ aggregates through the activation of microglial phagocytosis.Citation6 Besides the clearance, aducanumab is the only antibody that disrupts the Aβ aggregation process by inhibiting the secondary nucleation (formation of oligomers on the fibril surface) due to its affinity and binding stoichiometry to Aβ aggregates.Citation19,Citation31 These reasons made aducanumab the most promising drug for AD of the last decade.
Clinical Development and Regulatory History Overview
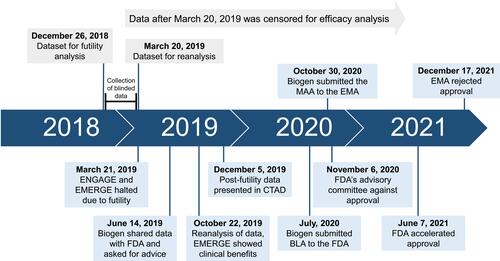
Aducanumab’s path to approval was slightly unconventional, due to a close collaboration between the sponsor (Biogen) and the FDA, during the whole process. The aducanumab clinical program had three essential studies to evaluate efficacy: PRIME, ENGAGE and EMERGE, also known as studies 103, 301, and 302, respectively.Citation19,Citation32 Next, we explore the clinical development of aducanumab until regulatory agencies’ decisions ().
Figure 1 Aducanumab timeline since futility analysis until regulatory agencies decisions.
Clinical Trials and the FDA Collaboration
Aducanumab moved to clinical trials after preclinical studies showed satisfactory brain penetration, target engagement, and a significant reduction of Aβ plaques in transgenic mice brains.Citation6,Citation33 The first Phase 1 trial began in 2011 (study 101, NCT01397539), aiming to evaluate the safety, tolerability, and pharmacokinetics (PK) of an ascending dose of aducanumab (0.3 mg/kg to 60 mg/kg). The study showed a reasonable safety profile and linear PK at doses ≤ 30mg/kg.Citation34 In 2012, a phase 1b randomized trial, denominated PRIME (study 103, NCT01677572), started with mild cognitive impairment (MCI) and mild AD patients who had brain Aβ plaques confirmed by positron emission tomography (PET) imaging. Participants received monthly intravenous infusions of aducanumab at doses 1, 3, 6, and 10 mg/Kg to examine the safety, tolerability, PK, and pharmacodynamics. Biogen also performed an exploratory clinical evaluation in PRIME. Results showed that ARIA was the most common side effect, increasing with dose and ApoE4 genotype (a genetic risk factor for AD), and revealed a significant time and dose-dependent reduction of brain Aβ plaques. In addition, researchers reported a dose-dependent slowing of cognitive deterioration, measured by two dementia rating scales.Citation6 The PRIME was the first trial to demonstrate a possible correlation between the reduction of brain Aβ and clinical benefits by an anti-Aβ antibody (proof of concept).Citation19
These promising results prompted the start of phase 3 trials under a special protocol assessment with the FDA.Citation19 Therefore, in 2015, Biogen initiated simultaneously two identical, large, global, double-blind, randomized, placebo-controlled, phase 3 studies: ENGAGE (study 301, NCT02477800) and EMERGE (study 302, NCT02484547). The studies aimed to assess efficacy and safety in MCI and mild AD patients with a positive amyloid PET scan for 18 months. The two studies enrolled 3285 participants from 20 countries and were supposed to run until 2022.Citation19,Citation35
However, on March 21, 2019, Biogen announced the early termination of both phase 3 trials (). The decision was made after an interim analysis had demonstrated that EMERGE and ENGAGE were unlikely to reach their primary efficacy endpoint, the slowing of cognitive decline.Citation36 This futility analysis was performed using pooled data from both studies, collected up to December 26, 2018 (when approximately 50% of participants completed week 78 of the trials). Though, if the trials had been independently assessed (using non-pooled data), EMERGE would not have met the futility criteria.Citation19,Citation35,Citation37 Given this observation, the sponsor decided to collect three additional months of blinded data, between the date cutoff of futility analysis and the public futility announcement (). The reanalysis of this larger dataset showed that the high-dose arm in EMERGE met the primary endpoint, while ENGAGE was a failed study. In June 2019, Biogen shared this data with the FDA asking for advice on the validity of the results and their interpretation, given the premature discontinuation. After four months of analysis, the FDA considered that data are interpretable and reliable for further conclusions, but efficacy data collected after March 20, 2019, should be censored.Citation19
On October 22, 2019, Biogen formally announced that aducanumab showed a statistically significant clinical benefit in EMERGE, reaching primary and secondary endpoints. In contrast, the ENGAGE trial did not meet any of the endpoints. Despite that, Biogen’s researchers considered that ENGAGE is not a negative study, as a subset of patients who received sufficient exposure to the high-dose of aducanumab had similar benefits to those reported in EMERGE (trials data will be explored later in this article).Citation38 Based on these results, and with PRIME trial providing additional evidence of effectiveness, the FDA considered that a marketing application for aducanumab was reasonable. Thus, Biogen submitted a Biologics License Application (BLA) to the FDA in July 2020 ().Citation19
In short, the BLA of aducanumab was based on data from 3 trials. EMERGE is a robust positive trial that has proven a cognitive decline reduction. ENGAGE is a partially discordant study whose post hoc analysis supports the EMERGE results. PRIME is a phase Ib trial whose clinical exploratory assessment demonstrated a positive outcome. The partially divergent results and the absence of further studies raised a considerable debate about the decisions of regulatory agencies.
FDA Decision
On November 6, 2020, the Peripheral and Central Nervous System (PCNS) Drugs Advisory Committee met to evaluate the clinical data of aducanumab. None of the members voted in favor of aducanumab’s approval (10 against, 1 abstention). They considered that the results of the studies were conflicting, and the data presented did not show sufficient evidence of clinical efficacy.Citation19,Citation39 Contrary to the independent committee decision, the FDA granted accelerated approval to aducanumab on June 7, 2021, leading to the resignation of three members of the PCNS committee.Citation20,Citation40 The accelerated approval pathway allows patients to have early access to drugs that target a serious disease and which provide a meaningful improvement over current treatments. This approval is based on a surrogate endpoint that is thought to predict clinical benefit, and not on clinical outcomes. In this case, the FDA believes that the reduction of brain Aβ plaques is a biomarker reasonably likely to predict the slowing of cognitive decline. Furthermore, the accelerated approval program requires a post-approval trial to confirm clinical benefits. If the trial fails to show clinical benefits or the risks outweigh the benefits, aducanumab approval will be withdrawn.Citation5,Citation41
EMA and PMDA Decisions
In Europe, aducanumab was under review at the EMA since October 2020 ().Citation14,Citation42 On December 17, 2021, the EMA’s Committee for Medicinal Products for Human Use recommended the rejection of the marketing application for Aduhelm™.Citation22,Citation43,Citation44 According to the performed studies, EMA experts considered that this drug did not show a clear signal of efficacy nor a satisfactory safety profile to treat patients in the early stage of AD.Citation22 In contrast to the FDA, they considered that reduction of brain Aβ aggregates is not an acceptable biomarker to predict clinical benefit. At the time of press, Biogen is appealing against EMA’s decision.Citation23,Citation44 Shortly after the EMA’s decision, the Japanese Medicines agency also denied the approval of aducanumab. For the PMDA, trials results are inconclusive and they require additional data before considering Aduhelm™ approval.Citation23,Citation24
Ongoing Studies and Confirmatory Trial
Currently, Biogen is running two clinical studies: EMBARK and ICARE-AD. The EMBARK (NCT04241068) is a phase 3b, open-label, extension study enrolling patients that previously participated in aducanumab studies (PRIME, EVOLVE, ENGAGE and EMERGE). This study intends to assess the safety and efficacy of aducanumab after prolonged treatment interruption.Citation45,Citation46 On the other hand, ICARE-AD (NCT05097131) is an observational study designed after aducanumab’s approval. It is a prospective cohort aiming to collect safety and efficacy data in clinical practice (Phase 4).Citation47,Citation48 Moreover, Biogen is still designing the FDA’s required phase 4 confirmatory study, a new global, placebo-controlled clinical trial named ENVISION. The company intends to start recruiting patients in May 2022, and the study should be completed in 2026.Citation49,Citation50
Data from ENGAGE and EMERGE Clinical Trials
To date, full clinical trials data have not been published in a peer-reviewed journal. The results presented below were obtained from Biogen’s presentation at the Alzheimer’s Disease Conference (CTAD) in December 2019 and from the combined FDA and Biogen briefing document that emerged from the PCNS Drugs Advisory Committee meeting.Citation19,Citation32,Citation35 Therefore, we cannot exclude any potential bias introduced by the sponsor.
The ENGAGE (1647 patients) and EMERGE (1638 patients) were two phase 3 trials identically designed to assess aducanumab’s safety and efficacy in MCI and mild AD patients. Participants were randomized 1:1:1 in placebo and two-dose regimens of aducanumab: low-dose (3 and 6 mg/kg for Apolipoprotein E (ApoE) ε4 carriers and non-carriers, respectively) and high-dose (6 and 10 mg/kg for ApoE ε4 carriers and non-carriers, respectively). After protocol version 4 (PV4), the high-dose in ApoE ε4 carriers increased to 10 mg/kg.Citation19,Citation39
Efficacy
The main objective of these studies was to assess the efficacy of aducanumab in reducing cognitive decline. Biogen used four recognized clinical efficacy scales to measure it. The primary endpoint was the change in Clinical Dementia Rating-Sum of Boxes (CDR-SB) from baseline to week 78. Secondary objectives were to evaluate the changes in Mini-Mental State Examination (MMSE), Alzheimer’s Disease Assessment Scale-Cognitive Subscale (13-item version) (ADAS-Cog 13), and Alzheimer’s Disease Cooperative Study-Activities of Daily Living Inventory-Mild Cognitive Impairment version (ADCS-ADL-MCI). In addition, sub-studies were conducted to analyze the changes in amyloid PET, tau PET, and in CSF (cerebrospinal fluid) biomarkers such as phosphorylated tau (p-tau), total tau (t-tau), and 42-amino acid form of amyloid β (Aβ42).Citation19
The efficacy data are presented in . Both studies showed a reduction in brain Aβ, measured by PET, in all treatment groups compared to placebo (p<0.0001). This reduction was dose-dependent. However, only the high-dose arm of EMERGE showed a statistically significant benefit on CDR-SB (p<0.05) and all the secondary outcomes: MMSE, ADAS-Cog 13 and ADCS-ADL-MCI (p<0.05). ENGAGE did not demonstrate significant differences between the patients treated with aducanumab and placebo on primary and secondary efficacy outcomes. Moreover, the results of the low-dose arm were numerically better than the high-dose ().
Table 1 Efficacy and Biomarker (Amyloid PET) Final Data at Week 78 in ENGAGE and EMERGE Studies
Besides amyloid PET, the other sub-studies (tau PET, p-tau, t-tau, and Aβ42) were performed on a small number of subjects, with less than 10% of the studies participants undergoing these analyses.Citation37 Tau PET was evaluated using combined data from ENGAGE and EMERGE trials, comprising only 37 patients. As presented in , aducanumab produced a dose-related reduction in brain tau levels in frontal (p<0.05), medial temporal (p<0.001), and temporal (p<0.05) composite brain regions. The medial temporal region experienced the highest reduction of tau levels, with statistical significance in both low and high-dose administration of aducanumab (p=0.0012 and p=0.0005, respectively). The cingulate, parietal, and occipital composites did not show a significant difference relative to placebo, but numeric data were not provided.Citation19,Citation51 In addition, the changes in CSF biomarkers (p-tau, t-tau, Aβ1-42) were not presented in sufficient detail for both trials. Despite that, available data demonstrated that aducanumab produced a significant dose-dependent reduction of CSF p-tau and t-tau in EMERGE trial. In ENGAGE, p-tau and t-tau also decreased in the aducanumab-treated patients, but the low-dose arm showed a higher reduction than the high-dose arm.Citation19,Citation35,Citation37 Finally, the Aβ42 analysis is unavailable for ENGAGE, but in EMERGE, aducanumab increased the CSF Aβ42 levels in a dose-response relationship.Citation19
Table 2 Changes from Baseline in Tau PET
Safety
In terms of safety, ARIA was the most common adverse event in ENGAGE and EMERGE. ARIAs are abnormal findings detected in brain magnetic resonance imaging (MRI) and can present as brain edema or sulcal effusions (ARIA-E) and as intracerebral hemorrhage often accompanied by superficial hemosiderin deposits (ARIA-H).Citation52 Although the mechanism leading to ARIA remains not fully understood, studies suggest that Aβ clearance causes damage to vessel walls which increases the cerebrovascular permeability and the risk of hemorrhage.Citation52,Citation53 ARIA has been associated with anti-Aβ antibodies therapies and was reported in the phase 1b trial of aducanumab (PRIME).Citation6,Citation52,Citation54–Citation56
Salloway et al published the combined safety data from the ENGAGE and EMERGE trials in December 2021.Citation21 However, we presented the ARIA incidence for each study independently in . In general, around 40% of the patients taking the aducanumab high-dose developed ARIA (ARIA-E or ARIA-H), compared to 10% on placebo (). Although most of the patients were asymptomatic (ARIA was detected only in MRI), some patients experienced symptoms such as headache, dizziness, visual disturbance, and nausea.Citation21,Citation35 ARIA-E was the most common adverse event, occurring in approximately 35% of the patients in the high-dose group (35.7% and 34.4% for ENGAGE and EMERGE, respectively) compared with 3% in the placebo group (3% and 2.2% for ENGAGE and EMERGE, respectively) (). The ARIA-E incidence increased with aducanumab dose and was higher among ApoE4 carriers than in non-carriers, in the aducanumab-treated patients ().
Table 3 Summary of ARIA Incidence in ENGAGE and EMERGE Studies, in the Placebo-Controlled Period
Moreover, brain microhemorrhages were the most common type of ARIA-H in both trials (17.7% and 18.9% in the high-dose group of ENGAGE and EMERGE, respectively) (). According to Salloway et al’s analysis, ARIA-H are more frequent in patients who also had ARIA-E (approximately 40% of ARIA-E patients had ARIA-H). Finally, despite the high incidence, ARIA episodes typically resolved in 4–16 weeks, and less than 1% of patients treated with aducanumab experienced severe ARIA symptoms.Citation21
Discordant Results and Post Hoc Analysis
The divergent outcomes of the trials are the main controversy over aducanumab approval. Investigators consider that two identically designed studies, not fully completed, whose results directly contradict each other cannot prove the efficacy of aducanumab.
Biogen researchers performed a post hoc analysis of data to understand the differences between the ENGAGE and EMERGE trials. They found two possible explanations for these differences: duration of exposure to high-dose aducanumab and imbalance of rapid disease progressors. Firstly, exposure to a high dose of aducanumab is seen as the critical variable for the different outcomes. In total, 29% of the patients in EMERGE received the full possible 14 doses of 10 mg/kg of aducanumab compared to 22% of the patients in ENGAGE.Citation19,Citation35,Citation37 Several factors contributed to this exposure discrepancy, such as the beginning of the trials, the timing of implementation of PV4 (allowed ApoE ε4 carriers to receive 10 mg/kg), and the timing of futility analysis. Moreover, Biogen presented a subset of data limited to patients exposed to PV4 who received 14 infusions of 10 mg/kg of aducanumab (). In this analysis, ENGAGE subset of patients showed a reduction in cognitive decline, measured by CDR-SB, similar to EMERGE (−0.48 and −0.53 for the high-dose arm of ENGAGE and EMERGE, respectively) (). The second factor that explains the difference between the trials was the higher number of rapid progression patients in the high dose arm of ENGAGE compared to EMERGE (9 and 5 patients, respectively). Exploration showed that excluding the rapid progressors, the CDR-SB changes from 0.03 (2%), see , to −0.09 (−6%) in the high-dose of ENGAGE.Citation19,Citation32 The other groups were not affected by rapid progressors.
Table 4 Changes in CDR-SB in Intention to Treat (ITT) Population Compared to Post-Protocol Version 4 (PV4) Population Who Received 14 High-Dose Aducanumab Treatments for ENGAGE and EMERGE Studies at Week 78
Although this examination appears to confirm that ENGAGE trial supports the positive results of EMERGE, post-hoc analyzes do not have the same predictive power as the primary assessments due to the possible introduction of bias. Knopman et al mentioned in their review that although plausible, post hoc explanations are insufficient to justify a claim of efficacy for aducanumab.Citation37 The same author states that the variance in placebo decline is an alternative explanation for the different results of the trials. The placebo group declined 1.56 points for CDR-SB in ENGAGE, while in EMERGE, the decline was 1.74 points (). The higher placebo decline in EMERGE could explain the statistically significant clinical benefit in the high-dose arm.Citation37 Identically, the higher placebo decline in the PV4 patient’s subgroup compared to the original intention to treat (ITT) population in ENGAGE (1.79 vs 1.56 points in CDR-SB) is a potential reason for the better outcomes and not necessarily the exposure to high-dose of aducanumab ().Citation37 However, we should note that the cognitive performance of placebo groups is not consistent, as it differs from the other efficacy endpoints. In ADAS-Cog 13 evaluation, placebo decline was similar in both trials, but in MMSE the decline was higher in ENGAGE than in EMERGE ().Citation19
Furthermore, the duration of exposure to high-dose aducanumab does not explain why the low-dose arm in ENGAGE was numerically better than the high-dose, despite none of the participants being exposed to 10 mg/kg of aducanumab (). In fact, the high-dose arm in ENGAGE was worse than the placebo in CDR-SB and MMSE measures (p=0.8330 and p=0.8106, respectively) (). Further, Knopman et al pointed out that the significantly better results in the low-dose arms of PV4 patient’s subset appear to contradict the argument that the exposure to 10 mg/kg dose of aducanumab is the main responsible for clinical efficacy ().Citation37
Finally, researchers have raised some questions about ARIA management. Although Biogen mentioned that bias due to ARIA is not apparent, some authors still consider that the sporadic unblinding due to ARIA episodes management could have impacted the results. The incidence of ARIA was higher in aducanumab-treated patients, and all the four efficacy outcomes may be subject to bias when drug assignment is known.Citation19,Citation32,Citation57
Biomarkers Analysis
The results showed a clear target engagement of aducanumab, with a significant dose-dependent reduction of Aβ in PET. However, there is a lack of correlation between Aβ PET changes and cognitive changes, measured by CDR-SB, for high-dose patients.Citation19 This observation raises doubts about the biomarker-based approval of aducanumab by the FDA. Similarly, the tau biomarkers also decreased with aducanumab treatment, but the correlation between tau changes and CDR-SB is not significant in the high dose.Citation19 Though, the small sample size of tau substudies (p-tau, t-tau, and tau PET) limits the conclusions due to possible statistical noise.
Bearing this last point in mind, Biogen asked independent researchers to analyze stored plasma samples from 1815 ENGAGE and EMERGE trials participants. Data presented at the 2021 CTAD conference showed that aducanumab produced a time and dose-dependent reduction of plasma p-tau. Moreover, the results demonstrated that plasma p-tau reduction had a statistically significant correlation with less clinical decline on all four efficacy outcomes measures and with the lowering of Aβ plaques.Citation58
In short, aducanumab appears to have an effect on both main pathological features of AD (Aβ and tau protein). Further, this analysis showed that p-tau has the potential to be a better surrogate endpoint for cognition than Aβ plaques.
Controversy
After aducanumab’s approval, several experts manifested disagreement with the decision of the FDA.Citation11–Citation13 In this section, we will explore the controversial points of aducanumab.
Firstly, as mentioned above, the conflicting results of phase 3 trials are the main core of the controversy. The data available and the post hoc analysis do not provide sufficient evidence to support the clinical efficacy of aducanumab. The FDA should have required a third phase 3 trial with a high-dose of aducanumab before approval.Citation37,Citation59 Secondly, there is no evidence that Aβ reduction correlates with clinical benefits, so the majority of experts consider that Aβ plaques are not a valid surrogate endpoint.Citation10,Citation60 Moreover, recent studies showed that tau accumulation correlates better with cognitive impairment than Aβ.Citation61,Citation62 Thirdly, the close collaboration between the FDA and the sponsor could have impacted the objectivity of the FDA’s decision.Citation63,Citation64 Furthermore, the AD´s advocacy groups exerted much pressure on the FDA, defending that a marginal benefit of aducanumab would be significant to patients and caregivers.Citation65 Fourthly, the FDA’s decision passes to physicians the unfair role of removing false hopes from patients.Citation59 Fifthly, aducanumab approval could prejudice scientific development, leading pharmaceutical companies to abandon other targets associated with AD, redirecting their efforts towards amyloid pathology and using an unproven biomarker to obtain approval. Currently, two new monoclonal anti-Aβ antibodies are under review by the FDA: lecanemab (Eisai/Biogen) and donanemab (Eli Lilly’s). Gantenerumab also gained new interest after aducanumab approval.Citation39,Citation65
In addition, there is also a substantial debate about the cost-effectiveness and the safety of aducanumab. Initially, aducanumab had an annual cost of US$ 56,000 per person, but Biogen reduced the price to half at the beginning of 2022. Despite that, the value remains much higher than the $2500-$8300 predicted for aducanumab to be cost-effective.Citation11,Citation66–Citation68 Further, given the ARIA high incidence under trials conditions (40% high dose aducanumab vs 10% in placebo), there are some concerns about whether the benefits outweigh the risks in the clinical practice. Importantly, monitoring the ARIA with MRI scans will increase the cost of treatment and the complexity of infrastructures needed.Citation39
Finally, there was controversy over the FDA prescribing label for aducanumab because it was initially approved for anyone with AD, despite the trials only enrolled MCI and mild AD patients.Citation13 Only after a month, the FDA narrowed the indication to MCI and mild AD.Citation12,Citation51 Moreover, the prescribing information does not require a positive amyloid biomarker (amyloid PET or CSF biomarkers) to confirm the diagnosis of AD.Citation39,Citation51 This fact appears to be contradictory, once all participants of ENGAGE and EMERGE had a positive amyloid PET and being Aβ plaques the aducanumab target. Therefore, an expert panel published a use recommendation for aducanumab to cover these critical issues and guide clinicians. We explore it in the next section.Citation69
Use Recommendations of Aducanumab
The Expert Panel, headed by Cummings, recommends using aducanumab in patients with a diagnosis of MCI and mild AD. In addition, they consider mandatory a positive amyloid PET or CSF biomarkers consistent with AD and a score ≥ 21 in MMSE (or equivalent cognitive test) to initiate treatment. ApoE genotype screening is optional, despite the high risk of ARIA in ApoE4 carriers.Citation69
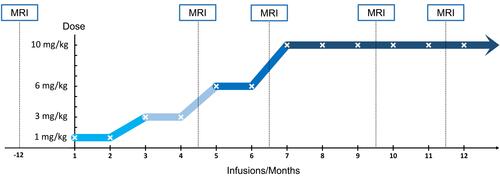
The panel also requires aducanumab titration to the target dose of 10 mg/kg over 6 months, and this dose is continued for the future (). Aducanumab is administered as an intravenous infusion over 45–60 minutes every month, and the titration aims to minimize the risk of ARIA. To monitor ARIA, they recommend a brain MRI one year before the first administration of aducanumab and prior to the 5th (before initiating 6 mg/kg), 7th (before the target dose of 10 mg/kg), and the 12th infusions (after 6 doses of 10 mg/kg). An additional MRI should be considered before the 10th infusion, given the high incidence of ARIA in 10 mg/kg dose (). Patients with ARIA symptoms or moderate to severe asymptomatic ARIA should suspend or discontinue the treatment. Finally, the expert panel does not recommend aducanumab treatment in preclinical and moderate to severe AD patients because no data is available for these AD stages.Citation69
Figure 2 Dose titration of aducanumab from 1 mg/kg to 10 mg/kg and magnetic resonance imaging monitoring.
Challenges and Opportunities
Despite controversies, aducanumab is a new drug for a disease that significantly impacts individuals and society. Its approval generated challenges and opportunities for the management of AD.
The high cost of aducanumab is probably the most challenging point surrounding its approval, especially because aducanumab treatment is not time-limited. Its questionable efficacy and the initial price of US$ 56,000 led many insurances and hospitals to refuse to cover the cost of the treatment. Consequently, few patients had access to Aduhelm™ in the first six months, with disappointing sales for Biogen. After the rejection of EMA, Biogen announces the reduction of the price to $28,200 at the beginning of 2022, expecting to improve patient’s access to aducanumab through an increase in insurance coverage.Citation67
However, on April 7, 2022, the Centers for Medicare and Medicaid Services (CMS) restricted aducanumab coverage only to clinical trials (randomized controlled trials).Citation70,Citation71 There has been a considerable debate since the announcement that CMS planned to limit Aduhelm™ coverage.Citation72 The CMS decision includes the whole class of anti-Aβ antibodies, which also affect lecanemab, donanemab, and gantenerumab.Citation73,Citation74 Moreover, this decision could also interfere with private insurance coverage because most follow the CMS’s orientations.Citation74 Furthermore, Biogen had to adapt its confirmatory study (ENVISION) to be covered by the CMS.Citation50
In addition, aducanumab approval created challenges for current and future AD clinical trials. Firstly, patients will want to drop out of investigational drugs trials for the aducanumab.Citation39 Secondly, it could be challenging to recruit and keep patients in placebo-controlled trials.Citation12,Citation13
Moving to the opportunities, aducanumab’s effect on downstream tau pathology could finally empower the combination therapies approach (anti-tau and anti-amyloid drugs). Given the complexity of AD pathology, combined therapy is gaining strength in the scientific community. Having one anti-Aβ immunotherapy approved, with the demonstrated effect of tau pathology, could open the door for clinical trials targeting more than one toxic protein.Citation75
Further Implications of Aducanumab Approval
Aducanumab approval will have a major impact on AD therapy. The FDA’s decision can redirect the investigation efforts towards the amyloid hypothesis, a therapeutic approach that would be practically “dead” in the face of aducanumab’s failure.
Furthermore, this accelerated approval will also have significant implications in other therapeutic areas, namely in oncology which have a high proportion of drugs approved by the accelerated pathway. Lythgoe et al published a critical paper covering this subject.Citation76 The authors stated that the FDA’s decision suggests that any biomarker can be used as a surrogate endpoint to submit for accelerated approval, regardless of whether or not it is validated. Additionally, aducanumab’s approval showed that despite negative clinical trials or negative advisory committee opinions, drugs can receive accelerated approvals. Thus, an exponential increase in accelerated approvals of anti-cancer drugs is expected in the coming years, and some of them may have dubious efficacy. Finally, the authors indicated that the large time given to Biogen to complete the confirmatory trial (9 years) could set a precedent for companies to delay the completion of this trial.Citation76 As a result, some experts are now questioning the utility of FDA´s accelerated approvals, showing concerns about its increasing frequency, and suggesting reform of this pathway.Citation77
Conclusion
Aducanumab has been the most promising drug for AD of the last decade. However, the divergent results between ENGAGE and EMERGE and its post hoc analysis did not show sufficient evidence of clinical benefit. In addition, there is no reliable evidence that correlates Aβ plaques reduction with clinical efficacy to support the FDA approval. Some authors mention that a third randomized, placebo-controlled, phase 3 clinical trial is necessary to prove aducanumab efficacy.Citation37 Moreover, at best, aducanumab has a modest benefit in AD, which coupled with its high cost and adverse events (ARIA) raise questions about whether the benefits outweigh the risk and the cost burden to healthcare systems. Therefore, the recent decision of CMS has been seen as a correction to the original approval of the FDA by many experts, and we share that vision.Citation73,Citation74 In this article, we tried to give a balanced and objective perspective on the data and the trajectory of aducanumab. However, in our opinion, the EMA and the PMDA took the right decision by refusing to approve aducanumab.
Despite the controversy, it is clear that aducanumab reduces significantly the Aβ in the brain, one of AD’s hallmarks. Further, the results also showed that aducanumab acts on the second AD’s hallmark, decreasing tau brain levels. This observation could become a turning-point for the approach therapy to AD. Researchers are concluding that both Aβ and tau protein has a major role in the neurodegeneration process and they interfere with each other.Citation78 Thus, a combination therapy targeting the accumulation of Aβ aggregates (upstream pathology) and the intracellular tau accumulation (downstream pathology) have the potential to change the natural progress of AD.Citation75 The combination therapy is now a necessity.Citation79
In conclusion, we hope that aducanumab approval does not redirect the focus of research only to Aβ immunotherapies. Targeting AD in multiple pathways could be the most effective way to have a truly disease-modifying therapy for this epidemic of our century.
Acknowledgments
The authors acknowledge National Funds by FCT - Foundation for Science and Technology (UID/Multi/00709/2019), and FEDER funds through the POCI - COMPETE 2020 - Operational Programme Competitiveness and Internationalization in Axis I - Strengthening research, technological development and innovation (Project POCI-01-0145- FEDER-007491) and “Programa Operacional do Centro, Centro 2020” (ICON project - Interdisciplinary Challenges On Neurodegeneration; CENTRO-01-0145-FEDER-000013).
Disclosure
The authors report no conflicts of interest in this work.
References
- Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021;17(3):327–406. doi:10.1002/alz.12328.
- World Health Organization. Global action plan on the public health response to dementia 2017–2025. Geneva:World Heal Organ; 2017: 27. Available from: http://www.who.int/mental_health/neurology/dementia/action_plan_2017_2025/en/. Accessed May 14, 2022.
- Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol. 2018;25(1):59–70. doi:10.1111/ene.13439
- Deture MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14(1):1–18. doi:10.1186/s13024-019-0333-5
- U.S. Food and Drug Administration (FDA). FDA grants accelerated approval for Alzheimer’s drug. FDA News Release; 2021. Available from: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug. Accessed November 14, 2021.
- Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537(7618):50–56. doi:10.1038/nature19323
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608. doi:10.15252/emmm.201606210
- Pietro IB, Ippati S, Watling M. Should drug discovery scientists still embrace the amyloid hypothesis for Alzheimer’s disease or should they be looking elsewhere? Expert Opin Drug Discov. 2020;15(11):1241–1251. doi:10.1080/17460441.2020.1793755
- Salloway S, Farlow M, McDade E, et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat Med. 2021;27(7):1187–1196. doi:10.1038/s41591-021-01369-8
- Sturchio A, Dwivedi AK, Young CB, et al. High cerebrospinal amyloid-β 42 is associated with normal cognition in individuals with brain amyloidosis. EClinicalMedicine. 2021;38:100988. doi:10.1016/j.eclinm.2021.100988
- Landmark Alzheimer’s MA. drug approval confounds research community. Nature. 2021;594(7863):309–310. doi:10.1038/d41586-021-01546-2
- Rubin R. Recently approved Alzheimer drug raises questions that might never be answered. JAMA. 2021;326(6):469. doi:10.1001/jama.2021.11558
- Walsh S, Merrick R, Milne R, Brayne C. Aducanumab for Alzheimer’s disease? BMJ. 2021;374. doi:10.1136/bmj.n1682.
- Nisticò R, Borg JJ. Aducanumab for Alzheimer’s disease: a regulatory perspective. Pharmacol Res. 2021;171:105754. doi:10.1016/j.phrs.2021.105754
- Alzheimer’s Forum Association. Aducanumab approved to treat Alzheimer’s disease. Alzforum; 2021. Available from: https://www.alzforum.org/news/research-news/aducanumab-approved-treat-alzheimers-disease. Accessed November 11, 2021.
- Hooker JM. FDA Approval of aducanumab divided the community but also connected and united it. ACS Chem Neurosci. 2021;12(15):2716–2717. doi:10.1021/acschemneuro.1c00393
- Cummings J, Aisen P, Lemere C, Atri A, Sabbagh M, Salloway S. Aducanumab produced a clinically meaningful benefit in association with amyloid lowering. Alzheimers Res Ther. 2021;13(1):1–3. doi:10.1186/s13195-021-00838-z
- Alzheimer’s Forum Association. FDA advisory committee throws cold water on aducanumab filing. Alzforum; 2020. Available from: https://www.alzforum.org/news/community-news/fda-advisory-committee-throws-cold-water-aducanumab-filing. Accessed November 11, 2021.
- U.S. Food and Drug Administration (FDA). Combined FDA and applicant PCNS drugs advisory committee briefing document. Peripheral and Central Nervous System (PCNS) Drugs Advisory Committee Meeting. Online; 2020. Available from: https://www.fda.gov/media/143502/download. Accessed November 14, 2021.
- Mahase E. Three FDA advisory panel members resign over approval of Alzheimer’s drug. BMJ. 2021;373:n1503. doi:10.1136/bmj.n1503
- Salloway S, Chalkias S, Barkhof F, et al. Amyloid-related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol. 2021. doi:10.1001/jamaneurol.2021.4161
- European Medicines Agency (EMA). Refusal of the marketing authorisation for Aduhelm (aducanumab). EMA; 2021. Available from: https://www.ema.europa.eu/documents/smop-initial/refusal-marketing-authorisation-aduhelm-aducanumab_en.pdf. Accessed December 17, 2021.
- Lythgoe MP, Jenei K, Prasad V. Regulatory decisions diverge over aducanumab for Alzheimer’s disease. BMJ. 2022;376. doi:10.1136/bmj-2021-069780.
- Eisai. Japan’s first committee on new drugs of the pharmaceutical affairs and food sanitation council seeks additional data; aducanumab remains under review. Eisai Global; 2021. Available from: https://www.eisai.com/news/2021/news2021101.html. Accessed April 9, 2022.
- Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179(2):312–339. doi:10.1016/j.cell.2019.09.001
- Vaz M, Silvestre S. Alzheimer’s disease: recent treatment strategies. Eur J Pharmacol. 2020;887:173554. doi:10.1016/j.ejphar.2020.173554
- Penke B, Szucs M, Bogár F. Oligomerization and conformational change turn monomeric β-amyloid and tau proteins toxic: their role in Alzheimer’s pathogenesis. Molecules. 2020;25(7):1659. doi:10.3390/molecules25071659
- Cline EN, Bicca MA, Viola KL, Klein WL. The amyloid-β oligomer hypothesis: beginning of the third decade. J Alzheimers Dis. 2018;64(s1):S567–S610. doi:10.3233/JAD-179941
- Chen XQ, Mobley WC. Alzheimer disease pathogenesis: insights from molecular and cellular biology studies of oligomeric Aβ and tau species. Front Neurosci. 2019;13:(JUN):659. doi:10.3389/fnins.2019.00659
- Arndt JW, Qian F, Smith BA, et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci Rep. 2018;8(1):1–16. doi:10.1038/s41598-018-24501-0
- Linse S, Scheidt T, Bernfur K, et al. Kinetic fingerprints differentiate the mechanisms of action of anti-Aβ antibodies. Nat Struct Mol Biol. 2020;27(12):1125–1133. doi:10.1038/s41594-020-0505-6
- U.S. Food and Drug Administration (FDA). Aducanumab for the treatment of Alzheimer’s disease: clinical overview of efficacy. Peripheral and Central Nervous System (PCNS) Drugs Advisory Committee Meeting. Online; 2020. Available from: https://www.fda.gov/media/143504/download. Accessed November 14, 2021.
- Bussiere T, Weinreb PH, Dunstan RW, et al. Differential in vitro and in vivo binding profiles of BIIB037 and other anti-abeta clinical antibody candidates. Neurodegener Dis. 2013;11(Suppl1):2576.
- Ferrero J, Williams L, Stella H, et al. First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer’s disease. Alzheimers Dement Transl Res Clin Interv. 2016;2(3):169–176. doi:10.1016/j.trci.2016.06.002
- Biogen. EMERGE and ENGAGE topline results: two phase 3 studies to evaluate aducanumab in patients with early Alzheimer’s disease. Clinical Trials on Alzheimer’s Disease (CTAD). San Diego; 2019. Available from: https://investors.biogen.com/static-files/ddd45672-9c7e-4c99-8a06-3b557697c06f. Accessed November 14, 2021.
- Biogen. Biogen and eisai to discontinue phase 3 ENGAGE and EMERGE trials of aducanumab in Alzheimer’s disease. Biogen; 2019. Available from: https://investors.biogen.com/news-releases/news-release-details/biogen-and-eisai-discontinue-phase-3-engage-and-emerge-trials. Accessed November 8, 2021.
- Knopman DS, Jones DT, Greicius MD. Failure to demonstrate efficacy of aducanumab: an analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement. 2021;17(4):696–701. doi:10.1002/alz.12213
- Biogen. Biogen plans regulatory filing for aducanumab in Alzheimer’s disease based based on new analysis of larger dataset from phase 3 studies. Biogen. 2019. Available from: https://investors.biogen.com/news-releases/news-release-details/biogen-plans-regulatory-filing-aducanumab-alzheimers-disease. Accessed November 20, 2021.
- Tampi RR, Forester BP, Aducanumab: AM. Evidence from clinical trial data and controversies. Drugs Context. 2021;10:1–9. doi:10.7573/dic.2021-7-3
- Belluck P, Robbins R. Three F.D.A. advisers resign over approval of Alzheimer’s drug. The New York Times; 2021. Available from: https://www.nytimes.com/2021/06/10/health/aduhelm-fda-resign-alzheimers.html. Accessed April 9, 2022.
- U.S. Food and Drug Administration (FDA). Accelerated Approval; 2021. Available from: https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/accelerated-approval. Accessed April 10, 2022.
- Biogen. European medicines agency accepts Biogen’s aducanumab marketing authorization application for Alzheimer’s disease. Biogen; 2020. Available from: https://investors.biogen.com/news-releases/news-release-details/european-medicines-agency-accepts-biogens-aducanumab-marketing. Accessed November 23, 2021.
- Mahase E. Aducanumab: European agency rejects Alzheimer’s drug over efficacy and safety concerns. BMJ. 2021;375:n3127. doi:10.1136/bmj.n3127
- Biogen. Update on regulatory review of aducanumab in the European Union. Biogen. Available from: https://investors.biogen.com/news-releases/news-release-details/update-regulatory-submission-aducanumab-european-union.; 2021. Accessed April 9, 2022.
- Castrillo-Viguera C, Chalkias S, Burkett P, et al. EMBARK: a Phase 3b, open-label, single-arm, safety study to evaluate the long-term safety and efficacy of aducanumab in eligible participants with Alzheimer’s disease (2448). Neurology. 2021;96(15Supplement):2448.
- ClinicalTrials.gov. A study to evaluate safety and tolerability of aducanumab in participants with Alzheimer’s disease who had previously participated in the aducanumab studies 221AD103, 221AD301, 221AD302 and 221AD205; 2020. Available from: https://clinicaltrials.gov/ct2/show/NCT04241068?term=aducanumab&draw=2&rank=3. Accessed November 13, 2021.
- Biogen. ADUHELM ICARE AD-US study, the first real-world observational phase 4 study in Alzheimer’s disease at AAIC 2021. Biogen; 2021. Available from: https://investors.biogen.com/news-releases/news-release-details/biogen-and-eisai-announce-design-aduhelm-icare-ad-us-study-first. Accessed November 13, 2021.
- ClinicalTrials.gov. An observational study of aducanumab-avwa in participants with Alzheimer’s disease in the US - full text view - ClinicalTrials.gov; 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT05097131?term=aducanumab&draw=2&rank=10. Accessed November 13, 2021.
- Biogen. Update on the phase 4 confirmatory study of ADUHELM®. Biogen; 2021. Available from: https://investors.biogen.com/news-releases/news-release-details/update-phase-4-confirmatory-study-aduhelmr. Accessed December 17, 2021.
- Biogen. Update on the Phase 4 ENVISION confirmatory study of ADUHELM® | biogen. Biogen; 2022. Available from: https://investors.biogen.com/news-releases/news-release-details/update-phase-4-envision-confirmatory-study-aduhelmr. Accessed January 29, 2022.
- U.S. Food and Drug Administration (FDA). ADUHELM: full prescribing information; 2021. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761178s003lbl.pdf. Accessed November 14, 2021.
- Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease — one peptide, two pathways. Nat Rev Neurol. 2020;16(1):30–42. doi:10.1038/s41582-019-0281-2
- Sperling RA, Jack CR, Black SE, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7(4):367–385. doi:10.1016/j.jalz.2011.05.2351
- Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):322–333. doi:10.1056/nejmoa1304839
- Avgerinos KI, Ferrucci L, Kapogiannis D. Effects of monoclonal antibodies against amyloid-β on clinical and biomarker outcomes and adverse event risks: a systematic review and meta-analysis of Phase III RCTs in Alzheimer’s disease. Ageing Res Rev. 2021;68:101339. doi:10.1016/j.arr.2021.101339
- Penninkilampi R, Brothers HM, Eslick GD. Safety and efficacy of anti-amyloid-β Immunotherapy in Alzheimer’s disease: a systematic review and meta-analysis. J Neuroimmune Pharmacol. 2017;12(1):194–203. doi:10.1007/s11481-016-9722-5
- Gleason A, Ayton S, Bush AI. Unblinded by the light: amyloid-related imaging abnormalities in Alzheimer’s clinical trials. Eur J Neurol. 2021;28(1):e1. doi:10.1111/ene.14484
- Biogen. New phase 3 data show positive correlation between ADUHELMTM treatment effect on biomarkers and reduction in clinical decline in Alzheimer’s disease | biogen. Biogen; 2021. Available from: https://investors.biogen.com/news-releases/news-release-details/new-phase-3-data-show-positive-correlation-between-aduhelmtm?cid=osm-lkdn-ctad-um-alz-111121. Accessed November 28, 2021.
- Fleck LM. Alzheimer’s and aducanumab: unjust profits and false hopes. Hastings Cent Rep. 2021;51(4):9–11. doi:10.1002/hast.1264
- Alexander GC, Knopman DS, Emerson SS, et al. Revisiting FDA approval of Aducanumab. N Engl J Med. 2021;385(9):769–771. doi:10.1056/nejmp2110468
- La JR, Visani AV, Baker SL, et al. Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci Transl Med. 2020;12:524. doi:10.1126/scitranslmed.aau5732
- Leuzy A, Smith R, Cullen NC, et al. Biomarker-based prediction of longitudinal tau positron emission tomography in Alzheimer disease. JAMA Neurol. 2021;79(2):149–158. doi:10.1001/jamaneurol.2021.4654
- Mukhopadhyay S, Banerjee D. A primer on the evolution of aducanumab: the first antibody approved for treatment of Alzheimer’s disease. J Alzheimers Dis. 2021;83(4):1537–1552. doi:10.3233/JAD-215065
- Alexander GC, Emerson S, Kesselheim AS. Evaluation of aducanumab for Alzheimer disease: scientific evidence and regulatory review involving efficacy, safety, and futility. J Am Med Assoc. 2021;325(17):1717–1718. doi:10.1001/jama.2021.3854
- Alzheimer’s Forum Association. Aducanumab approved to treat Alzheimer’s disease. Alzforum; 2021. Available from: https://www.alzforum.org/news/research-news/aducanumab-approved-treat-alzheimers-disease. Accessed January 6, 2022.
- Institute for Clinical and Economic Review. ICER issues statement on the FDA’s approval of aducanumab for Alzheimer’s disease. ICER; 2021. Available from: https://icer.org/news-insights/press-releases/icer-issues-statement-on-The-fdas-approval-of-aducanumab-for-alzheimers-disease/. Accessed November 11, 2021.
- Biogen. Biogen announces reduced price for ADUHELM® to improve access for patients with early Alzheimer’s disease | biogen. Biogen; 2021. Available From: https://investors.biogen.com/news-releases/news-release-details/biogen-announces-reduced-price-aduhelmr-improve-access-patients. Accessed January 9, 2022.
- Whittington MD, Campbell JD, Rind D, Fluetsch N, Lin GA, Pearson SD. Cost-effectiveness and value-based pricing of aducanumab for patients with early Alzheimer disease. Neurology. 2022;98(9):e968–e977. doi:10.1212/WNL.0000000000013314
- Cummings J, Aisen P, Apostolova LG, Atri A, Salloway S, Weiner M. Aducanumab: appropriate use recommendations. J Prev Alzheimers Dis. 2021;8(4):398–410. doi:10.14283/jpad.2021.41
- Centers for Medicare & Medicaid Services. Monoclonal antibodies directed against amyloid for the treatment of Alzheimer’s disease; 2022. Available from: https://www.cms.gov/newsroom/press-releases/cms-finalizes-medicare-coverage-policy-monoclonal-antibodies-directed-against-amyloid-treatment. Accessed April 11, 2022.
- Belluck P. Medicare officially limits coverage of aduhelm to patients in clinical trials. The New York Times; 2022. Available from: https://www.nytimes.com/2022/04/07/health/aduhelm-medicare-alzheimers.html. Accessed April 10, 2022.
- Butcher L. Mixed response to the CMS decision on coverage for aducanumab. Neurol Today. 2022;22(3):1,16–17. doi:10.1097/01.nt.0000821676.02301.7a
- Association AF. On aduhelm, medicare agency gets pressure from all sides | ALZFORUM. Alzforum; 2022. Available from: https://www.alzforum.org/news/community-news/aduhelm-medicare-agency-gets-pressure-all-sides. Accessed January 27, 2022.
- Association AF. CMS plans to limit aduhelm coverage to clinical trials | ALZFORUM. Alzforum; 2022. Available from: https://www.alzforum.org/news/research-news/cms-plans-limit-aduhelm-coverage-clinical-trials. Accessed January 27, 2022.
- Bednar MM. Combination therapy for Alzheimer’s disease and related dementias. In: Progress in Molecular Biology and Translational Science. Academic Press; Vol. 168. 2019:289–296. doi:10.1016/bs.pmbts.2019.10.001
- Lythgoe MP, Prasad V. How the US Food and Drug Administration’s approval of aducanumab for Alzheimer’s disease has implication for oncology and beyond. Eur J Cancer. 2021;157:68–70. doi:10.1016/j.ejca.2021.08.012
- Gyawali B, Ross JS, Kesselheim AS. Fulfilling the mandate of the US Food and Drug Administration’s accelerated approval pathway: the need for reforms. JAMA Intern Med. 2021;181(10):1275–1276. doi:10.1001/jamainternmed.2021.4604
- Silvestro S, Valeri A, Mazzon E. Aducanumab and its effects on tau pathology: is this the turning point of amyloid hypothesis? Int J Mol Sci. 2022;23(4):2011. doi:10.3390/ijms23042011
- Association AF. Aduhelm lowers tau; registry to track real-world performance | ALZFORUM. Alzforum; 2021. Available from: https://www.alzforum.org/news/conference-coverage/aduhelm-lowers-tau-registry-track-real-world-performance. Accessed February 1, 2022.