
Abstract
Background
Our previous study has shown that cadmium (Cd) exposure is not only a risk factor for nasopharyngeal carcinoma (NPC), but also correlated with the clinical stage and lymph node metastasis. However, the underlying molecular events of Cd involved in NPC progression remain to be elucidated.
Purpose
The objective of this study was to decipher how Cd impacts the malignant phenotypes of NPC cells.
Methods
NPC cell lines CNE-1 and CNE-2 were continuously exposed with 1 μM Cd chloride for 10 weeks, designating as chronic Cd treated NPC cells (CCT-NPC). MTT assay, colony formation assay and xenograft tumor growth were used to assess cell viability in vitro and in vivo. Transwell assays were performed to detect cell invasion and migration. The protein levels of E-cadherin, N-cadherin, Vimentin as well as β-catenin and casein kinase 1α(CK1α) were measured by Western blot. Immunofluorescence staining was used to observe the distribution of filament actin (F-actin), β-catenin and CK1α. The mRNA levels of downstream target genes of β-catenin were detected by RT-PCR. Wnt/β-catenin signaling activity was assessed by TOPFlash/FOPFlash dual luciferase report system. MS-PCR was used to detect the methylation status of CK1α. Finally, the activation of Wnt/β-catenin pathway and cell biological properties were examined following treatment of CCT-NPC cells with 5-aza-2-deoxy-cytidine(5-aza-CdR).
Results
CCT-NPC cells showed an increase in cell proliferation, colony formation, invasion and migration compared to the parental cells. Cd also induced cytoskeleton reorganization and epithelial-to-mesenchymal transition. Upregulation and nuclear translocation of β-catenin and increased luciferase activity accompanied with transcription of downstream target genes were found in CCT-NPC cells. Treatment of CCT-CNE1 cells with 5-aza-CdR could reverse the hypermethylation of CK1α and attenuate the cell malignancy.
Conclusion
These results support a role for chronic Cd exposure as a driving force for the malignant progression of NPC via epigenetic activation of the Wnt/β-catenin pathway.
Introduction
Cd, a ubiquitous carcinogenic pollutant, has long been recognized as a toxic metal. Occupational inhalation, cigarette smoke, food and drinking water, or ambient air are the primary routes to Cd exposure.Citation1 Cd has a very long biological half-life ranging from 15 to 40 years, and is retained in the liver and kidneys. In 1993, Cd and its inorganic compounds were classified as Group 1 carcinogens by the International Agency for Research on Cancer for causing lung cancer.Citation2,Citation3
Occupational Cd exposure has been associated with elevated risk for lung and prostate cancers.Citation3,Citation4 Nevertheless, enhanced cancer risk may not be restricted to comparatively high occupational exposure.Citation5 For the general population, daily exposures are under low level conditions. Numerous data from cohort or cross-sectional studies have shown that environmentally relevant dietary exposure to Cd contributes to the development of cancer of the prostate, lung, genitourinary, breast, endometrium, pancreas, urinary bladder and colon cancers, as well as hepatocellular carcinoma.Citation6–Citation11 Furthermore, a putative carcinogenic role of Cd has been validated by in vitro and in vivo models.Citation11–Citation17
NPC is a unique malignancy that arises from the epithelium of the nasopharynx with a high prevalence in east and Southeast Asia, especially in the Guangdong and Guangxi provinces in southern China.Citation18 The unique ethnic and geographical distribution of NPC indicates its unusual etiology. Three major etiologic factors, genetic susceptibility, Epstein–Barr virus infection and environmental factors, have been identified as being involved in NPC pathogenesis, alone or in synergy.Citation18,Citation19 It has been acknowledged that environmental exposures serve as a driving force in tumor development and progression.Citation20 But to date, only nitrosamine, polycyclic aromatic hydrocarbons and nickel are regarded as environmental risk factors in the development and progression of NPC.Citation18 Recent epidemiological data from Khlifi et al and our laboratory suggested a correlation between NPC and blood levels of Cd in Tunisian and Chinese Teochew populations, southeast of China.Citation21,Citation22 In addition, our results illustrated that Cd burden was positively associated with clinical stage and node grade,Citation22 suggesting Cd burden may contribute to NPC progression. Nonetheless, a cause-and-effect association between chronic Cd exposure and the malignant progression of NPC has not been established.
Mechanistically, oxidative stress, one of the primary mechanisms involved in heavy-metal-mediated carcinogenesis, has been implicated in Cd carcinogenesis and causes most of the genotoxic events such as DNA strand breaks, chromosomal aberration and gene mutations.Citation23 However, direct interaction of Cd with DNA is minimal. Epigenetic alterations, including hypermethylation, have been suggested to the predominant molecular processes involved in Cd-induced carcinogenesis,Citation24–Citation26 and also contribute to NPC carcinogenesis.Citation27,Citation28 A few signaling pathways have been identified deregulated by DNA methylation in NPC, including the MAPK, Hedgehog, TGF-β and Wnt signaling.Citation29 Promoter methylation-induced silence of some Wnt inhibitors has been linked with the aberrant activation of Wnt signaling and transcription of its downstream targets.Citation30
The Wnt/β-catenin pathway has emerged as a key signaling pathway promoting malignancy in mouse kidney and triple-negative breast cancer cells with chronic Cd exposure.Citation31,Citation32 However, the precise mechanism by which Cd mediates Wnt signaling has not been elucidated, especially epigenetic regulation of some core components of this pathway. CK1α, consisting of the “destruction complex” with Axin, APC, Ser/Thr kinases glycogen synthase kinase 3β for phosphorylating and proteolytically degrading cytoplasmic β-catenin, is known to be a key factor determining β-catenin stability and transcriptional activity in tumor cells.Citation19 Since downregulation of CK1α in melanoma cells correlated with promoter methylation and induction of β-catenin signaling to promote metastasis,Citation33 we hypothesized that chronic Cd exposure promoted NPC cell growth and metastatic potential through activation of the Wnt/β-catenin signaling pathway by epigenetic modulation of CK1α.
This is the first study to reveal the stimulative effect of chronic Cd exposure on malignant progression of NPC. In particular, we suggest an epigenetic mechanism involved in Cd carcinogenesis by upregulation of Wnt/β-catenin signaling.
Materials and methods
Cell lines and cell culture
CNE-1 and CNE-2 cell lines were a gift from Dr. Ya Cao (Xiangya Hospital, Hunan, People’s Republic of China) and maintained in our laboratory.Citation34,Citation35 Cells were maintained in RPMI-1640 medium (HIMEDIA, Mumbai, India) supplemented with 10% FBS (HyClone, Logan, UT, USA) at 37°C and 5% CO2. Cd chloride (purity 99%; Sigma, St. Louis, MO, USA) was dissolved in double-distilled water to make a 1M stock solution. For cytotoxicity assessment, cells were placed in 96-well plates for 24 hours and then incubated with various concentrations of Cd (1 nM, 1 μM and 1 mM) for 72 hours. For the chronic exposure experiment, cell lines were continuously exposed to a non-toxic level (1 μM) of Cd for up to 10 weeks (23 passages). To distinguish Cd-exposed cells from their parental cells, the cells exposed to Cd for 10 weeks were designated as CCT-NPC cells including CCT-CNE1 and CCT-CNE2 cells. We used Cd-free cultured CNE-1 and CNE-2 cells as controls. This study was performed with the approval of the Human Ethical Committee of the Cancer Hospital of Shantou University Medical College.
Cell treatment
To investigate the mechanism responsible for the Wnt/β-catenin signaling activation and CK1α downregulation, CCT-CNE1 and CCT-CNE2 cells (1×106) were treated with 0, 5, 10, or 50 μM 5-aza-CdR (Santa Cruz, Dallas, Texas, USA) for 48 hours, after which cells were harvested and genomic DNA, total RNA and total protein were extracted, and then subjected to methylation specific-PCR (MS-PCR), RT-PCR and Western blot analysis, respectively. Simultaneously, the cell characteristics were evaluated by MTT assays and transwell assay. Control cultures were treated under similar experimental conditions in the absence of 5-aza-CdR. To further identify the modulating role of CK1α in Wnt/β-catenin pathway activation induced by Cd, CK1α was silenced by small interfering RNA (siRNA) in CCT-CNE1 cells. The sequences of the siRNAs used to suppress CK1α were as follows: forward: 5-CUCAGGAUUAAACCAGUUATT-3 and reverse: 5-UAACUGGUUUAAUCCUGAGTT-3. Both the siRNA and control sequences were ordered from GenePharma Co., Ltd. (Suzhou, People’s Republic of China). Transfections were performed using Lipofectamine 3,000 (Invitrogen, Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the manufacturer’s instructions. After that, the β-catenin protein level and mRNA levels of its downstream genes were determined by Western blot and RT-PCR, respectively. The analysis was repeated three times.
MTT assay
The 3-(4,5-dimethylthiazol-2-yl)–2,5-diphenyltetrazolium bromide (MTT) assay was conducted to quantify cell viability by the protocol described previously.Citation36 Briefly, exponentially growing cells (1×104 cells/well) in 100 μL medium were seeded in 96-well plates. The dye crystals were dissolved in 100 μL DMSO and the absorbance was measured with a Multiskan MK3 reader (Thermo Fisher Scientific, Inc.) at 492 nm. Three independent experiments were performed in triplicate wells.
Colony formation assay
Exponentially growing cells were suspended in RPMI-1640 medium and seeded in 6-well plates at a density of 200 cells per well. The plates were maintained at 37°C in a humidified incubator with 5% CO2 for two weeks. After fixation in paraformaldehyde, the colonies were stained with Giema for 10 minutes and then counted using a light microscope. The cloning efficiency (%)=(number of colonies formed)/(number of cells added)×100. All groups were assessed in triplicate.
Cell migration and invasion assay
Cell migration and invasion were measured using transwell chambers (8 μm pore size; Corning Incorporated, Corning, NY, USA) according to methods described previously.Citation37 600 μL 10% FBS-supplemented RPMI-1640 medium was added to the lower chamber. The invasion chambers were pre-coated with 50 μL Matrigel solution. 1×105 cells were layered in the upper chambers and incubated at 37°C for 24 hours (migration assay) and 48 hours (invasion assay). Cells adhering to the lower surface of the membrane were fixed by methanol, stained with hematoxylin-eosin (migration) or Giemza (invasion) and counted under a light microscope. Experiments were independently performed in triplicate.
Western blot
Western blot was performed as previously describedCitation37 using monoclonal antibodies against β-actin, β-catenin, (1:1,000; Cell Signaling Technology, Beverly, MA, USA), CK1α (1:1,000; Cell Signaling Technology) as well as E-cadherin, N-cadherin and Vimentin (1:1,000; Cell Signaling Technology), at 4°C overnight followed by rinsing and addition of an anti-rabbit/mouse secondary antibody (Gene Tech, Shanghai, People’s Republic of China) at 1:1,000 dilution for 1 hour. Protein concentrations were determined with a BCA Protein Assay Kit (Beyotime, Shanghai, People’s Republic of China).
Immunofluorescence analysis
Each group of cells was washed and treated with 0.5% Triton X-100 for 20 minutes. After blocking with goat serum for 30 minutes, the fixed and blocked cells were incubated with primary anti-β-catenin (rabbit antihuman, 1:100; Cell Signaling Technology), CK1α (rabbit anti human, 1:100; (Cell Signaling Technology) and actin filament antibodies overnight at 4°C. After washing with PBS, cells were incubated with secondary antibodies (1:2,000; ZSGB-BIO, Beijing, People’s Republic of China) conjugated with rhodamine (Santa Cruz, Dallas, Texas, USA) for 1 hour at 37°C. Actin filament was stained with rhodamine phalloidin (Cytoskeleton, Denver, CO, USA) for 30 minutes. Finally, nuclei were stained with DAPI (Beyotime). Fluorescence images were then taken with a fluorescence microscope (Olympus Corporation, Tokyo, Japan).
RT-PCR
Total RNA was extracted from untreated and Cd-treated CNE-1 and CNE-2 cells using TRIzol (Invitrogen, Grand Island, NY, USA) and cDNA was prepared using reverse transcriptase PCR kit (Takara, Shiga, Japan). RT-PCR was conducted as previously describedCitation37 using primers designed with Primer Express Software version 2.0 (Applied Biosystems, Forster City, Canada): cyclin D1 forward 5-TGTCCTACTACCGCCT CACA-3 and reverse 5-CAGGGCTTCGATCTGCTC-3; cyclin E forward 5-AAAAGGTTTCAGGGTATCAG-3 and reverse 5-TGTGGGTCTGTATGTTGTG-3; c-myc forward 5-GCCCCTCAACGTTAGCTTCA-3 and reverse 5-TTCCAGATATCCTCGCTGGG-3; c-jun forward 5-AAGAACTCGGACCTCCTCAC-3 and reverse 5-CTCCTGCTCATCTGTCACG-3; β-actin forward 5-AGCGAGCATCCCCCAAAGTT-3 and reverse 5-GGGCACGAAGGCTCATCATT-3. Gene expression relative to β-actin was determined by the comparative CT method (2−ΔΔCT). All experiments were performed in triplicate.
Dual luciferase assay
CNE-1 and CCT-CNE1 cells were seeded in 24-well plates overnight and then transiently transfected with TOPflash reporter plasmid (400 ng/well; Millipore, MA, USA) and Renilla luciferase plasmid (100 ng/well; Promega, Fitchburg, WI, USA) by Lipofectamine 3,000 (Invitrogen, Camarillo, CA, USA). Luciferase activity was measured at 48 hours after transfection by the Dual-Luciferase Reporter Assay System (Pro-mega), and normalized to Renilla luciferase relative light unit values. Three independent experiments were performed.
MS-PCR
Genomic DNA was isolated from cell lines with different treatments to detect the methylation status of the CK1α in a promoter CpG island. MS-PCR was conducted as described previously.Citation36 The primer sets were as follows: unmethylated forward (5′-TGTGTAGTTAGTAGGAGTTGTAGTGT-3), unmethylated reverse (5′-AAAAAT-CAACAACAAAAAAACAAA-3′), and methylated forward (5′-TTGCGTAGTTAGTAG-3′), methylated reverse (5′-AAATCGACAACGAAAAAACGA-3′), which amplified 116- and 115 bp products respectively.
Tumor xenografts
All animal studies were approved by the Animal Ethics Committee of Shantou University Medical College and followed the guidelines of the Animal Laboratory Center. Four-week-old BALB/c nude mice were purchased from Vital River (Beijing, People’s Republic of China) and maintained under pathogen-free conditions according to standard institutional guidelines. CNE-1 and CCT-CNE1 cells were harvested at a concentration of 3×106 cells/mL. For tumor xenograft experiments, mice were injected subcutaneously in the right axilla with 100 μL of cell suspension (n=5 per group). Tumor volumes (widthCitation2×length×0.5) were obtained by serial caliper measurement every 3 days. At 28 days after injection, the mice were euthanized and tumors were removed and weighed.
Statistical analysis
All the statistical procedures were performed with SPSS software. Measurement data are presented as mean ± SD. Statistical significance was assessed using a two-tailed Student’s t-test. P<0.05 was considered statistically significant.
Results
NPC cells exhibit increased cell growth in vitro and in vivo after chronic Cd exposure
Before chronic exposure, a non-cytotoxic concentration for treatment of NPC cells was first identified. In 72 hours of exposure, neither 1 nM nor 1 μM Cd reduced cell survival in either CNE-1 or CNE-2 cell lines, whereas 1 mM Cd exerts a cytotoxic effect such that viable cells are rarely found at 24 hours (). This indicates that micromolar exposure is not acutely toxic, and even improves cell viability slightly compared to nanomolar exposure. Based on continuous Cd exposure previously shown in other research,Citation32,Citation38 1 μM was selected for our chronic exposure concentration in NPC. After 10 weeks of exposure to low level of Cd (1 μM), MTT assay showed that cell viability was significantly increased in CCT-CNE1 and CCT-CNE2 cells compared to the parental cells (, P<0.05). Also, the colony formation capacity of CCT-CNE1 and CCT-CNE2 cells was markedly increased, 1.48 and 1.54-fold, respectively (, P<0.01). We further explored tumorigenesis of CCT-CNE1 cells in vivo. At 10 days after injection, CCT-CNE1 xenograft tumors exhibited increased growth compared to CNE-1 transplanted controls ( and ). These results collectively illustrate that CCT-NPC cell lines acquire a more proliferative phenotype, both in vitro and in vivo.
Figure 1 Cell proliferation, colony formation and xenograft tumor growth in CCT-NPC or parental cells.
Notes: (A) MTT assays for acute exposure to Cd (1 nM, 1 µM and 1 mM). (B) MTT assays following 1 µM Cd treatment for 10 weeks. (C) Effects of chronic Cd exposure on the colonogenic ability in CNE-1/CNE-2 and CCT-CNE1/CCT-CNE2 cells (N=3). (D) Gross appearance of xenograft tumors at 28 days after CNE-1 or CCT-CNE1 cells injection. (E). Tumor growth curves and weight data in transplanted nude mice with CNE-1 and CCT-CNE1 through four weeks; data are mean (± SD) tumor volume (N=5). Each assay was performed in triplicate. *P<0.05; **P<0.01; ***P<0.001, compared with the parental cells.
Abbreviation: CCT-NPC, chronic cadmium-treated nasopharyngeal carcinoma.
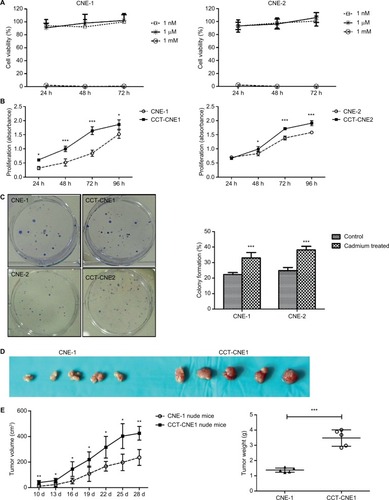
Chronic Cd exposure promotes NPC cell invasion and migration
Transwell assays were performed to determine the effect of Cd on cell aggressiveness. The results showed that the invasive capacity of CCT-CNE1 and CCT-CNE2 cells was markedly increased 1.46 (P<0.01) and 1.40-(P<0.01) fold of the parental controls, respectively (). Analogously, CCT-CNE1 and CCT-CNE2 displayed robust migration compared to their parental cell lines, as the number of transmigrated cells was 1.30 and 1.37 times greater than that of CNE-1 (P<0.001) and CNE-2 (P<0.01) cells, respectively ().
Figure 2 CCT-NPC cells acquired metastasis-associated phenotype.
Notes: (A) The gross view of cell invasion assay stained with Giemza and corresponding quantitative analyses of the results for CCT-NPC cells and the control cells. Magnification 200×. (B) The gross view of cell migration assay stained with hematoxylin-eosin and corresponding quantitative analyses of the results for CCT-NPC cells and the control cells. Magnification 200×. (C) Immuno-fluorescence staining for actin filament in CCT-NPC cells and the parental cells. Actin filament cytoskeleton was stained with rhodamine conjugated phalloidin (red) and nuclei with DAPI (blue). Magnification 400×. (D) Expression of the EMT markers E-cadherin, vimentin and N-cadherin in CCT-NPC and NPC cell lines. *P<0.05; **P<0.01; ***P<0.001, compared with the parental cells. *P<0.05; **P<0.01; ***P<0.001, compared with the parental cells.
Abbreviations: CCT-NPC, chronic cadmium-treated nasopharyngeal carcinoma; EMT, epithelial–mesenchymal transition.
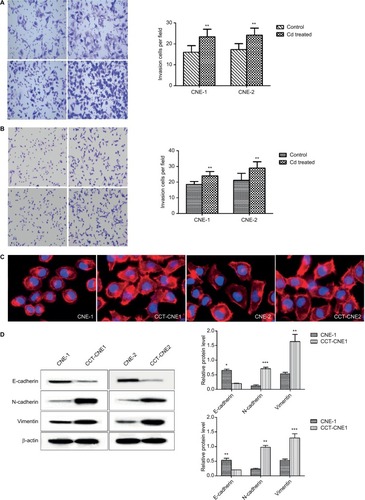
Chronic Cd exposure induces cytoskeleton reorganization and promotes EMT
It is commonly accepted that the dynamic reorganization of the actin cytoskeleton as well as EMT are prerequisites for cancer cells to gain invasive and metastatic properties.Citation39,Citation40 Actin filament is one of the most important components of the cytoskeleton and changes in intracellular actin structures are a key step in cellular migration and invasion and closely related to EMT.Citation39,Citation41–Citation43 Therefore, immunofluorescence analysis was used to study the influence of Cd on the actin filament distribution. Both CCT-CNE1 and CCT-CNE2 cells exhibited more dense, highly labeled microtubule network compared to the parental cell lines, displaying actin filament assembly and formation of migratory membrane protrusions (), which provides evidence of cytoskeleton reorganization. It is widely accepted that tumor invasiveness and metastasis are also caused by motility and EMT. Hence, the expression of the epithelial marker E-cadherin and mesenchymal markers N-cadherin and vimentin was determined by Western blot. As expected, both CCT-CNE1 and CCT-CNE2 had increased vimentin and N-cadherin and decreased E-cadherin expression compared to controls, suggesting that chronic Cd treatment promotes NPC EMT ().
Chronic Cd treatment induces activation of the Wnt/β-catenin pathway
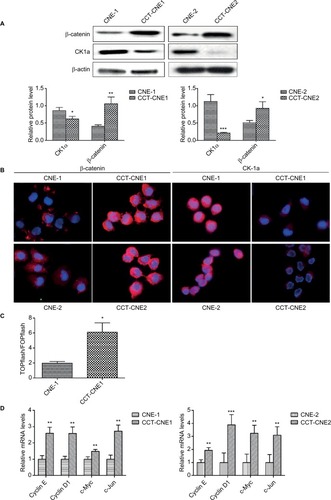
Before exploring whether the Wnt signaling pathway was activated in CCT-NPC cell lines, we detected the protein expression of β-catenin and CK1α by Western blot. Compared to primary NPC cells, a remarkable upregulation of β-catenin was found in both CCT-CNE1 and CCT-CNE2 cell lines, whereas the protein level of CK1α was reduced in CCT-NPC cells (). These were corroborated by immunofluorescence microscopy that exhibited a more diffused intense signal of β-catenin, preferentially localized in the nucleus in CCT-NPC cells, compared to the controls with a weak plaque-like signal, predominately in the cytoplasm. Conversely, CK1α was strongly expressed in the cytoplasm and nucleus of parental cells, whereas a weak signal was observed in CCT-NPC cells (). TOP/FOPflash luciferase reporter gene assay was further performed to assess the activation of Wnt/β-catenin signaling. There was a 3.11 fold increase of luciferase activity in CCT-CNE1 cells compared to CNE-1 cells, suggesting increased TCF/LEF-mediated transcription following Cd exposure (). This was further confirmed by the observation of elevated transcription of well-known Wnt target genes including cyclin D1, cyclinE, c-Myc and C-jun ().These results indicate that the Wnt/β-catenin signaling was activated in response to continuous low-level Cd exposure in NPC cells.
Figure 3 Cd treatment activates Wnt/β-catenin signaling and aberrant methylation of the CK1α promoter in NPC cell lines.
Notes: (A) Western blot analysis of total β-catenin and CK1α in cCd-treated NPC cells and controls; β-actin was used as a loading control. (B) Immunofluorescence staining patterns of β-catenin and CK1α in NPC and CCT-NPC cells. Nuclei were stained with DAPI (magnification 400×). (C) Luciferase reporter assays using TOPflash/FOPflash reporter plasmids to assess the activity of Wnt/β-catenin. (D) RT-PCR analysis of relative transcript levels of the β-catenin target genes cyclin E, cyclin D1, c-Myc and c-Jun. *P<0.05; **P<0.01; ***P<0.001.
Abbreviations: CCT-NPC, chronic cadmium-treated nasopharyngeal carcinoma; CK1α, casein kinase 1α; RT-PCR, reverse transcription-PCR.
Hypermethylation of CK1α plays a mediating point for Wnt pathway activation and malignant progression in CCT-NPC cells
It has been suggested that epigenetic inactivation of negative Wnt/β-catenin signaling regulators contributes to aberrant activation of this signaling pathway in NPC tumorigenesis.Citation44 Besides, aberrant DNA methylation plays an important role in Cd-induced carcinogenesis.Citation24 Given that the DNA hyper-methylation induced by Cd is responsible for the reduction of CK1α, thereby activating the Wnt/β-catenin pathway, we next analyzed the methylation status of CK1α. The results of MSP assays revealed that the CK1α promoter region is highly methylated in both CCT-CNE1 and CCT-CNE2 cells, whereas only slight methylation was detected in their parental cells (), suggesting that the decreased expression of CK1α is attributed to hypermethylation of the promoter CpG island induced by Cd in CCT-NPC cells.
Figure 4 Hypermethylation of CK1α induces a switch in Wnt/β-catenin signaling and malignant progression in CCT-NPC cells.
Notes: (A) Methylation status of the CK1α promoter analyzed by MS-PCR. (B) Western blot analyses of CK1α and β-catenin in CCT-CNE1 cells following treatment with 50 µM 5-aza-CdR for 48 hours. (C) RT-PCR analysis of relative transcript levels of β-catenin target genes following treatment of cells with 5-aza-CdR (50 µM). (D) Western blot and RT-PCR analyses of the effect of CK1α depletion and (or) 5-aza-CdR treatment on β-catenin expression and downstream gene transcription in CCT-CNE1 cells. (E) MTT assays for cell viability of CCT-CNE1 and CCT-CNE2 cells with increasing concentrations of 5-aza-CdR treatment. (F) Invasion and migration ability of 5-aza-CdR-treated CCT-CNE1 cells. *P<0.05; **P<0.01; ***P<0.001, compared with parental cells. #P>0.05, compared with CK1α treated cells.
Abbreviations: CCT-NPC, chronic cadmium-treated nasopharyngeal carcinoma; CK1α, casein kinase 1α; MS-PCR, methylation specific-PCR; RT-PCR, reverse transcription-polymerase chain reaction.
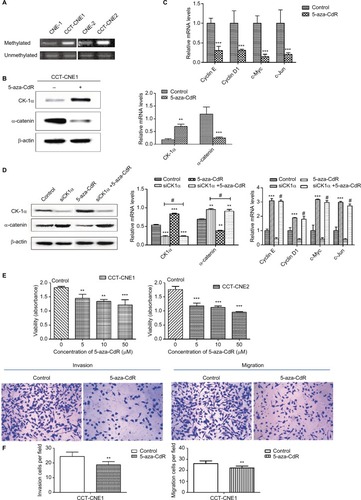
Then we treated CCT-CNE1 cells with 5-aza-CdR to address the hypothesis that hypermethylated CK1α induced by Cd is involved in the Wnt/β-catenin signaling activation and malignant progression of CCT-NPC cells. As expected, the expression of CK1α was found to be restored by 5-aza-CdR, while the β-catenin protein level () as well as the transcription of target genes including cyclin D1, cyclinE, c-Myc and C-jun was downregulated following 5-aza-CdR treatment (). Additionally, in order to exclude the nonspecific effects of 5-aza-CdR, we knocked down the CK1α with RNA interference assay. The data show that the knockdown of CK1α blocked the downregulation effect of 5-aza-CdR on β-catenin protein level and its downstream target genes’ mRNA level (). We think these data strongly support that hypermethylation of CK1α induces a switch in Wnt/β-catenin signaling in CCT-CNE1 cells. Finally, we found that treatment of CCT-NPC cells with 5-aza-CdR suppressed cell proliferation (), invasion and migration (). In summary, these results highlight that the hypermethylation of the promoter of CK1α induced by Cd may serve as a master governor of Wnt/β-catenin signaling pathway activation to promote malignant phenotypes.
Discussion
In the present study, CNE-1 and CNE-2 cells were continuously exposed to 1 μM Cd, a non-toxic level, for up to 10 weeks to mimic chronic low-level Cd exposure. It has been revealed that Cd can trigger a hormesis-like response characterized by a low-dose stimulation and a high-dose inhibition in human cells of mammary, prostate, embryo lung fibroblast and embryonic kidney.Citation45–Citation48 In the present study, exposure of CNE-1 and CNE-2 cells to Cd at concentration of 1 μM for 10 weeks also exerts a stimulatory role in cell proliferation in vitro. Furthermore, injection of nude mice with CCT-CNE1 cells induced marked increase in xenograft tumor volume compared to the parental cells. These findings are consistent with previous reports that long-term exposure to low concentrations of Cd induces cell proliferation and tumor growth and thus promotes malignant transformation in human lung cells, bronchial epithelial cells and prostate epithelial cells.Citation38,Citation49,Citation50
NPC is a malignant tumor with high rates of local invasion and distant metastasis. With considerable improvement in radiotherapy technology and advances in multimodal treatment over the past three decades, excellent local control for NPC can generally be achieved, but distant metastasis becomes the major pattern of treatment failure for NPC.Citation51 Metastasis, a major feature of malignant tumors, is a multi-step process including dissemination, migration, intravasation, extravasation, and colonization to form secondary tumors.Citation52 Previous studies have suggested that chronic exposure of human bronchial, lung, prostate or breast epithelial cells to Cd induces malignant transformation with hyperproliferation and increased potential to invade and migrate.Citation49,Citation53,Citation54 Recently, Wei And Shaikh reported that prolonged Cd treatment in triple-negative breast cancer cells stimulates cell proliferation, adhesion, cytoskeleton reorganization as well as migration and invasion.Citation32 In this study, we describe for the first time that chronic low-dose Cd exposure not only confers NPC cells a growth advantage in vitro and in vivo, but also stimulates metastasis-associated phenotype, as evidenced by enhanced invasion and migration, cytoskeleton reorganization, upregulation of mesenchymal markers N-cadherin and vimentin as well as repression of epithelial marker E-cadherin. The present study provides experimental evidence for the findings of our preliminary human study that Cd seems to be a risk factor for NPC and may promote the occurrence and development of this disease.
The Wnt/β-catenin pathway has been implicated in a number of cancers including NPC, lung cancer, colorectal cancer, melanoma, and leukemia. Abnormalities of the Wnt/β-catenin pathway also have been indicated to be involved in Cd nephro-carcinogenesis,Citation31,Citation55–Citation57 in which Cd enhances nuclear translocation of β-catenin in human renal epithelial cells followed by binding to TCF/LEF and inducing target genes transcription to upregulate cell proliferation and survival.Citation56 Similarly, our study shows that chronic Cd treatment induces the protein expression and nuclear translocation of β-catenin and upregulates luciferase activity as well as the transcription of the downstream genes including cyclin D1, cyclin E, c-myc and c-jun, suggesting chronic Cd exposure elicits the activation of the Wnt/β-catenin signaling pathway. Since Wnt/β-catenin pathway is believed to play a pivotal role in multiple malignancies including cell proliferation, EMT and migratory process,Citation58–Citation63 our results strongly suggest that chronic Cd exposure induces activation of the Wnt/β-catenin signaling to accelerate cell proliferation, invasion, migration and EMT in NPC cells.
Although Wnt/β-catenin signaling has been documented to participate in Cd carcinogenicity, little is known regarding the precise mechanism of how Cd mediates Wnt/β-catenin signaling. Mutations in some components of Wnt/β-catenin pathway, such as mutation of β-catenin at position Ser45 or mutations in APC or Axin, have been described in cancers and considered as inappropriate activation of the Wnt pathway.Citation64 However, present consensus is that the direct muta-genic effect of Cd is weak or just restricted to comparatively high-concentration exposure.Citation5 Chronic Cd exposure has been linked with increases in DNA methyltransferase activity and global 5mC and aberrant DNA methylation of some DNA repair genes or tumor suppressor genes, such as RASSF1A and p16 in Cd-induced malignant transformation.Citation50,Citation65 Aberrant Wnt/β-catenin signaling is also a critical component of NPC and most NPC tumors exhibit Wnt/β-catenin pathway protein dysregulation. It has been shown that decreased expression of the Wnt inhibitory factor, an endogenous Wnt antagonist, is silenced via promoter hypermethylation in NPC cell lines.Citation66
CK1α, a component of Wnt/β-catenin signaling pathway, has been proposed as a negative regulator of this pathway by phosphorylation of β-catenin at Ser45. Inhibition or down-regulation of CK1α leads to an accumulation of cytoplasmic β-catenin.Citation67 A recent study identified CK1α was a novel tumor suppressor in melanoma cells as evidence that knockdown of CK1α enhanced the invasive capacity of melanoma cells and this effect was overexpression of CK1α in metastatic melanoma cells resulted in suppression of Wnt/β-catenin signaling and reduction of cell growth and metastasis.Citation33 Surprisingly, very little is known about whether Cd activates Wnt/β-catenin signaling pathway by targeting CK1α. Results from this study demonstrate that chronic Cd exposure induces decreased expression of CK1α due to the hypermethylated promoter region in NPC. And we also found that 5-aza-CdR treatment restores the expression of CK1α and induces β-catenin degradation and thus blocks the transcription of downstream genes (cyclin D1, cyclinE, c-Myc and C-jun). Furthermore, knockdown of CK1α by siRNA before 5-aza-CdR treatment suppressed 5-aza-CdR-induced alterations of β-catenin level and downstream genes transcription in CCT-CNE1 cells. It appears that CK1α acts as a target for Cd-induced hyper-methylation and induces a switch in Wnt/β-catenin signaling pathway. Moreover, further functional analyses indicate that treatment with demethylation agent in CCT-NPC cells causes impaired cell proliferation, invasion and migration. These findings are supportive of our hypothesis that methylation of CK1α induced by Cd might be responsible for the induction of malignant phenotype in NPC cells ().
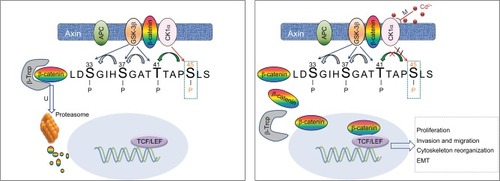
Figure 5 Hypothesized model for the induction of Cd on Wnt/β-catenin signaling and malignant progression in NPC cells.
Notes: Chronic low-dose Cd treatment of NPC cells induces CK1α promoter hypermethylation that downregulates CK1α expression, leading to accumulation and nuclear translocation of β-catenin thereby activating Wnt/β-catenin signaling to promote malignancy.
Abbreviations: APC, adenomatous polyposis coli; CK1α, casein kinase α; EMT, epithelial–mesenchymal transition; GSK-3β, glycogen synthase kinase 3β; NPC, nasopharyngeal carcinoma; TCF/LEF, T-cell factor/lymphoid enhancer factor; β-Trcp, β-transducin repeats-containing proteins.
Conclusion
Taken together, this study highlights for the first time, to our knowledge, that NPC cells exposed to chronic low-dose Cd acquired enhanced malignant progression, including more proliferative and aggressive characteristics, at least in part, by activating the Wnt/β-catenin pathway via DNA methylation of CK1α in promoter CpG islands.
Author contributions
Lin Peng, Fan Zhang and Jiong-Yu Chen carried out the experiments. Lin Peng and Fan Zhang wrote the original draft. Yi-Teng Huang reviewed and revised the manuscript. Fan Zhang contributed to the statistical analysis. Yi-Teng Huang conceived and designed the work. Jiong-Yu Chen and Xia Huo supervised the overall project. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Abbreviations
| Cd | = | Cadmium |
| NPC | = | nasopharyngeal carcinoma |
| CCT-NPC | = | chronic cadmium-treated nasopharyngeal carcinoma |
| EMT | = | epithelial–mesenchymal transition |
| CK1α | = | casein kinase 1α |
| 5-aza-CdR | = | 5-aza-2-deoxy-cytidine |
| APC | = | adenomatous polyposis coli |
| RT-PCR | = | reverse transcription-PCR, MS- PCR, methylation-specific PCR |
| CpG | = | methylated cytosine-guanine. TCF/LEF, T-cell factor/lymphoid enhancer factor |
Acknowledgments
This work was supported by funds from The National Natural Science Foundation of China (No. 81602886), The Natural Science Foundation of Guangdong province (No. 2016A030313062), and Science and Technology Planning Project of Shantou City, People’s Republic of China (No. 2015-113). We thank Dr. Stanley Lin for his constructive comments and Prof. Yukun Cui for his kindly gift of luciferase plasmid.
Disclosure
The authors report no conflicts of interest in this work.
References
- FaroonOAshizawaAWrightSToxicological Profile for CadmiumAtlanta, GAAgency for Toxic Substances and Disease Registry2012
- IARCBeyllium, cadmium, Hg, and exposure in the glass manufacturing industryIARC Monogr Eval Carcinog Risks Hum19935819
- StaynerLSmithRThunMSchnorrTLemenRA quantitative assessment of lung cancer risk and occupational cadmium exposureIARC Sci Publ1992118447455
- KolonelLWinkelsteinWCadmium and prostatic carcinomaLancet1977280371269100
- HartwigACadmium and cancerMet Ions Life Sci20131149150723430782
- SatarugSLong-term exposure to cadmium in food and cigarette smoke, liver effects and hepatocellular carcinomaCurr Drug Metab201213325727122455552
- JulinBWolkABergkvistLBottaiMAkessonADietary cadmium exposure and risk of postmenopausal breast cancer: a population-based prospective cohort studyCancer Res20127261459146622422990
- JulinBWolkAJohanssonJEAnderssonSOAndrénOAkessonADietary cadmium exposure and prostate cancer incidence: a population-based prospective cohort studyBr J Cancer2012107589590022850555
- MezynskaMBrzóskaMMEnvironmental exposure to cadmium-a risk for health of the general population in industrialized countries and preventive strategiesEnviron Sci Pollut Res Int20182543211323229230653
- AkessonAJulinBWolkALong-term dietary cadmium intake and postmenopausal endometrial cancer incidence: a population-based prospective cohort studyCancer Res200868156435644118676869
- EriksenKTHalkjærJSørensenMDietary cadmium intake and risk of breast, endometrial and ovarian cancer in Danish postmenopausal women: a prospective cohort studyPLoS One201496e10081524963789
- CartularoLLaulichtFSunHKluzTFreedmanJHCostaMGene expression and pathway analysis of human hepatocellular carcinoma cells treated with cadmiumToxicol Appl Pharmacol2015288339940826314618
- StrumylaiteLKregzdyteRBoguseviciusAPoskieneLBaranauskieneDPranysDAssociation between cadmium and breast cancer risk according to estrogen receptor and human epidermal growth factor receptor 2: epidemiological evidenceBreast Cancer Res Treat2014145122523224692081
- Il’yasovaDSchwartzGGCadmium and renal cancerToxicol Appl Pharmacol20052072179186
- KidoTHondaRTsuritaniIIshizakiMYamadaYNogawaKHigh urinary cadmium concentration in a case of gastric cancerBr J Ind Med19894642882713285
- Feki-TounsiMHamza-ChaffaiACadmium as a possible cause of bladder cancer: a review of accumulated evidenceEnviron Sci Pollut Res Int20142118105611057324894749
- García-EsquinasEPollanMTellez-PlazaMCadmium exposure and cancer mortality in a prospective cohort: the strong heart studyEnviron Health Perspect2014122436337024531129
- ChuaMLKWeeJTSHuiEPChanATCNasopharyngeal carcinomaLancet2016387100221012102426321262
- LiuCLiYSemenovMControl of beta-catenin phosphorylation/degradation by a dual-kinase mechanismCell2002108683784711955436
- ParsaNEnvironmental factors inducing human cancersIran J Public Health2012411119
- KhlifiROlmedoPGilFRisk of laryngeal and nasopharyngeal cancer associated with arsenic and cadmium in the Tunisian populationEnviron Sci Pollut Res Int20142132032204224022098
- PengLWangXHuoXBlood cadmium burden and the risk of nasopharyngeal carcinoma: a case-control study in Chinese Chaoshan populationEnviron Sci Pollut Res Int20152216123231233125903187
- EngströmKSVahterMJohanssonGChronic exposure to cadmium and arsenic strongly influences concentrations of 8-oxo-7,8-dihydro-2′-deoxyguanosine in urineFree Radic Biol Med20104891211121720153423
- WangBLiYShaoCTanYCaiLCadmium and its epigenetic effectsCurr Med Chem201219162611262022471978
- TakiguchiMAchanzarWEQuWLiGWaalkesMPEffects of cadmium on DNA-(Cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformationExp Cell Res2003286235536512749863
- WangBLiYTanYLow-dose Cd induces hepatic gene hypermethylation, along with the persistent reduction of cell death and increase of cell proliferation in rats and micePLoS One201273e3385322457795
- DaiWCheungAKKoJMComparative methylome analysis in solid tumors reveals aberrant methylation at chromosome 6p in nasopharyngeal carcinomaCancer Med2015471079109025924914
- KimJSKimSYLeeMKimSHKimSMKimEJRadioresistance in a human laryngeal squamous cell carcinoma cell line is associated with DNA methylation changes and topoisomerase II αCancer Biol Ther201516455856625719218
- DaiWZhengHCheungAKLungMLGenetic and epigenetic landscape of nasopharyngeal carcinomaChin Clin Oncol2016521627121876
- LiLZhangYFanYCharacterization of the nasopharyngeal carcinoma methylome identifies aberrant disruption of key signaling pathways and methylated tumor suppressor genesEpigenomics20157215517325479246
- ChakrabortyPKScharnerBJurasovicJMessnerBBernhardDThévenodFChronic cadmium exposure induces transcriptional activation of the Wnt pathway and upregulation of epithelial-to-mesenchymal transition markers in mouse kidneyToxicol Lett20101981697620478370
- WeiZShaikhZACadmium stimulates metastasis-associated phenotype in triple-negative breast cancer cells through integrin and β-catenin signalingToxicol Appl Pharmacol2017328708028527916
- SinnbergTMenzelMKaeslerSSuppression of casein kinase 1alpha in melanoma cells induces a switch in beta-catenin signaling to promote metastasisCancer Res201070176999700920699366
- YeXXLiuCBChenJYTaoBHZhi-YiCThe expression of cyclin G in nasopharyngeal carcinoma and its significanceClin Exp Med2012121212421688120
- HongCQRanYGChenJYWuXYouYJStudy on promoter methylation status of E-cadherin gene in nasopharyngeal carcinoma cell linesZhonghua Bing Li Xue Za Zhi201039853253621055032
- WuXZhuangYXHongCQClinical importance and therapeutic implication of E-cadherin gene methylation in human ovarian cancerMed Oncol201431810024973953
- LiXYLinYCHuangWLZoledronic acid inhibits proliferation and impairs migration and invasion through downregulating VEGF and MMPs expression in human nasopharyngeal carcinoma cellsMed Oncol201229271472021431960
- SonYOWangLPoyilPCadmium induces carcinogenesis in BEAS-2B cells through ROS-dependent activation of PI3K/AKT/GSK-3β/β-catenin signalingToxicol Appl Pharmacol2012264215316022884995
- SunBOFangYLiZChenZXiangJRole of cellular cytoskeleton in epithelial-mesenchymal transition process during cancer progressionBiomed Rep20153560361026405532
- PasquierJAbu-KaoudNAl ThaniHRafiiAEpithelial to Mesenchymal Transition in a Clinical PerspectiveJ Oncol2015201579218226425122
- NürnbergAKitzingTGrosseRNucleating actin for invasionNat Rev Cancer201111317718721326322
- JacquemetGHamidiHIvaskaJFilopodia in cell adhesion, 3D migration and cancer cell invasionCurr Opin Cell Biol201536233126186729
- ShankarJMessenbergAChanJUnderhillTMFosterLJNabiIRPseudopodial actin dynamics control epithelial-mesenchymal transition in metastatic cancer cellsCancer Res20107093780379020388789
- YingYTaoQEpigenetic disruption of the WNT/beta-catenin signaling pathway in human cancersEpigenetics200945307312
- HaoCHaoWRole of ERK in the hormesis induced by cadmium chloride in HEK293 cellsWei Sheng Yan Jiu201140451752221861362
- SchmidtCMChengCNMarinoAKonsoulaRBarileFAHormesis effect of trace metals on cultured normal and immortal human mammary cellsToxicol Ind Health2004201–5576815807409
- JiangGDuanWXuLSongSZhuCWuLBiphasic effect of cadmium on cell proliferation in human embryo lung fibroblast cells and its molecular mechanismToxicol In Vitro200923697397819573589
- GaddipatiJPRajeshkumarNVGroveJCLow-Dose Cadmium Exposure Reduces Human Prostate Cell Transformation in Culture and Up-Regulates Metallothionein and MT-1G mRNANonlinearity Biol Toxicol Med20031219921219330122
- PersonRJTokarEJXuYOrihuelaRNgalameNNWaalkesMPChronic cadmium exposure in vitro induces cancer cell characteristics in human lung cellsToxicol Appl Pharmacol2013273228128823811327
- Benbrahim-TallaaLWaterlandRADillALWebberMMWaalkesMPTumor suppressor gene inactivation during cadmium-induced malignant transformation of human prostate cells correlates with overexpression of de novo DNA methyltransferaseEnviron Health Perspect2007115101454145917938735
- LeeAWMaBBNgWTChanATBbMWtNManagement of Nasopharyngeal Carcinoma: Current Practice and Future PerspectiveJ Clin Oncol201533293356336426351355
- CoghlinCMurrayGICurrent and emerging concepts in tumour metastasisJ Pathol2010222111520681009
- Benbrahim-TallaaLTokarEJDiwanBADillALCoppinJFWaalkesMPCadmium malignantly transforms normal human breast epithelial cells into a basal-like phenotypeEnviron Health Perspect2009117121847185220049202
- AchanzarWEDiwanBALiuJQuaderSTWebberMMWaalkesMPCadmium-induced malignant transformation of human prostate epithelial cellsCancer Res200161245545811212230
- ThévenodFChakrabortyPKThe role of Wnt/beta-catenin signaling in renal carcinogenesis: lessons from cadmium toxicity studiesCurr Mol Med201010438740420455852
- ChakrabortyPKLeeWKMolitorMWolffNAThévenodFCadmium induces Wnt signaling to upregulate proliferation and survival genes in sub-confluent kidney proximal tubule cellsMol Cancer2010910220459685
- ThévenodFWolffNABorkULeeWKAbouhamedMCadmium induces nuclear translocation of beta-catenin and increases expression of c-myc and Abcb1a in kidney proximal tubule cellsBiometals200720580782017136310
- AnastasJNMoonRTWNT signalling pathways as therapeutic targets in cancerNat Rev Cancer2013131112623258168
- YangXLiLHuangQWnt signaling through Snail1 and Zeb1 regulates bone metastasis in lung cancerAm J Cancer Res20155274875525973312
- NovellasdemuntLAntasPLiVSTargeting Wnt signaling in colorectal cancer. A Review in the Theme: Cell Signaling: Proteins, Pathways and MechanismsAm J Physiol Cell Physiol20153098C511C52126289750
- DimeoTAAndersonKPhadkePA novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancerCancer Res200969135364537319549913
- TetsuOMcCormickFBeta-catenin regulates expression of cyclin D1 in colon carcinoma cellsNature1999398672642242610201372
- JeongWJYoonJParkJCRas stabilization through aberrant activation of Wnt/β-catenin signaling promotes intestinal tumorigenesisSci Signal20125219ra3022494971
- PolakisPWnt signaling in cancerCold Spring Harb Perspect Biol201245a00805222438566
- LuevanoJDamodaranCA review of molecular events of cadmium-induced carcinogenesisJ Environ Pathol Toxicol Oncol201433318319425272057
- LinYCYouLXuZWnt signaling activation and WIF-1 silencing in nasopharyngeal cancer cell linesBiochem Biophys Res Commun2006341263564016427602
- AmitSHatzubaiABirmanYAxin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathwayGenes Dev20021691066107612000790