
Abstract
Background
Understanding the genomic determinants associated with metastasis in colorectal cancers (CRCs) provides crucial clues for improving patient care.
Patients and methods
In this study, we performed whole-exome sequencing as well as RNA sequencing analyses on five pairs of primary and liver metastasized samples from CRC patients together with blood/normal control samples for each pair.
Results
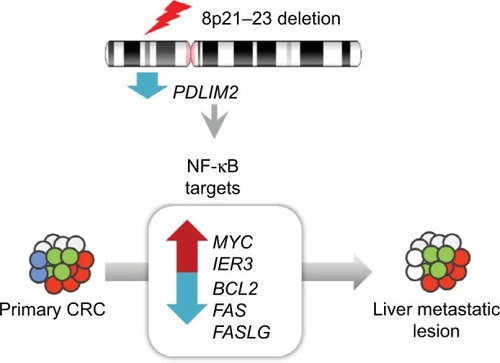
We identified genomic deletions in the region of 8p21-23 (q value <0.01) from analysis of recurrent regions with copy number variations in both primary and matched metastatic lesions. Consistent with this result, we found significantly decreased expression levels of all 12 genes (ADAMDEC1, C8orf80, CLDN23, EPHX2, GFRA2, NEFL, NEFM, PDLIM2, PTK2B, SCARA5, SLC18A1 and STMN4) located within this region (adjusted P<0.01). Notably, the mRNA levels of PDLIM2, a key regulator of well-known cancer-associated genes including the proto-oncogene c-MYC, an early response gene IER3, and regulators of apoptosis such as BCL2, FAS, and FASLG, were highly downregulated in tumors compared to normal tissues.
Conclusion
Taken together, our findings uncovered various genomic alterations potentially leading to metastasis in CRC and provide important insights into the development of potential therapeutic targets for preventing metastatic progression of CRC.
Introduction
Colorectal cancer (CRC) is one of the most common malignancies and a leading cause of cancer-related death worldwide.Citation1 The survival outcome of CRC has steadily improved; however, metastasis is the major cause of death in patients with CRC and the survival rate of these patients remains low. Metastasis is found in ~20% of CRC patients at diagnosis, and ~50% of patients develop metastasis during the disease course.Citation2 Metastasis seems to be caused by various factors, including genomic alterations.Citation3–Citation7 Therefore, identification of key factors, especially crucial genomic determinants, associated with the metastatic process plays a significant role in the development of therapeutic strategies for effective treatment of patients with metastatic CRC (mCRC).
Recent revolutionary progress of next-generation sequencing technology has facilitated identification of various genomic alterations associated with metastasis in CRCs. A recent genomic analysis with 230 cancer-associated gene panels showed a high degree of concordance for recurrent mutations such as APC, TP53, and KRAS between primary and metastatic tumors in CRC.Citation8 In addition, targeted exome sequencing of 750 cancer-associated genes demonstrated that broad regions of allelic imbalance and focal copy changes are generally shared between the primary tumor and metastases.Citation9 In addition, RNA sequencing (RNA-seq) is a powerful tool for quantifying, comparing, and annotating the transcriptome with high resolution.Citation10 Using RNA-seq, comparative analyses of gene expression profiles can identify alterations of genes associated with metastatic potential.Citation11 Despite several previous efforts, the associations between genomic alterations and their functional impact on the induction of metastasis from primary CRCs in patients have not yet been clearly addressed.
Here, we performed whole-exome sequencing (WES) and RNA-seq with five pairs of primary and metastatic samples from patients with metastasis of CRC and examined comprehensive somatic alterations, copy number variations (CNVs), and gene expression changes in these patient samples. Especially, we identified a genomic region harboring significant alterations and enriched copy number deletions (in three of five pairs) occurring in both primary and metastatic samples. Gene expression analysis identified major genes involved in this change including PDLIM2, which is associated with abnormal activation of the nuclear factor-kappa B (NF-κB) signaling pathway. Elucidation of involved target genes in this pathway might contribute to understanding of the molecular mechanism underlying tumorigenesis and metastasis of CRC.
Patients and methods
Patients and sample preparation
A total of five patients with mCRC who underwent surgery of primary CRC from February 2004 to January 2010 at Sam-sung Medical Center were included (). All patients had histologically confirmed primary adenocarcinoma and underwent surgery for the primary tumor and metastatic lesions.
Table 1 Patient’s characteristics of five CRCs
Specimens from normal colorectal tissue, primary tumor, and matched metastatic tumor were obtained from five CRC patients for RNA-seq (Table S1). In addition, venous blood samples were obtained from these patients. All patients provided written informed consent to participate, and the study was approved by Samsung Medical Center Institutional Review Board (no SMC 2013-04-021).
Isolation of genomic DNA and quality control
Formalin-fixed paraffin-embedded (FFPE) sections were deparaffinized and lysed with proteinase K for 4 h, and genomic DNA was extracted using a Maxwell 16 CSC DNA FFPE kit and Maxwell MDx automation instrument (Promega Corporation, Fitchburg, WI, USA). DNA concentration and purity of all genomic DNA samples were measured using a Nanodrop 8000 UV-Vis spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) and Qubit 2.0 Fluorometer (Thermo Fisher Scientific). To estimate DNA degradation, DNA median size and ΔCt value were measured by 2200 TapeStation Instrument and real-time polymerase chain reaction (PCR) (Agilent Technologies, Santa Clara, CA, USA). To improve sequencing quality, we used strict sample quality thresholds for extracted DNA: 1) purity: 260/280>1.8, 260/230>1.8; 2) total amount >250 ng; and 3) degradation: ΔCt value <2.0 or DNA median size >0.35 kb.
Exome sequencing
Genomic DNA (1 µg) from each FFPE sample was sheared by Covaris S220 (Covaris, MA, USA) and used for construction of a library with SureSelect XT Human All Exon v5 and the SureSelect XT reagent kit, HSQ (Agilent Technologies) according to the manufacturer’s protocol. This kit is designed to enrich 335,756 exons of 21,058 genes, covering ~71 Mb of the human genome. After the enriched exome libraries were multiplexed, the libraries were sequenced on a HiSeq 2500 sequencing platform (Illumina, San Diego, CA, USA). Briefly, a paired-end DNA sequencing library was prepared through gDNA shearing, end-repair, A-tailing, paired-end adaptor ligation, and amplification. After hybridization of the library with bait sequences for 16 h, the captured library was purified and amplified with an index barcode tag and the quality and quantity of the library were measured. Sequencing of the exome library was carried out using the 100 bp paired-end mode of the TruSeq Rapid PE Cluster kit and TruSeq Rapid SBS kit (Illumina).
Whole transcriptome sequencing
Library construction for whole transcriptome sequencing was performed using a Truseq RNA sample preparation v2 kit (Illumina). Isolated total RNA (2 µg) was used in a reverse transcription reaction with poly(dT) primers with SuperScriptTM II reverse transcriptase (Thermo Fisher Scientific) according to the manufacturer’s protocols. Briefly, an RNA-seq library was prepared through cDNA amplification, end-repair, 3¢ end adenylation, adapter ligation, and amplification. Quality and quantity of the library were measured using Bioanalyzer and Qubit. Sequencing of the transcriptome library was carried out using the 100 bp paired-end mode of the TruSeq Rapid PE Cluster kit and TruSeq Rapid SBS kit.
Exome-seq data analysis
Sequencing reads were aligned to the UCSC hg19 reference genome (downloaded from http://genome.ucsc.edu) using Burrows-Wheeler Aligner,Citation12 version 0.6.2 with default settings. PCR duplications were marked by Picard tools 1.8 (http://picard.sourceforge.net/), followed by data cleanup with GATK and variant identification with GATK-2.2.9.Citation13 Point mutations were identified using the MuTect tool (https://github.com/broadinstitute/mutect) with paired samples. Perl script and Annovar were used to annotate variants. CNVs were identified by EXCAVATOR.Citation14 Significant focal somatic copy number alterations were summarized by GISTIC analysis.Citation15
RNA-seq data analysis
The reads from the FASTQ files were mapped against the hg19 human reference genome using TopHat Version 2.0.6 (http://tophat.cbcb.umd.edu/). Raw read counts mapped to genes were measured with the BAM format file by HTSeq Version 0.6.1.Citation16 A total of 18,161 coding genes were subjected to the measurement of transcript abundance, and genes with low expression were filtered out based on the criteria of maximum read count >20 across all samples. Read counts were normalized by the Trimmed Mean of M-values normalization method. Differentially expressed genes were identified using the DESeq R package (www.huber.embl.de/users/anders/DESeq/). Differentially expressed genes for each patient sample were extracted using the Cufflink and Cuffdiff analysis pipeline (http://cole-trapnell-lab.github.io/cufflinks/). Gene set enrichment tests were performed using the GAGE R toolCitation17 to identify significant pathways that are likely to be involved in primary CRC and its metastasis.
Clonality analysis
Subclones were obtained by clustering cancer cell fractions (CCFs) using PyClone, which can deconvolve the tumor into subclones using a hierarchical Bayesian clustering model.Citation18 The input data were generated with somatic single-nucleotide variants (SNVs) detected by MuTect and CNVs corresponding to SNVs. Step filtering of the SNVs with large credible intervals in the previous stage was applied to remove those with noninformative posterior distributions. The subclones were compared between primary tumors and each lymph node using the density plot of CCFs.
Results
Sequencing of patients with mCRC
We carried out WES and RNA-seq on five pairs of blood samples together with primary and matched mCRC tumor samples (Table S1 and ). After WES, we obtained a total of 1.0 billion paired-end reads from 15 samples including blood and matched primary tumors and metastases. Based on sequence analysis, 85.94±4.37% of the total reads were uniquely aligned to the human genome reference. The average mean target depth for all samples was 119.2x (SD 24.7). Sequencing reads covered whole exome regions with at least 99.2% coverage. We obtained a total of 565 million paired-end reads from RNA-seq. Across all RNA-seq data from five samples, 82.68±3.13% of total reads were uniquely aligned to the human genome reference.
Figure 1 Schematic diagram of the workflow of CRC genome and transcriptome analysis using NGS.
Exome and transcriptome profiles show similar concordance rates between primary and metastatic lesions
We first identified somatic mutations for each primary and paired metastatic lesion from WES data. There was overlap in the number of mutations detected from primary and metastatic regions (114 primary, 176 metastatic, and 63 shared mutations for CRC1; 142, 201, and 78 for CRC2; 79, 30, and 10 for CRC3; 126, 76, and six for CRC4; and 143, 172, and 81 for CRC5; Figure S1). An average of 36.9% of somatic mutations of primary tumors (corresponding to 36.3% of metastatic lesions) were concordant in primary and matched metastatic tumors (CRC1: 55.3% of primary lesions and 35.8% of metastatic lesions; CRC2: 54.9 and 38.8%; CRC3: 12.7 and 33.3%; CRC4: 4.8 and 7.9%; CRC5: 56.6 and 47.1%; ). Somatic mutations in APC and TP53, known to be highly mutated genes in CRC, were identified in both primary and metastasized lesions of CRC5 and CRC1 (Table S2). In addition, we found that MAP2K4 and TCF7L2 were mutated in both lesions of these samples. MAP2K4 is known to function as a tumor suppressor.Citation19 Consistent with this notion, the MAP2K4 R355T mutation located within the kinase domain was also found in several other cancer types including colorectal, lung adenocarcinoma, melanoma, and ovarian cancers.Citation20 As a previous study showed that mutant TCF7L2 is closely associated with aberrant activation of the Wnt signaling pathway in CRC, we speculate that the TCF7L2 R363W mutation can affect the transcriptional repressors of cell growth-promoting genes.Citation21
Figure 2 Genome and transcriptome changes (including mutations, CNVs, and expressed genes) found in primary tumor only, in metastasis (Meta) only, and in both.
Abbreviations: CCF, cancer cell fraction; CNVs, copy number variations; CRC, colorectal cancer.
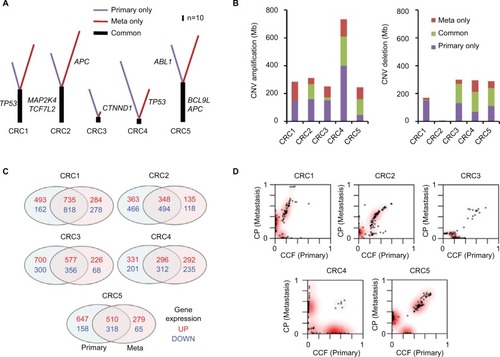
Next, we examined CNVs in five paired CRC samples by analyzing WES data (Table S3 and Figure S2). Copy number amplification and deletion regions were compared between primary and metastasis samples (). A total of 2,557 Mb of genomic regions were altered in primary samples of five CRC patients (amplifications of 1,355 Mb and deletions of 1,202 Mb), and 3,280 Mb of genomic regions were altered in metastasis samples (amplifications of 1,826 Mb and deletions of 1,454 Mb). Overall, 456 of 1,355 Mb with copy number alterations (33.7%) in the primary samples were concordantly amplified and 473 Mb of 885 Mb (53.4%) were concordantly deleted.
By data analyses of RNA-seq results, genes showing significantly altered mRNA expression were selected based on the cutoff of high and low levels of log2 ratio ≥2.0 and ≤–2.0, respectively, for each individual primary–metastasis pair (). On average, 1,000 genes from five primary tumors were found to be highly expressed; among them, 493.2 (49.2%) overlapped with highly expressed genes in metastatic lesions. For each primary tumor, an average of 717 genes were found to be downregulated and 459.6 (64.1%) of them over-lapped with the expression in metastatic lesions. These results show that the pattern of gene expression changes was more concordant than that of mutations and CNVs between primary and metastasis samples. Among 291 selected genes showing significantly altered expression between normal and primary tumors (adjusted [adj] P-value <0.0001; Figure S3A), only five (NKX2-3, GATA4, VIP, EDN1, and MMP3) genes showed significant differences in the levels of mRNA expression between primary tumors and liver metastases (Figure S3B), suggesting that these genes might be involved in metastasis in these patients. Notably, the expression of GATA4 and EDN1 was upregulated (Figure S3C), whereas that of NKX2-3, VIP, and MMP3 was significantly downregulated (Figure S3D) in metastatic samples. Among them, VIP has been reported as a cytokine in an early metastatic stage of prostate cancer through the involvement of the NF-κB/MMPs-RECK/E-cadherin pathway.Citation22 This suggests that this gene may also be involved in the metastatic cascade of CRC through those pathways.
To further investigate the functional importance of differential mRNA expression, we performed gene set enrichment analysis with RNA-seq datasets and identified signaling pathways that were specifically induced in primary tumors (adj P-value <0.0001; Figure S4). Notably, we found high enrichment of immune-related pathways, including “T-cell receptor signaling”, “B-cell receptor signaling”, “cell adhesion molecules”, “antigen processing and presentation”, and “chemokine signaling pathway“ in primary tumors, suggesting that several signaling pathways related to the immune response play a crucial role in metastatic spread of tumor cells. Specifically, the expression levels of eight genes (CD247, CD3D, CD3E, CD3G, CD8A, CHP2, ITK, and PIK3CG) belonging to T-cell receptor signaling were significantly lower in three CRC datasets (Figure S5A). Seven genes (BTK, CD79A, CHP2, CR2, PIK3CG, PLCG2, and PRKCB) belonging to B-cell receptor signaling were also significantly downregulated in primary tumor samples (Figure S5B). The expression pattern of those genes in CRC appears to be different from that in other types of cancer (Figure S5C and D). Importantly, the patient group with a low level of expression in these sets of these genes showed poor clinical outcome (Figure S5E).
We also examined the tumor clonality of each primary and matched metastatic tumor. All of the CRCs had common subclones between primary and metastatic lesions as well as additional subclones in metastatic lesions (). CRC2 and CRC4 had more subclones in metastatic lesions than the other patients, indicating clonal evolution with more heterogeneity.
8q23.1 copy number deletions are correlated with decreased expression of corresponding genes
Next, we examined focal somatic copy number alterations by performing GISTIC analysisCitation15 and showed that the 8q23.1 region was significantly deleted (q value <0.01) in both primary and metastatic lesions (). Consistent with these data, out of 650 significantly altered genes (adj P-value <0.01) identified in both primary and metastatic lesions, we found that the mRNA expression levels of 12 genes (ADAMDEC1, C8orf80, CLDN23, EPHX2, GFRA2, NEFL, NEFM, PDLIM2, PTK2B, SCARA5, SLC18A1, and STMN4) located within the 8q23.1 region were greatly decreased ( and Table S4), suggesting that this genomic region might be highly dysregulated in the course of CRC progression and possibly associated with liver metastasis.Citation23 In addition, we found four chromosomal regions – 2q36.1, 8q21.11, 17p13.2, and 20p13 – that were commonly amplified in primary and metastatic lesions (Figure S6).
Figure 3 Copy number variations in primary tumors and liver metastases.
Abbreviations: CRC, colorectal cancer; DEG, differentially expressed genes.
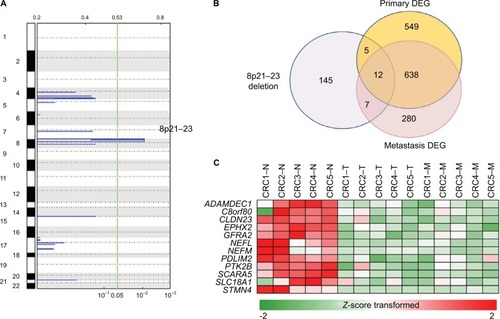
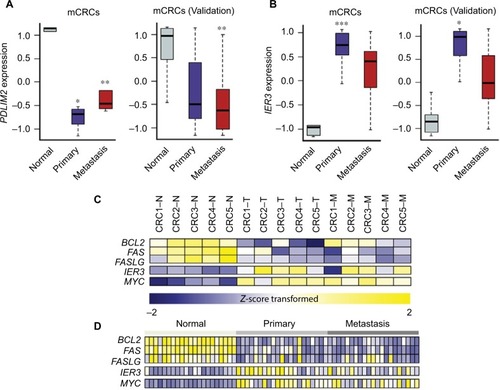
Among 12 genes in the 8q23.1 region, PDLIM2 was of great interest because its expression was most significantly downregulated in both primary tumors (log2 fold change [FC] =−2.37; adj P=7.87e−4) and metastatic lesions (log2 FC =−1.76; adj P=9.14e−3) (). Furthermore, recent analysis of 276 CRC patient samples revealed a strong correlation between copy number alteration of PDLIM2 and its gene expression (The Cancer Genome Atlas) (Figure S7).Citation24 The expression level of PDLIM2 was also decreased in an independent set of mCRCs as a validation data set (). PDLIM2 is known to function as an essential terminator of transcription factor NF-κB activation by binding to p65 and subsequently promoting its ubiquitination.Citation25,Citation26 Consistent with these findings, PDLIM2 expression was found to be repressed in various human CRC cell lines.Citation27 Therefore, we further examined whether there was any correlation between PDLIM2 loss and changes in gene expression of NF-κB target genes in samples harboring PDLIM2 loss. Among 433 NF-κB target genes, the expression levels of 51 genes were significantly altered (adj P<0.01), indicating that a large number of genes (P=0.03; hypergeometric test) were affected (Table S5 and Figure S8). Notably, we found that, whereas apoptosis inducers including BCL2, FAS, and FASLG were significantly downregulated (log2 FC >2.0; adj P<0.001) in primary tumors or metastatic lesions (), the oncogene MYC was significantly upregulated (log2 FC >2.0; adj P<0.001) (). Furthermore, the expression level of an early response gene, IER3, was increased in primary tumors (log2 FC =2.60; adj P=0.04) but not in metastatic lesions (log2 FC =2.05; adj P=0.07) (). The expression level of IER3 was also decreased in primary tumors of an independent set of mCRCs as a validation test ().
Figure 4 Gene expression values (Z-score transformed) of PDLIM2, IER3, and other target genes of NF-kB in mCRCs.
Abbreviations: CRCs, colorectal cancers; mCRCs, metastatic CRCs.
Discussion
In this study, we cataloged various genomic alterations occurring in five paired primary and mCRC patient samples by WES and RNA-seq-based analyses. Notably, genomic deletions within the 8p21–23 region, which was previously shown to be altered in CRCs, were commonly identified in three pairs of CRC samples. Among 12 genes that were transcriptionally affected by this deletion, PDLIM2 was of great interest because several lines of evidence indicate that a lower level of PDLIM2 expression is closely associated with abnormal activation of the NF-κB signaling pathway.Citation25,Citation28,Citation29 Specifically, PDLIM2 appears to function as a key negative regulator of NF-κB activation by direct binding to NF-κB p65. Consistent with this notion, we found that the expression levels of several target genes of NF-κB including c-MYC, BCL2, FAS, and FASLG were significantly altered in the samples harboring deleted PDLIM2 compared to the normal matched samples. The significance of PDLIM2 expression in CRC is further supported by data showing reduced expression in various CRC cell lines, suggesting that PDLIM2 might function as a key negative regulator in CRC tumorigenesis.Citation27 Furthermore, NF-κB p65 has been shown to modulate immune responses and activation of RELA is positively associated with multiple types of cancer.Citation30–Citation33 Consistent with these findings, enrichment of immune-related signaling pathways was observed in these pairs of samples. Therefore, we speculate that a potential inhibitor of NF-κB such as bortezomib might be effective in the treatment of a subset of mCRC and recurrent CRC harboring PDLIM2 deletion.Citation34,Citation35
In addition, we propose that IER3, an early response gene, might play a crucial role in metastatic events during CRC progression (). IER3, also known as IEX-1, is known to function as a key regulator of various cellular responses including cell apoptosis, proliferation, differentiation, and tumorigenesis.Citation36 For example, in the absence of cellular stress, IER3 serves to enhance cell growth, whereas under various cellular stress conditions, it seems to induce apoptosis.Citation37 In addition, IER3 is transcriptionally regulated by multiple transcription factors including p53, NF-κB/rel, and c-Myc.Citation38 IER3 seems to disturb certain survival pathways involving NF-κB.Citation39 Several studies have demonstrated that abnormal IER3 expression is associated with various human tumors, although its mechanism of action is still unclear. In addition, previous reports suggested that IER3 is involved in regulating T-cell homeostasis during immune responsesCitation40,Citation41 and can prevent activation-induced death of T cells.Citation38
Figure 5 Genomic and transcriptomic changes in metastatic events during CRC progression.
Recently, Robinson et alCitation42 have presented comprehensively genomic landscape and microenvironment of metastatic cancer through WES and RNA-seq. One of their key findings is that proliferation signature appears to be activated, while immune response is inactivated in CRC. In regard to this point, our results agree well with their finding. Our data clearly demonstrated that immune-related signaling pathways are highly enriched (adj P-value <0.0001), and T- and B-cell receptor genes belonging to those pathways are significantly downregulated, indicating the feature of immune escape occurring in the metastatic tumors. In addition, we speculate that loss of PDLIM2 found in metastatic tumors could be a potential cause of these altered immune signaling pathways based on the findings from two recent reports showing that PDLIM2 is responsible for the epithelial–mesenchymal transition and immune response.Citation26,Citation28
Conclusion
We demonstrated comprehensive genomic and transcriptomic changes in patients with mCRC. By association between a specific copy number change and its affected pathway, we suggest markers involved in metastatic potential. We also successfully validated these findings in an independent set of mCRCs. In conclusion, PDLIM2 and IER3 could be potential therapeutic markers for observing tumor progression in patients with mCRC.
Acknowledgments
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI15C1593; YBC); by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT, & Future Planning (2017R1A2B1007347; J-GJ); and by the Korea Health Technology R&D Project through the KHIDI, funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI15C2353; JC). The biospecimens for this study were provided by Samsung Medical Center Biobank.
Disclosure
The authors report no conflicts of interest in this work.
References
- SiegelRMaJZouZJemalACancer statistics, 2014CA Cancer J Clin201464192924399786
- FengQLiangLRenLA specific KRAS codon 13 mutation is an independent predictor for colorectal cancer metachronous distant metastasesAm J Cancer Res20155267468825973306
- YamasakiMTakemasaIKomoriTThe gene expression profile represents the molecular nature of liver metastasis in colorectal cancerInt J Oncol200730112913817143521
- KiDHJeungHCParkCHWhole genome analysis for liver metastasis gene signatures in colorectal cancerInt J Cancer200712192005201217640062
- Rodriguez-SalasNDominguezGBarderasRClinical relevance of colorectal cancer molecular subtypesCrit Rev Oncol Hematol201710991928010901
- NystromHNarediPBerglundAPalmqvistRTavelinBSundMLiver-metastatic potential of colorectal cancer is related to the stromal composition of the tumourAnticancer Res201232125183519123225415
- NadalCMaurelJGasconPIs there a genetic signature for liver metastasis in colorectal cancer?World J Gastroenterol200713445832584417990349
- BrannonARVakianiESylvesterBEComparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesionsGenome Biol201415845425164765
- TanIBMalikSRamnarayananKHigh-depth sequencing of over 750 genes supports linear progression of primary tumors and metastases in most patients with liver-limited metastatic colorectal cancerGenome Biol2015163225808843
- OzsolakFMilosPMRNA sequencing: advances, challenges and opportunitiesNat Rev Genet2011122879821191423
- LeeJRKwonCHChoiYTranscriptome analysis of paired primary colorectal carcinoma and liver metastases reveals fusion transcripts and similar gene expression profiles in primary carcinoma and liver metastasesBMC Cancer20161653927461012
- LiHDurbinRFast and accurate short read alignment with Burrows-Wheeler transformBioinformatics200925141754176019451168
- McKennaAHannaMBanksEThe Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing dataGenome Res20102091297130320644199
- MagiATattiniLCifolaIEXCAVATOR: detecting copy number variants from whole-exome sequencing dataGenome Biol20131410R12024172663
- MermelCHSchumacherSEHillBMeyersonMLBeroukhimRGetzGGISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancersGenome Biol2011124R4121527027
- AndersSPylPTHuberWHTSeq – a Python framework to work with high-throughput sequencing dataBioinformatics201531216616925260700
- LuoWFriedmanMSSheddenKHankensonKDWoolfPJGAGE: generally applicable gene set enrichment for pathway analysisBMC Bioinformatics20091016119473525
- RothAKhattraJYapDPyClone: statistical inference of clonal population structure in cancerNat Methods201411439639824633410
- DavisSJChoongDYRamakrishnaMRylandGLCampbellIGGorringeKLAnalysis of the mitogen-activated protein kinase kinase 4 (MAP2K4) tumor suppressor gene in ovarian cancerBMC Cancer20111117321575258
- AhnYHYangYGibbonsDLMap2k4 functions as a tumor suppressor in lung adenocarcinoma and inhibits tumor cell invasion by decreasing peroxisome proliferator-activated receptor gamma2 expressionMol Cell Biol201131214270428521896780
- TangWDodgeMGundapaneniDMichnoffCRothMLumLA genome-wide RNAi screen for Wnt/beta-catenin pathway components identifies unexpected roles for TCF transcription factors in cancerProc Natl Acad Sci U S A2008105289697970218621708
- Fernandez-MartinezABBajoAMSanchez-ChapadoMPrietoJCCarmenaMJVasoactive intestinal peptide behaves as a pro-metastatic factor in human prostate cancer cellsProstate200969777478619189304
- Macartney-CoxsonDPHoodKAShiHJMetastatic susceptibility locus, an 8p hot-spot for tumour progression disrupted in colorectal liver metastases: 13 candidate genes examined at the DNA, mRNA and protein levelBMC Cancer2008818718590575
- Cancer Genome Atlas NetworkComprehensive molecular characterization of human colon and rectal cancerNature2012487740733033722810696
- TanakaTGrusbyMJKaishoTPDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunitNat Immunol20078658459117468759
- BoweRACoxOTAyllonVPDLIM2 regulates transcription factor activity in epithelial-to-mesenchymal transition via the COP9 signalosomeMol Biol Cell201425118419524196835
- QuZYanPFuJDNA methylation-dependent repression of PDZ-LIM domain-containing protein 2 in colon cancer and its role as a potential therapeutic targetCancer Res20107051766177220145149
- QuZFuJMaHPDLIM2 restricts Th1 and Th17 differentiation and prevents autoimmune diseaseCell Biosci2012212322731402
- FuJYanPLiSQuZXiaoGMolecular determinants of PDLIM2 in suppressing HTLV-I Tax-mediated tumorigenesisOncogene201029496499650720838382
- HaydenMSWestAPGhoshSNF-kappaB and the immune responseOncogene200625516758678017072327
- JinXWangZQiuLPotential biomarkers involving IKK/RelA signal in early stage non-small cell lung cancerCancer Sci200899358258918215193
- YanMXuQZhangPZhouXJZhangZYChenWTCorrelation of NF-kappaB signal pathway with tumor metastasis of human head and neck squamous cell carcinomaBMC Cancer20101043720716363
- HoeselBSchmidJAThe complexity of NF-kappaB signaling in inflammation and cancerMol Cancer2013128623915189
- MackayHHedleyDMajorPA phase II trial with pharmacodynamic endpoints of the proteasome inhibitor bortezomib in patients with metastatic colorectal cancerClin Cancer Res200511155526553316061869
- KozuchPSRocha-LimaCMDragovichTBortezomib with or without irinotecan in relapsed or refractory colorectal cancer: results from a randomized phase II studyJ Clin Oncol200826142320232618467723
- WuMXUstyugovaIVHanLAkilovOEImmediate early response gene X-1, a potential prognostic biomarker in cancersExpert Opin Ther Targets201317559360623379921
- SchillingDPittelkowMRKumarRIEX-1, an immediate early gene, increases the rate of apoptosis in keratinocytesOncogene200120557992799711753682
- WuMXRoles of the stress-induced gene IEX-1 in regulation of cell death and oncogenesisApoptosis200381111812510147
- ArltAKruseMLBreitenbroichMThe early response gene IEX-1 attenuates NF-kappaB activation in 293 cells, a possible counter-regulatory process leading to enhanced cell deathOncogene200322213343335112761504
- ZhangYSchlossmanSFEdwardsRAOuCNGuJWuMXImpaired apoptosis, extended duration of immune responses, and a lupus-like autoimmune disease in IEX-1-transgenic miceProc Natl Acad Sci U S A200299287888311782530
- ZhangYFinegoldMJPorteuFKantetiPWuMXDevelopment of T-cell lymphomas in Emu-IEX-1 miceOncogene200322446845685114534530
- RobinsonDRWuYMLonigroRJIntegrative clinical genomics of metastatic cancerNature2017548766729730328783718